Introduction
Atherosclerosis is characterized by the accumulation
of lipids, mainly cholesterol and cholesterol esters, and the
infiltration of inflammatory cells, particularly macrophages.
Recent experimental and clinical evidence suggest that inflammation
plays a significant role in lipid-mediated cell injury (e.g.,
atherosclerosis) (1–4). Retention of lipoproteins in the
artery wall is one of the primary events in atherosclerosis
(5,6). In the vessel wall, low-density
lipoprotein (LDL) is trapped by matrix proteoglycans and becomes
susceptible to various modifications that trigger local
inflammatory responses leading to recruitment of macrophages in the
arterial wall (7). Uptake of
modified LDL by macrophages leads to foam cell formation, which is
characteristic of early atherosclerotic lesions (8).
Cardiovascular risk is increased in chronic
inflammatory states, up to 33-fold in patients with renal failure
and 50-fold in patients with immune dysregulation (9–12).
These diseases are often associated with inflammatory stress with
elevated inflammatory markers and inflammatory cytokines. However,
most mechanisms linking atherosclerosis to inflammatory stress
remain to be identified. Inflammation and infection may affect the
plasma levels of cholesterol and lipoproteins by modulating the
synthesis and secretion of apolipoproteins, the activity of
lipolytic enzyme or the expression of lipoprotein receptors. Two of
the major determinants of plasma cholesterol levels are the result
of the activity of the scavenger receptor A (SR-A), mediating
uptake of modified LDL and LDL receptor (LDL-R), and mediating
uptake of native LDL in macrophages. SR-A is expressed in human
atherosclerotic lesions (13,14),
and they have been shown to contribute to the uptake of modified
LDL (15). Since this uptake of
cholesterol through the macrophage scavenger receptors is poorly
regulated, excess cholesterol accumulation leads to
macrophage-derived foam cell formation. Macrophages play a very
important role in the pathogenesis of atherosclerosis and are one
of the major cell types involved in foam cell formation (16). However, the role of LDL-R and SR-A
receptor in macrophages under inflammation remains unclear. The
present experiment set out to investigate the effects of LPS on
cholesterol accumulation in macrophages, and the expression of
LDL-R and SR-A genes and proteins in the lipopolysaccharide
(LPS)-stimulated macrophage-like RAW264.7 cell line in the presence
of a high dose of native LDL.
Materials and methods
Cell culture
The macrophage-like RAW264.7 cell line was cultured
in growth medium containing RPMI-1640 medium, 10% newborn bovine
serum, 2 mmol/l glutamine, 100 U/ml penicillin and 100 μg/ml
streptomycin. All experiments were carried out in serum-free
RPMI-1640 medium containing 0.2% BSA, 2 mmol/l glutamine, 100 U/ml
penicillin and 100 μg/ml streptomycin. All reagents for cell
culture and LPS were obtained from Sigma. LDL was isolated from
healthy human plasma by sequential ultra-centrifugation.
Enzyme-linked immunosorbent assay (ELISA)
for tumor necrosis factor α (TNF-α)
Cultures of RAW264.7 cells were washed twice with
PBS at 37°C and then overlaid with 1 ml of fresh, serum-free
RPMI-1640 medium. After an incubation of 24 h, an additional 1 ml
of medium with or without 100 ng/ml LPS was added. Cells were
incubated for the desired time periods. The concentrations of TNF-α
in cell culture medium were determined by an ELISA assay kit
(eBioscience, UK) according to the manufacturer’s instructions.
Measurement of intracellular
cholesterol
The method was based on the cholesterol enzymatic
assay described by Gallo et al and Gamble et
al(17,18). RAW264.7 cells in 6-well plates were
cultured in serum-free medium without (control) or with 100 μg/ml
LDL in the absence or presence of 100 ng/ml LPS for 24 h. Cells
were then washed twice in PBS, intracellular lipids were extracted
in isopropanol and dried under vacuum, and total cholesterol (TC),
free cholesterol (FC) and cholesterol ester (CE) content were
measured by enzymatic assay (CE = TC - FC) and normalized by total
cell proteins determined by the modified Lowry assay.
Oil Red O stain
RAW264.7 cells were incubated in serum-free medium
for 24 h. Then, these cells were differentially treated. After 24
h, cells were fixed using 5% formalin for 30 min at room
temperature, soaked in Oil Red O staining solution for 30 min at
room temperature and washed three times, followed by hematoxylin
stain to visualize the nucleus.
Total RNA isolation and semi-quantitative
RT-PCR
Total RNA was isolated from cultured cells using
TRIzol reagent (Promega, USA). Total RNA (500 ng) was used as a
template for RT using an RT kit from Toyoba. The RT reaction was
set up in a 20-μl mixture containing 50 mmol/l KCl, 10 mmol/l
Tris-HCl, 5 mmol/l MgCl2, 1 mmol/l of each
deoxynucleoside triphosphate, 2.5 μmol/l random hexamers, 20 units
RNAase inhibitor and 50 units Moloney-murine leukemia virus RT.
