Introduction
Small supernumerary marker chromosomes (sSMCs) have
been defined as structurally abnormal chromosomes that cannot be
identified or characterized unambiguously by conventional banding
cytogenetics alone, and are (in general) equal in size or smaller
than chromosome 20 of the same metaphase spread. As they are too
small for their chromosomal origin to be considered by traditional
banding techniques, molecular cytogenetic techniques (including
array-based comparative genomic hybridization) are required for
their characterization (1). sSMCs
are found in approximately 4.4/100,000 newborns (2) and they are found in three different
shapes: ring, inverted duplicated and centric minute (3). Partial trisomy 7 or partial monosomy
7 share some common clinical features, such as mental retardation,
growth deficiency and finger abnormalities as well as eye and ear
anomalies. In addition, certain patients present variable features,
such as triangular face, high frontal hairline, high arched palate,
clinodactyly and asymmetry of limbs (4).
Uniparental disomy (UPD), the abnormal inheritance
of both chromosomes from only one parent, has been described for
different human chromosomes. UPD can be associated with an sSMC as
a result of an original trisomy with consecutive trisomic rescue
(5).
Congenital adrenal hyperplasia (CAH) is one of the
most common autosomal recessive disorders, with an estimated
carrier frequency of 1 in 50 (6),
and is due to 21-hydroxylase deficiency (21-OHD), one of the
enzymes required for the synthesis of cortisol in the adrenal
cortex. Impaired 21-hydroxylase activity causes accumulation of
steroid precursors, which then flow into biosynthetic pathways
unaffected by the enzymatic block, resulting in the production of
excess androgens. When androgens are over-secreted from the adrenal
glands, the female fetus is virilized (7). Complete enzyme inactivation or low
but measurable enzyme activity leads to the salt wasting (SW) form,
3–7% residual enzyme activity leads to the simple virilizing type
and more than 30% residual enzyme activity leads to the non-classic
type (7). Approximately 75% of
patients with classic 21-OHD have the SW form, which is associated
with severe impairment of 21-hydroxylation of progesterone and
17-hydroxyprogesterone (17-OHP) (7).
The 21-hydroxylase gene, CYP21 (CYP21A2, OMIM No.
201910), is located on chromosome 6p21.3 within the HLA
histocompatibility complex in close proximity to the highly
homologous inactive pseudogene, CYP21P (CYP21A1P) (8,9).
Recombinations and conversions between CYP21A1P and CYP21A2 result
in the generation of dysfunctional CYP21A2 alleles (7,10).
Approximately 90% of CYP21A2 mutations are a result of
CYP21A1P-derived conversions and recombinations (7,11).
CYP21A2 genotyping can be useful in the diagnosis of CAH and can
also predict the phenotype in 80–90% of cases (12,13).
In this study, we present a female
pseudohermaphroditism/adrenogenital syndrome (AGS) case with a 45%
mosaicism for a small supernumerary ring chromosome 7 associated
with a homozygous mutation in the CYP21A2 gene.
Materials and methods
Case report
The patient is the sixth child of non-related
parents. Her mother was 35 and her father 49 years of age when she
was born. She is now 10 years old, 135 cm in height, and was
referred to a cytogenetic analysis due to a gender determination