Incubations were performed in a DNA Thermal Cycler (Eppendorf, USA)
for 10 min at room temperature, followed by 30 min at 42°C and 5
min at 99°C. Semi-quantitative PCR was performed in a DNA Thermal
Cycler (Eppendorf) using PCR Master mix (DongSheng Biotec Co.,
China). Thermal cycler conditions contained holds for 10 min at
95°C, followed by 30 cycles of 30 sec at 95°C, 30 sec at 51°C and
45 sec at 72°C. The PCR products were separated by electrophoresis
and the relative amount of mRNA was calculated using the Genegenus
system and GeneTools software version 3.05 (Synoptics, UK). GAPDH
served as the reference housekeeping gene. The following
oligonucleotide primers were used: SR-A upper
5′-TCAATGACAGCATCCCTTCC-3′, lower 5′-ATGTCCTCCTGTTGCTTTGC-3′; LDL-R
upper 5′-TTGCAGTAGAAGACTCAGGC-3, lower 5′-ATGATT TGCAGCGGAAGTGG-3′;
GAPDH upper 5′-ATTCAACGG CACAGTCAA-3′, lower
3′-TGAGGGTGAGAAGGTGGAA-5′.
Western blot analysis
Cells from a 150-ml culture bottle were pooled and
allowed to swell at 4°C in 300 μl of lysis buffer (10 mmol/l HEPES,
pH 7.9, 10 mmol/l KCL, 1.5 mmol/l MgCl2, 0.5 mmol/l
dithiothreitol, 0.4 Nonidet P-40, 0.5 μmol/l phenylmethylsulfonyl
fluoride and 1 μg/ml of antipain, leupeptin, bestatin and
chymostatin) and then passed through a 23-gauge needle 20 times
before centrifugation at 14,000 × g at 4°C for 15 min. The
supernatant from this spin was used as the whole cell extract.
Identical amounts of total protein from the whole cell extract were
denatured and then subjected to electrophoresis on a 5% stacking
and 8% separating SDS polyacrylamide gel in a Bio-Rad Mini Protein
apparatus. Electrophoretic transfer to nitrocellulose was
accomplished at 60 V, 200 mA for 2 h in 25 mmol/l Tris, pH 8.3, 192
mmol/l glycine, 0.1% SDS and 20% methanol. The membrane was then
blocked with 5% skimmed milk for 1 h at room temperature, followed
by two 5-min washes in PBST (phosphate-buffered saline/1% Tween).
The membrane was incubated with the first antibody (goat-anti SR-A,
or rabbit-anti LDL-R, rabbit-anti SREBP-2; Santa Cruz, USA) for 2 h
at 37°C in antibody dilution buffer (1% BSA in PBST). The
horseradish peroxidase-labeled secondary antibody (rabbit
anti-goat, goat-anti rabbit; Santa Cruz) was diluted in antibody
dilution buffer and then added to the membrane for 1 h at 37°C,
followed by three 5-min washes in PBST. Finally, detection
procedures were performed using ECL Advance Western Blotting
Detection kit, in a Genegnome system (Synoptics). Band intensity
volumes were measured by Quantity One software (Bio-Rad, UK).
Immunocytochemical staining
RAW264.7 cells were fixed using 5% formalin for 30
min at room temperature, and then washed three times, followed by
immunocytochemical staining according to the kit (Boshide
Bio-Project Co., Wuhan, China) instructions. The first antibody was
rabbit anti-rabbit LDL-R antibody. Phosphate-buffered saline (PBS)
substituted for the first antibody was used as the negative
control. Cells with brown granules appearing in the nuclei were
considered to be positively stained and indicated activation of
LDL-R.
Statistical analysis
In the experiments, data are expressed as the means
± standard deviation (SD). Comparisons between two groups were
performed by t-test. All analyses were carried out using the SPSS
software (version 13.0; SPSS Inc., Chicago, IL, USA). P-values
<0.05 were considered to indicate statistical significance.
Results
Expression levels of TNF-α in ELISA
We examined LPS-dependent TNF-α secretion in
RAW264.7 cells to determine whether LPS induces TNF-α secretion in
a time-dependent manner and whether LPS causes inflammatory stress.
LPS-treated RAW264.7 cells secreted TNF-α, with levels beginning to
increase at 4 h of incubation and reaching a maximum at 16 h and
lasting until 48 h.
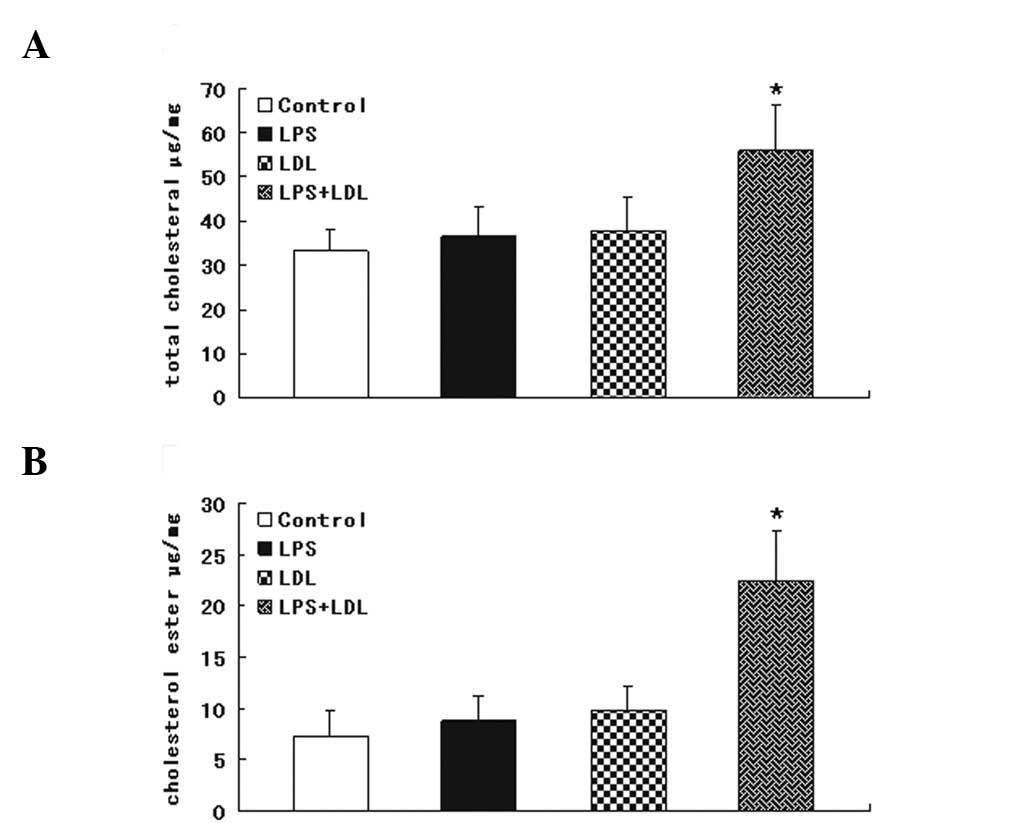
Intracellular cholesterol accumulation in
the cholesterol enzymatic assay
We examined the effect of LPS (100 ng/ml) on the
intracellular cholesterol content in RAW264.7 cells. Inflammation
resulted in a TC and CE accumulation and foam cell formation in
RAW264.7 cells, as evidence by the intracellular total cholesterol
assay (Fig. 1A) and cholesterol
ester assay (Fig. 1B).
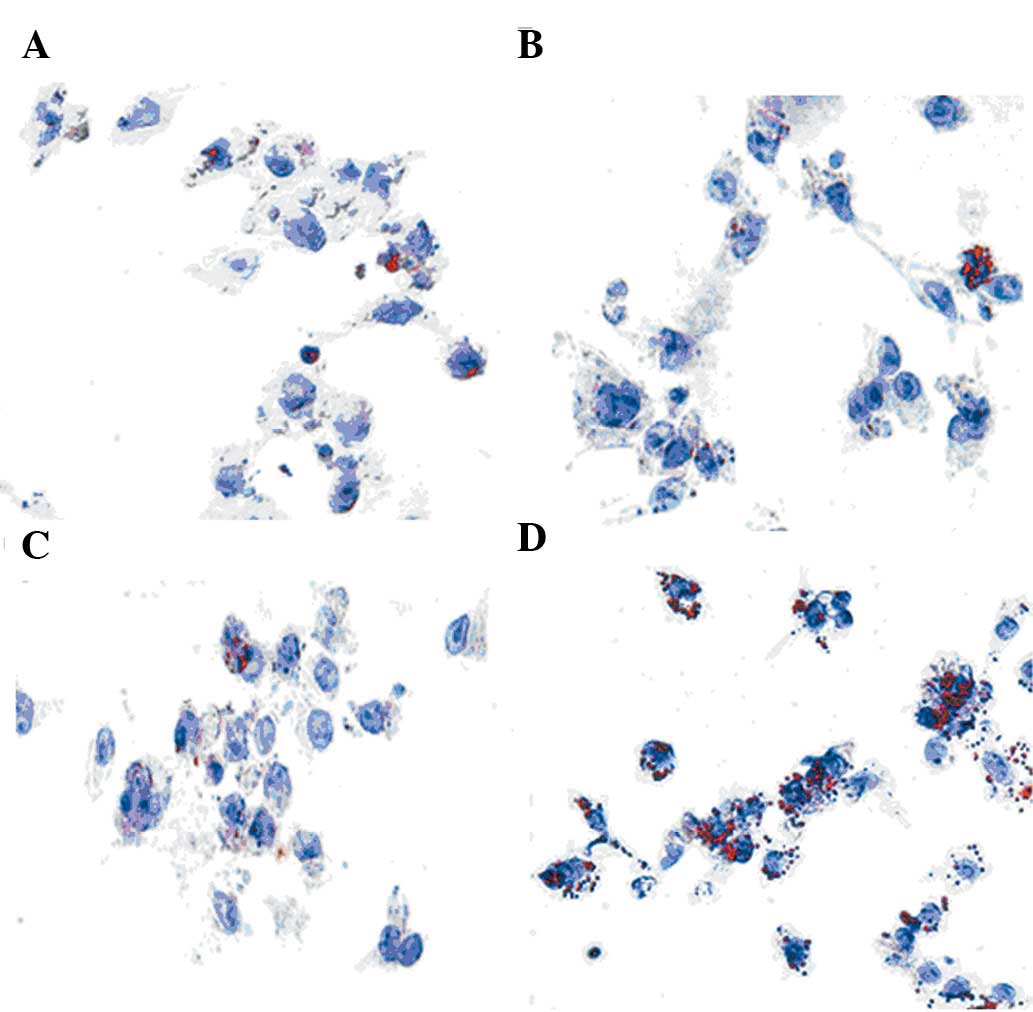
Intracellular cholesterol accumulation as