problem in July 2009. She was born in the 37th week of gestation
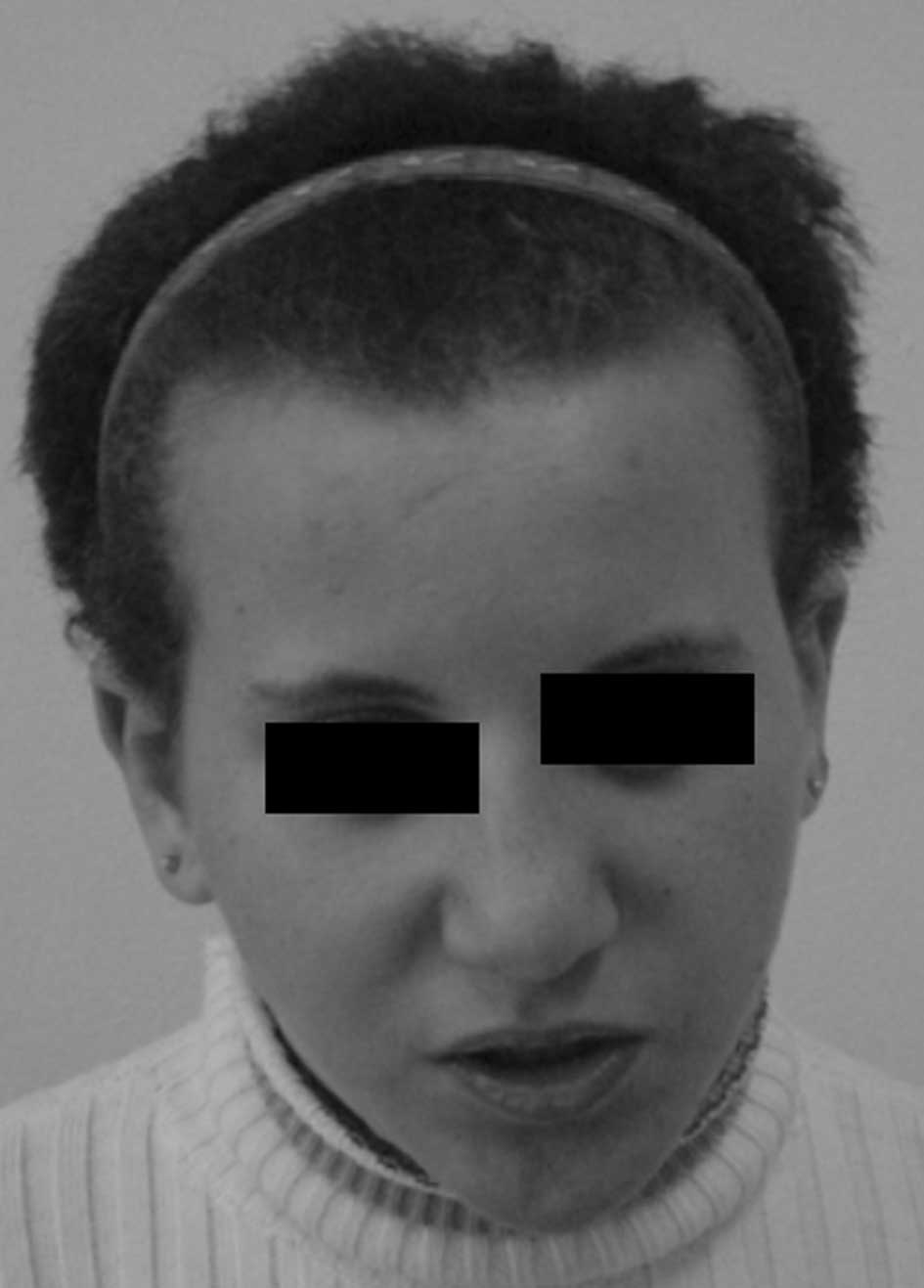
with a birth weight of 3,250 g. She has a facial dysmorphism
(triangular face, high frontal hairline, asymmetry of limbs
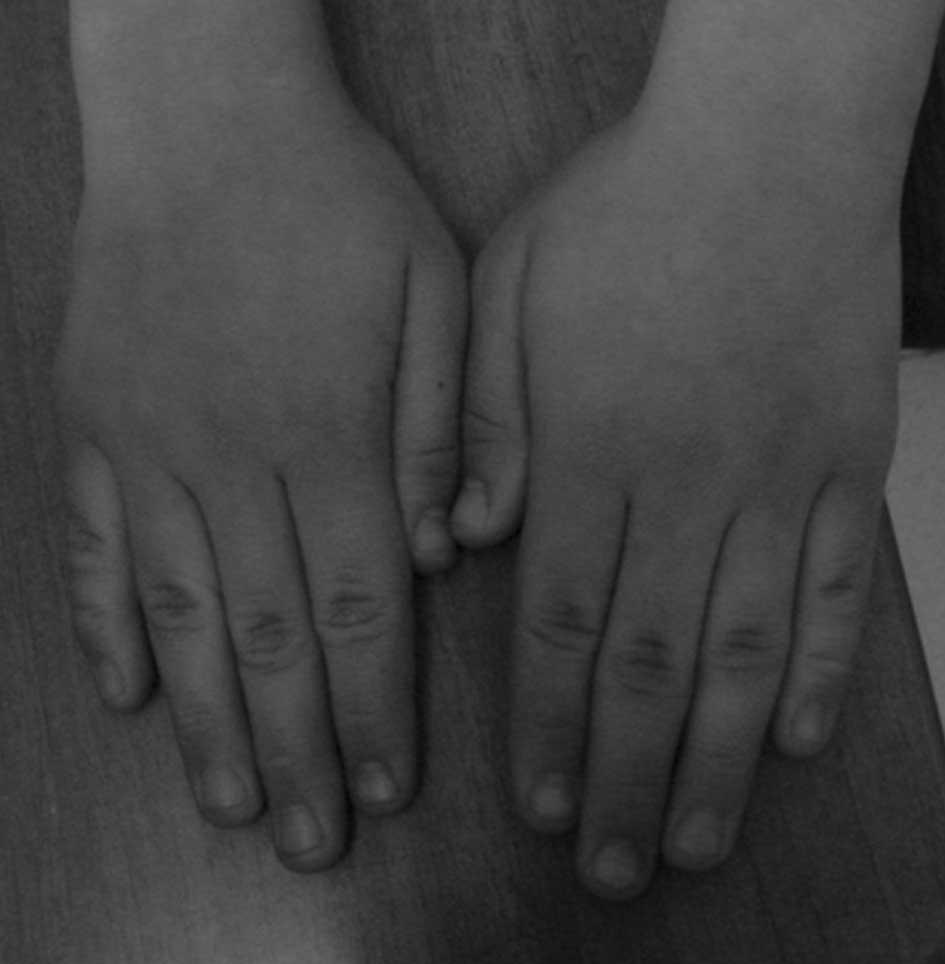
(Fig. 1) and long fingers
(Fig. 2). Her speech was delayed;
her first words were not until 2 years of age, and her language
skills were poor until 8 years, especially concerning her
performances on the expressive side. She has a uterus without
ovaries, dense hair distribution on her body (especially in the
pubic area), a large genital vagina; the clitoris is very
pronounced (5 cm in length) and lift cornet. However, she does not
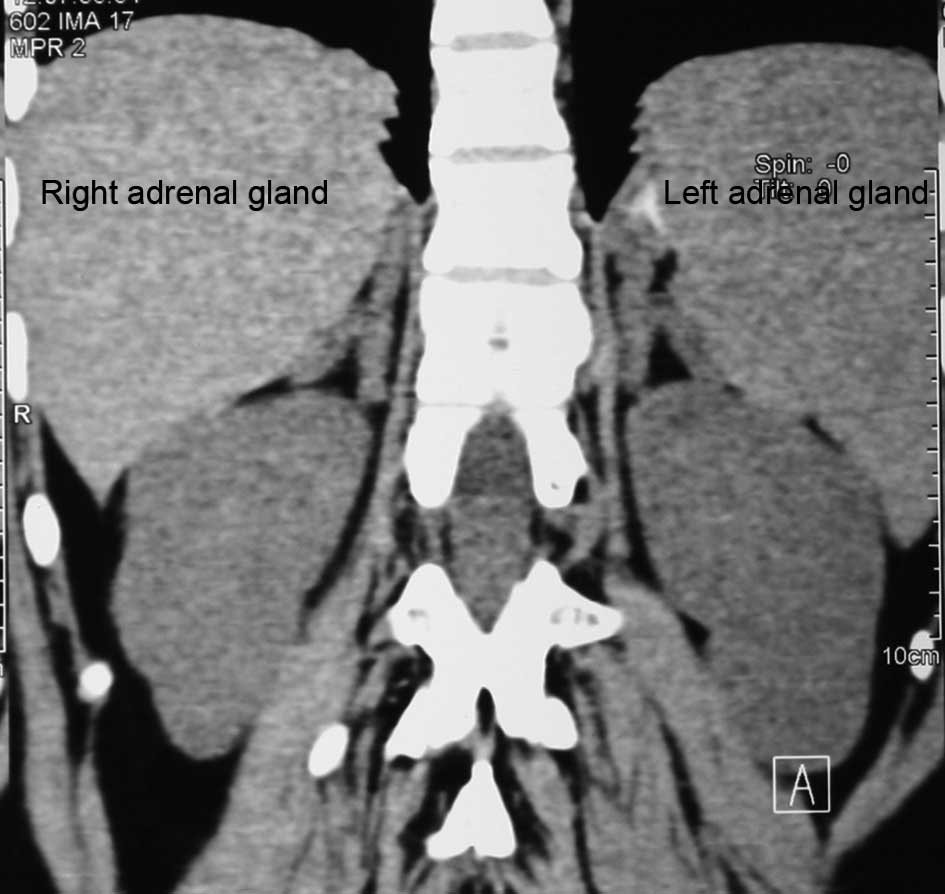
have testes. A CT scan showed enlargement of the adrenal glands:
left, 32×7 mm; right, 38×8 mm (normal adrenal gland size is 14×2
mm) (Fig. 3). The bone mass
densitometry (BMD) using the lunar prodigy advance system
(manufactured by GE Healthcare; analysis version 13.20), measured
at AP spin L1–L4 was 0.964 g/cm2 with a Z-score of 2.1,
which is significantly higher than normal limits for her age and
gender. The bone age was 18 years. The ACTH level was 228 pg/ml
(normal value <63), progesterone level was 5.35 ng/ml (normal
value <1.13), 17-OH progesterone level was 13.9 ng/ml (normal
value <2), 17-ketosteroid level was 58.37 mg/24 h (normal value
<14), δ-4 androstendion level was 4.4 ng/ml (normal value
<1), cortisol level was 5.6 μg/dl (normal value <25),
testosterone level was 210 ng/dl (normal value <100), LH level
was 2.2 mlU/ml in the follicular phase (normal value <11.6) and
FSH level was 6.3 mlU/ml in the follicular phase (normal value
<11.3).