assessed by Oil Red O staining
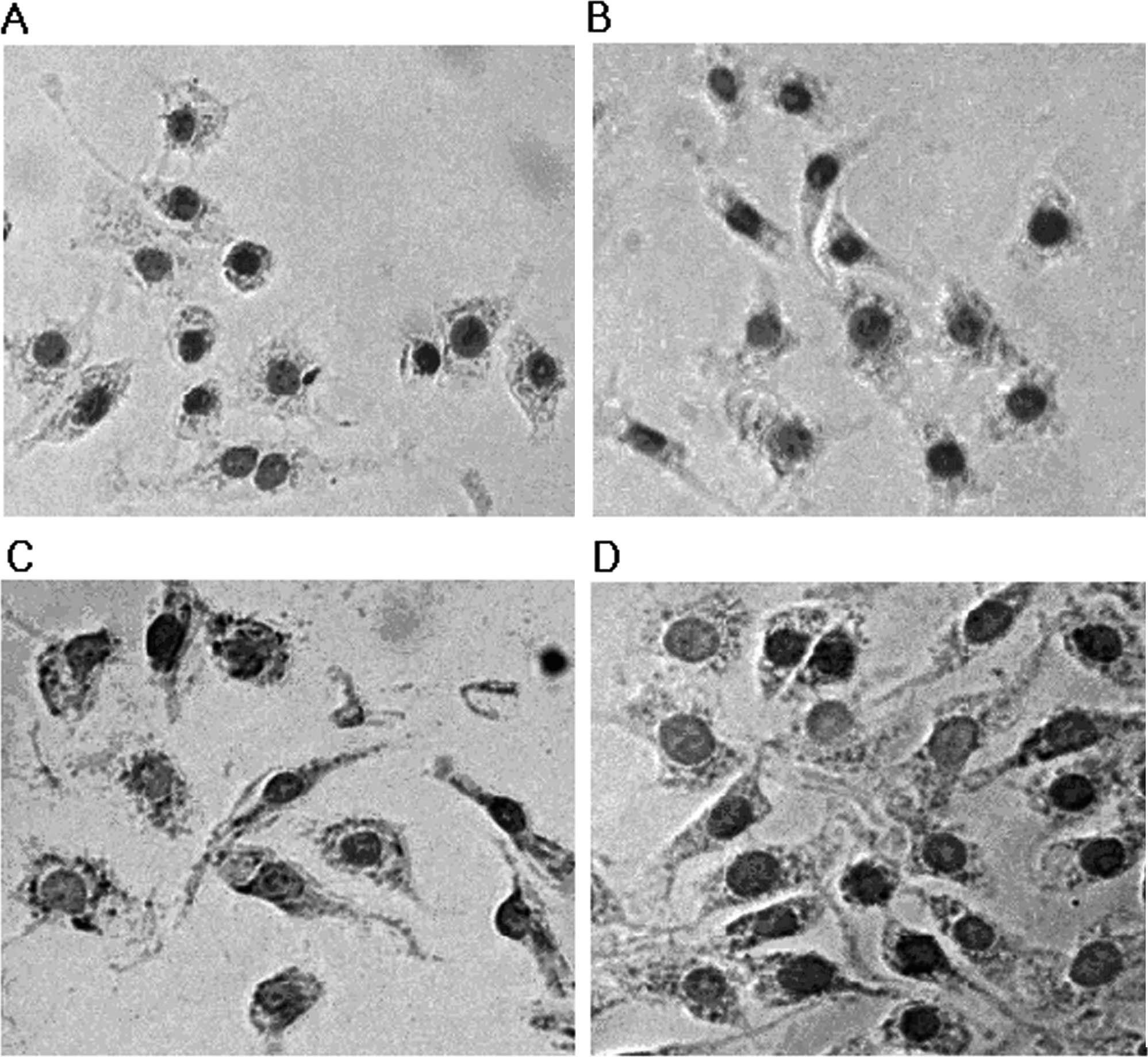
Staining of RAW264.7 cells with Oil Red O after
incubation with 100 μg/ml LDL or 100 ng/ml LPS alone showed a
slight increase in lipid droplets over that of the control group
(Fig. 2A-C). However, a
significant increase in cholesterol accumulation was observed in
RAW264.7 cells in the presence of 100 μg/ml LDL and 100 ng/ml LPS
together (Fig. 2D). These data
were consistent with the cholesterol enzymatic assay data.
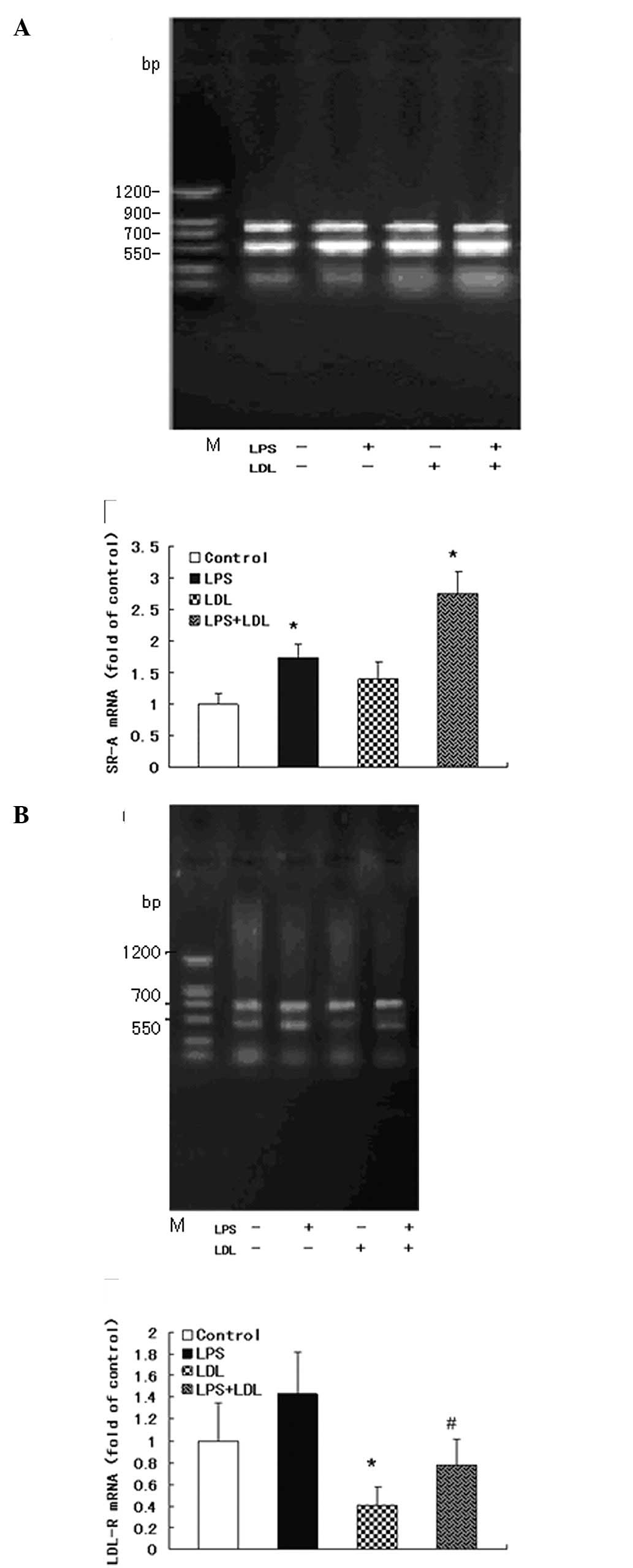
Expression levels of LDL-R and SR-A mRNA
in RT-PCR
To assess how LPS induces an increase in
intracellular cholesterol content, the mRNA and protein levels of
LDL-R and SR-A in RAW264.7 cells were evaluated. RAW264.7 cells
were stimulated with 100 μg/ml LDL, 100 ng/ml LPS, or with 100
μg/ml LDL and 100 ng/ml LPS, respectively, for up to 24 h. mRNA was
measured by semi-quantitative RT-PCR. Either LDL loading or LPS
increased SR-A mRNA expression in RAW264.7 cells (Fig. 3A). LDL loading inhibited LDL-R mRNA
expression and LPS disrupted this physiological feedback causing an
increase in LDL-R mRNA levels in RAW264.7 cells (Fig. 3B).
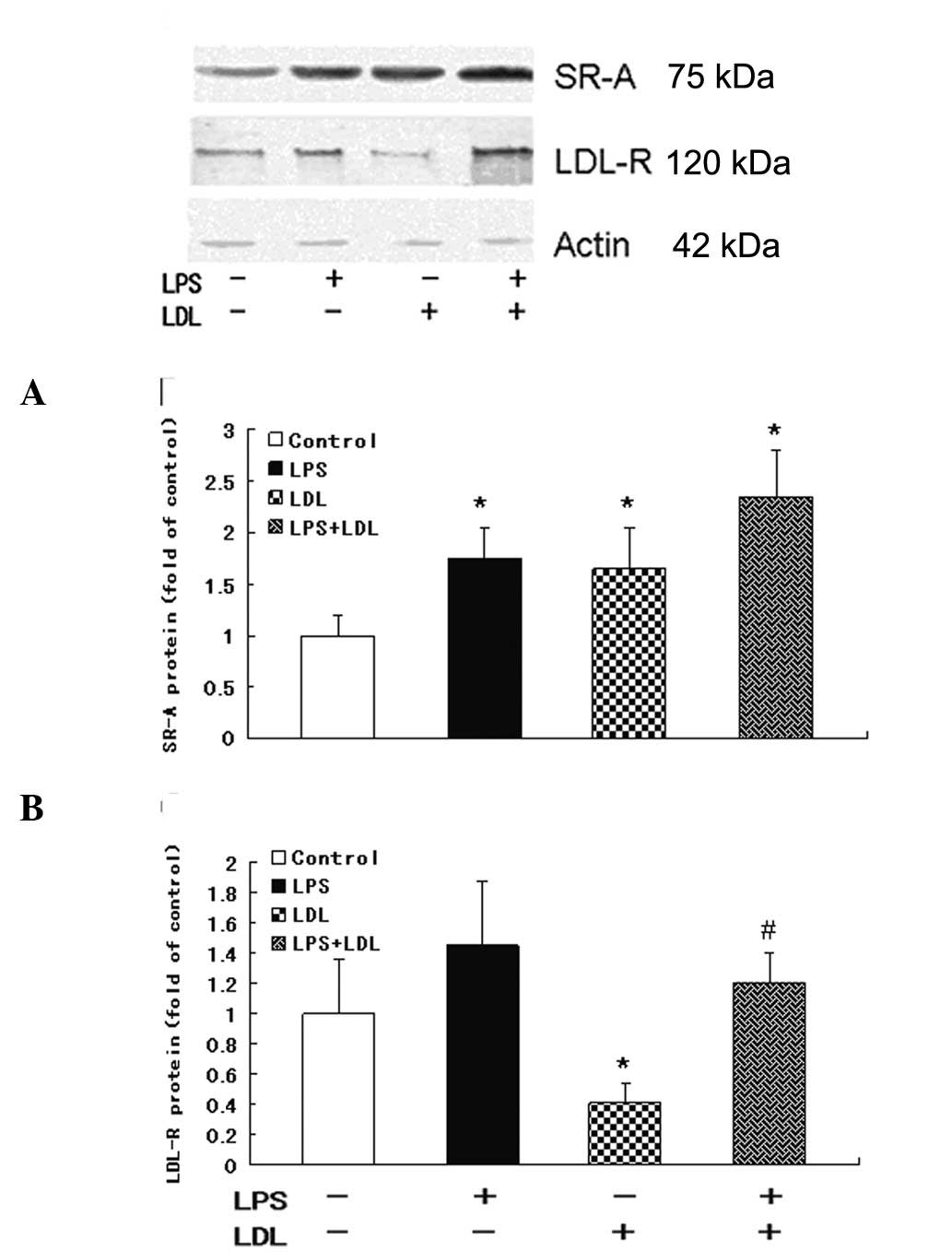
Western blot analysis of the expression
levels of LDL-R and SR-A protein
Western blotting demonstrated that LDL loading or
LPS increased SR-A protein expression in RAW264.7 cells (P<0.05)
(Fig. 4A). LDL loading inhibited
LDL-R protein levels (vs. control, P<0.05) and LPS increased
LDL-R levels in RAW264.7 cells (vs. LDL loading group, P<0.05)
(Fig. 4B). These data were
consistent with mRNA data.
Immunocytochemical analysis of LDL-R
protein expression
Immunocytochemical staining showed that LDL
inhibited LDL-R protein expression and LPS (100 ng/ml) overrode the
suppression of the LDL-R protein expression induced by LDL loading
(Fig. 5).
Discussion
As foam cell formation has long been deemed critical
to lesion development, the scavenger receptors involved in lipid
uptake have been causally implicated in the pathogenesis of
atherosclerosis. Several studies have shown that macrophages are
deemed to cause foam cells, have multiple functions, including
secretion and production of growth factors, cytokines, clearance of
particles in blood, and also play a key role in atherosclerosis by
expression of several different cell surface receptors that can be
modulated by inflammation, for cholesterol uptake and foam cell
formation (16,19). SR-A is an important cell surface
receptor for binding modified-LDL and plays a central role in
atherosclerosis. LDL-R is the primary receptor for binding and
internalization of plasma-derived LDL cholesterol and regulates
plasma LDL concentrations. With respect to macrophages, several
studies, although not all, have shown the effect of SR-A or LDL-R
alone on foam cell formation under inflammation, however, reports
on the synergic effect of LDL-R and SR-A on macrophage foam cell
formation are limited. In the present study, we mainly focus on
receptor-mediated macrophage-derived foam cell formation under
LPS-induced inflammatory stress.
SR-A, as a classical atherogenic receptor (20–22),
has been studied for decades. Scavenger receptor was coined to
describe the activity of macrophages which mediate the uptake of
modified forms of LDL. As a central feature of the pathology of
atherosclerosis, macrophage-derived foam cells are closely
resembled by the resultant lipid laden macrophages (23). The class A scavenger receptor was
shown to be a trimeric Type II membrane protein with a broad range
of polyanionic ligands, including components of Gram-positive
bacterial cell walls and modified forms of LDL, LPS of
Gram-negative bacteria (24).
Compared to macrophages from wild-type animals, macrophages of
SR-A-silenced mice exhibit 70–80% reduced uptake of acetylated LDL
(25). Certain studies also have
shown that inflammatory cytokines mediate the regulation of SR-A
and foam cell formation on macrophages in atherosclerosis (26), as well as LPS induces SR-A
expression in RAW264.7 cells (27). However, more and more studies
suggest that SR-A is not the only receptor responsible for
atherosclerosis.
It is well known that the high incidence of
atherosclerosis is correlated with the high concentration of LDL
and cholesterol in plasma. The activity of the LDL-R is another
major determinant of plasma cholesterol levels. The cellular uptake
and degradation of plasma is mediated by the LDL-R. It has been
reported that the activity of LDL-R is normally under tight
metabolic control via a feedback system, which is dependent on
intracellular cholesterol concentration (28–31).
The system maintains a constant level of cholesterol in cells by
controlling both the rate of cholesterol synthesis and the rate of
cholesterol uptake from LDL. The feedback regulation is controlled
through specific interactions of the sterol-regulatory element
(SRE)-1 of the LDL-R promoter and a family of SRE-binding proteins
(SREBPs), namely, SREBP-1 and SREBP-2 (32–35).
Moreover, a number of pro-inflammatory cytokines, such as
interleukin-6, oncostatin M and TNF-α, have been shown to increase
LDL-R gene expression under inflammatory stress (36–38).
We previously demonstrated in human vascular smooth muscle cells
and mesangial cells that inflammatory cytokines disrupt LDL-R
feedback regulation, allowing unregulated uptake of cholesterol in
the cells causing foam cell formation (39–42).
However, the role of the LDL-R in cholesterol accumulation in
macrophages remains to be elucidated.
In the present study, we investigated the effects of
native LDL that consist mostly of cholesterol of plasma in
circulation and macrophage-derived foam cells under inflammatory
stress, and evaluated the roles of SR-A and LDL-R in this process.
We demonstrated that LPS causes inflammatory stress and induces
TNF-α secretion in a time-dependent manner consequently. LPS
increased intracellular cholesterol accumulation in the RAW264.7
macrophage-like cell line (Fig. 1)
and caused foam cell formation (Fig.
2). To explore the molecular mechanisms, we further determined
the expression levels of LDL-R and SR-A mRNA and protein in
RAW264.7 cells and found that, under physiological conditions,
LDL-R was sensitive for downregulation of LDL loading.
Intracellular TC and CE contents remained at a relatively constant
level in the RAW264.7 cells. However, inflammation overrode this
tight feedback and caused foam cell formation via increased
expression of both SR-A and LDL-R (Figs. 3–5). The results of our study indicate that
the synergy of dysregulation of LDL-R under inflammatory stress and
upregulation of SR-A may contribute to macrophage-derived foam cell
formation.
In conclusion, our findings indicate that a novel
role for the synergy of upregulation of SR-A and dysregulation of
LDL-R plays a significant role in macrophage foam cell formation
under inflammatory stress.
Acknowledgements
The authors acknowledge the support of the National
Natural Science Foundation of China (nos. 30530360 and
30772098).
References
|
1
|
Libby P, Ridker PM and Hansson GK:
Inflammation in atherosclerosis: from pathophysiology to practice.
J Am Coll Cardiol. 54:2129–2138. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Galkina E and Ley K: Immune and
inflammatory mechanisms of atherosclerosis. Annu Rev Immunol.
27:165–197. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Krupinski J, Font A, Luque A, Turu M and
Slevin M: Angiogenesis and inflammation in carotid atherosclerosis.
Front Biosci. 13:6472–6482. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tahara N, Imaizumi T, Virmani R and Narula
J: Clinical feasibility of molecular imaging of plaque inflammation
in atherosclerosis. J Nucl Med. 50:331–334. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Williams KJ and Tabas I: The
response-to-retention hypothesis of early atherogenesis.
Arterioscler Thromb Vasc Biol. 15:551–561. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ylä-Herttuala S, Solakivi T, Hirvonen J,
et al: Glycosaminoglycans and apolipoproteins B and A-I in human
aortas: chemical and immunological analysis of lesion-free aortas
from children and adults. Arteriosclerosis. 7:333–340.
1987.PubMed/NCBI
|
|
7
|
Steinberg D, Parthasarathy S, Carew TE, et
al: Beyond cholesterol. Modifications of low-density lipoprotein
that increase its atherogenicity. N Engl J Med. 320:915–924.
1989.PubMed/NCBI
|
|
8
|
Li AC and Glass CK: The macrophage foam
cell as a target for therapeutic intervention. Nat Med.
8:1235–1242. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pennant M, Davenport C, Bayliss S,
Greenheld W, Marshall T and Hyde C: Community programs for the
prevention of cardiovascular disease: a systematic review. Am J
Epidemiol. 172:501–516. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bash LD, Erlinger TP, Coresh J,
Marsh-Manzi J, Folsom AR and Astor BC: Inflammation, hemostasis,
and the risk of kidney function decline in the Atherosclerosis Risk
in Communities (ARIC) Study. Am J Kidney Dis. 53:596–605. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sherer Y, Zinger H and Shoenfeld Y:
Atherosclerosis in systemic lupus erythematosus. Autoimmunity.
43:98–102. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Doria A: Atherosclerosis and lupus: what
we know and what we should know. J Rheumatol. 36:2380–2382. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ylä-Herttuala S, Rosenfeld ME,
Parthasarathy S, et al: Gene expression in macrophage-rich human
atherosclerotic lesions: 15-lipoxygenase and acetyl low density
lipoprotein receptor messenger RNA colocalize with oxidation
specific lipid-protein adducts. J Clin Invest. 87:1146–1152.
1991.
|
|
14
|
Kunjathoor VV, Febbraio M, Podrez EA, et
al: Scavenger receptors class A-I/II and CD36 are the principal
receptors responsible for the uptake of modified low density
lipoprotein leading to lipid loading in macrophages. J Biol Chem.
277:49982–49988. 2002. View Article : Google Scholar
|
|
15
|
Suzuki H, Kurihara Y, Takeya M, et al: A
role for macrophage scavenger receptors in atherosclerosis and
susceptibility to infection. Nature. 386:292–296. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shibata N and Glass CK: Regulation of
macrophage function in inflammation and atherosclerosis. J Lipid
Res. 50:S277–S281. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gallo LL, Atasoy R, Vahouny GV and
Treadwell CR: Enzymatic assay for cholesterol ester hydrolase
activity. J Lipid Res. 19:913–916. 1978.PubMed/NCBI
|
|
18
|
Gamble W, Vaughan M, Kruth HS and Avigan
J: Procedure for determination of free and total cholesterol in
micro- or nanogram amounts suitable for studies with cultured
cells. J Lipid Res. 19:1068–1070. 1978.PubMed/NCBI
|
|
19
|
Saha P, Modarai B, Humphries J, Mattock K,
Waltham M, Burnand KG and Smith A: The monocyte/macrophage as a
therapeutic target in atherosclerosis. Curr Opin Pharmacol.
9:109–118. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jozefowski S, Arredouani M, Sulahian T and
Kobzik L: Disparate regulation and function of the class A
scavenger receptors SR-AI/II and MARCO. J Immunol. 175:8032–8041.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Neyen C, Plüddemann A, Roversi P, Thomas
B, Cai L, van der Westhuyzen DR, Sim RB and Gordon S: Macrophage
scavenger receptor A mediates adhesion to apolipoproteins A-I and
E. Biochemistry. 48:11858–11871. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Greaves DR and Gordon S: The macrophage
scavenger receptor at 30 years of age: current knowledge and future
challenges. J Lipid Res. 50:S282–S286. 2009.PubMed/NCBI
|
|
23
|
Osto E, Kouroedov A, Mocharla P, Akhmedov
A, Besler C, Rohrer L, von Eckardstein A, Iliceto S, Volpe M,
Lüscher TF and Cosentino F: Inhibition of protein kinase Cbeta
prevents foam cell formation by reducing scavenger receptor A
expression in human macrophages. Circulation. 118:2174–2182. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Plüddemann A, Neyen C and Gordon S:
Macrophage scavenger receptors and host-derived ligands. Methods.
43:207–217. 2007.
|
|
25
|
Van Berkel TJ, Van Eck M, Herijgers N,
Fluiter K and Nion S: Scavenger receptor classes A and B: their
roles in atherogenesis and the metabolism of modified LDL and HDL.
Ann NY Acad Sci. 902:113–126. 2000.PubMed/NCBI
|
|
26
|
Li K, Yao W, Zheng X and Liao K: Berberine
promotes the development of atherosclerosis and foam cell formation
by inducing scavenger receptor A expression in macrophage. Cell
Res. 19:1006–1017. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Moore KJ and Freeman MW: Scavenger
receptors in atherosclerosis: beyond lipid uptake. Arterioscler
Thromb Vasc Biol. 26:1702–1711. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Goldstein JL and Brown MS: The LDL
receptor. Arterioscler Thromb Vasc Biol. 29:431–438. 2009.
View Article : Google Scholar
|
|
29
|
Mazière C and Mazière JC: Activation of
transcription factors and gene expression by oxidized low-density
lipoprotein. Free Radic Biol Med. 46:127–137. 2009.PubMed/NCBI
|
|
30
|
Issandou M: Pharmacological regulation of
low density lipoprotein receptor expression: current status and
future developments. Pharmacol Ther. 111:424–433. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vainio S and Ikonen E: Macrophage
cholesterol transport: a critical player in foam cell formation.
Ann Med. 35:146–155. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sato R: Sterol metabolism and SREBP
activation. Arch Biochem Biophys. 501:177–181. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Osborne TF and Espenshade PJ: Evolutionary
conservation and adaptation in the mechanism that regulates SREBP
action: what a long, strange tRIP it’s been. Genes Dev.
23:2578–2591. 2009.PubMed/NCBI
|
|
34
|
Bengoechea-Alonso MT and Ericsson J: SREBP
in signal transduction: cholesterol metabolism and beyond. Curr
Opin Cell Biol. 19:215–222. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shimano H: SREBPs: physiology and
pathophysiology of the SREBP family. FEBS J. 276:616–621. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu J, Zhang F, Li C, Lin M and Briggs MR:
Synergistic activation of human LDL receptor expression by SCAP
ligand and cytokine oncostatin M. Arterioscler Thromb Vasc Biol.
23:90–96. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gierens H, Nauck M, Roth M, Schinker R,
Schürmann C, Scharnagl H, Neuhaus G, Wieland H and März W:
Interleukin-6 stimulates LDL receptor gene expression via
activation of sterol-responsive and Sp1 binding elements.
Arterioscler Thromb Vasc Biol. 20:1777–1783. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hansson GK: Inflammatory mechanisms in
atherosclerosis. J Thromb Haemost. 7(Suppl 1): 328–331. 2009.
View Article : Google Scholar
|
|
39
|
Ye Q, Chen Y, Lei H, Liu Q, Moorhead JF,
Varghese Z and Ruan XZ: Inflammatory stress increases unmodified
LDL uptake via LDL receptor: an alternative pathway for macrophage
foam-cell formation. Inflamm Res. 58:809–818. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen Y, Ruan XZ, Li Q, et al: Inflammatory
cytokines disrupt LDL-receptor feedback regulation and cause statin
resistance: a comparative study in human hepatic cells and
mesangial cell. Am J Physiol Renal Physiol. 293:F680–F687. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ruan XZ, Moorhead JF, Fernando R, Wheeler
DC, Powis SH and Varghese Z: Mechanisms of dysregulation of
low-density lipoprotein receptor expression in vascular smooth
muscle cells by inflammatory cytokines. Arterioscler Thromb Vasc
Biol. 26:1150–1155. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ruan XZ, Varghese Z, Powis SH and Moorhead
JF: Dysregulation of LDL receptor under the influence of
inflammatory cytokines: a new pathway for foam cell formation.
Kidney Int. 60:1716–1725. 2001. View Article : Google Scholar : PubMed/NCBI
|