Cytogenetics
Chromosome analysis using GTG-banding was performed
according to standard procedures (14). A total of 100 metaphases analyzed
from stimulated peripheral blood culture were analyzed. The
karyotype was described according to the International System for
Human Cytogenetic Nomenclature (15).
Molecular cytogenetics
Fluorescence in situ hybridization (FISH)
using LSI SRY (Yp11.3) SpectrumOrange/CEP X SpectrumGreen probe
(Abbott Molecular/Vysis, USA) and centromere specific multicolor
FISH (cenM-FISH) was performed (16,17).
Subsequently, a centromere-near multicolor FISH (subcenM-FISH)
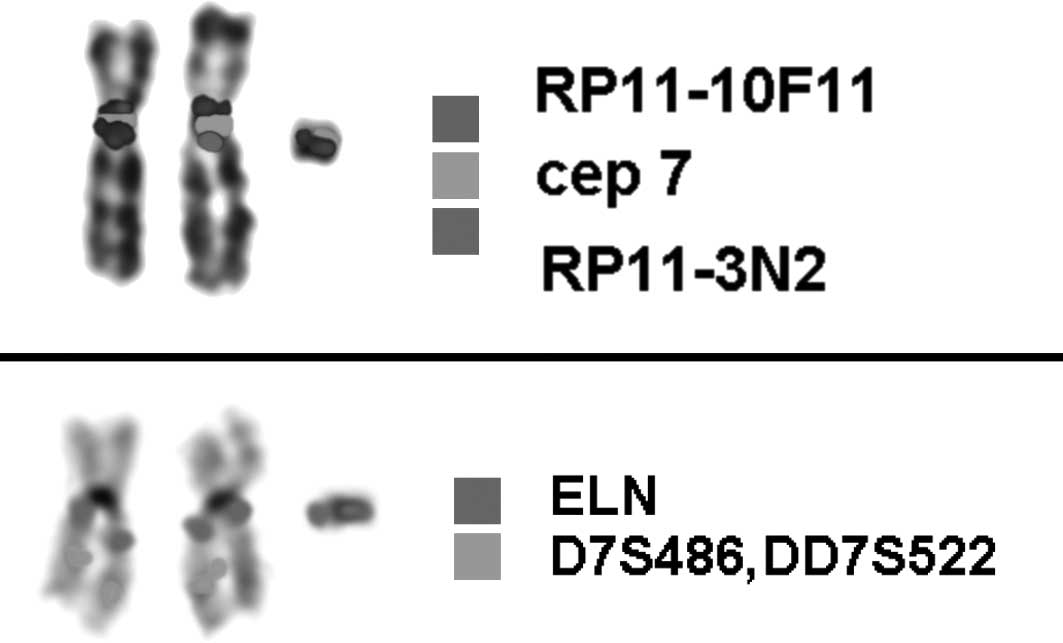
probe set for chromosome 7 (18)
was used to check for the presence of centromere-near euchromatic
material on the small marker chromosome. The applied subcenM-FISH
probe set consists of 2 partial chromosome painting (pcp) probes, 1
for the long and 1 for the short arm of chromosome 7, both
centromere-specific probes for chromosome 7 (D7Z1; Abbott
Molecular/Vysis) of 2 BAC probes (RP 11–10F11 specific for 7p11.2
and RP11–3N2 located in 7q11.21). Furthermore, a commercially
available probe for the ELN-gene in 7q11.23 together with a control
in 7q31 (D7S486, D7S522) was used to characterize the sSMC in
further detail. A total of 20 metaphase spreads were analyzed, each
using a fluorescence microscope (AxioImager.Z1 mot; Zeiss) equipped
with appropriate filter sets to discriminate between a maximum of 5
fluorochromes and the counterstain 4′,6-diamino-2-phenylindole
(DAPI). Image capturing and processing were carried out using an
ISIS imaging system (MetaSystems, Altlussheim, Germany).
Results
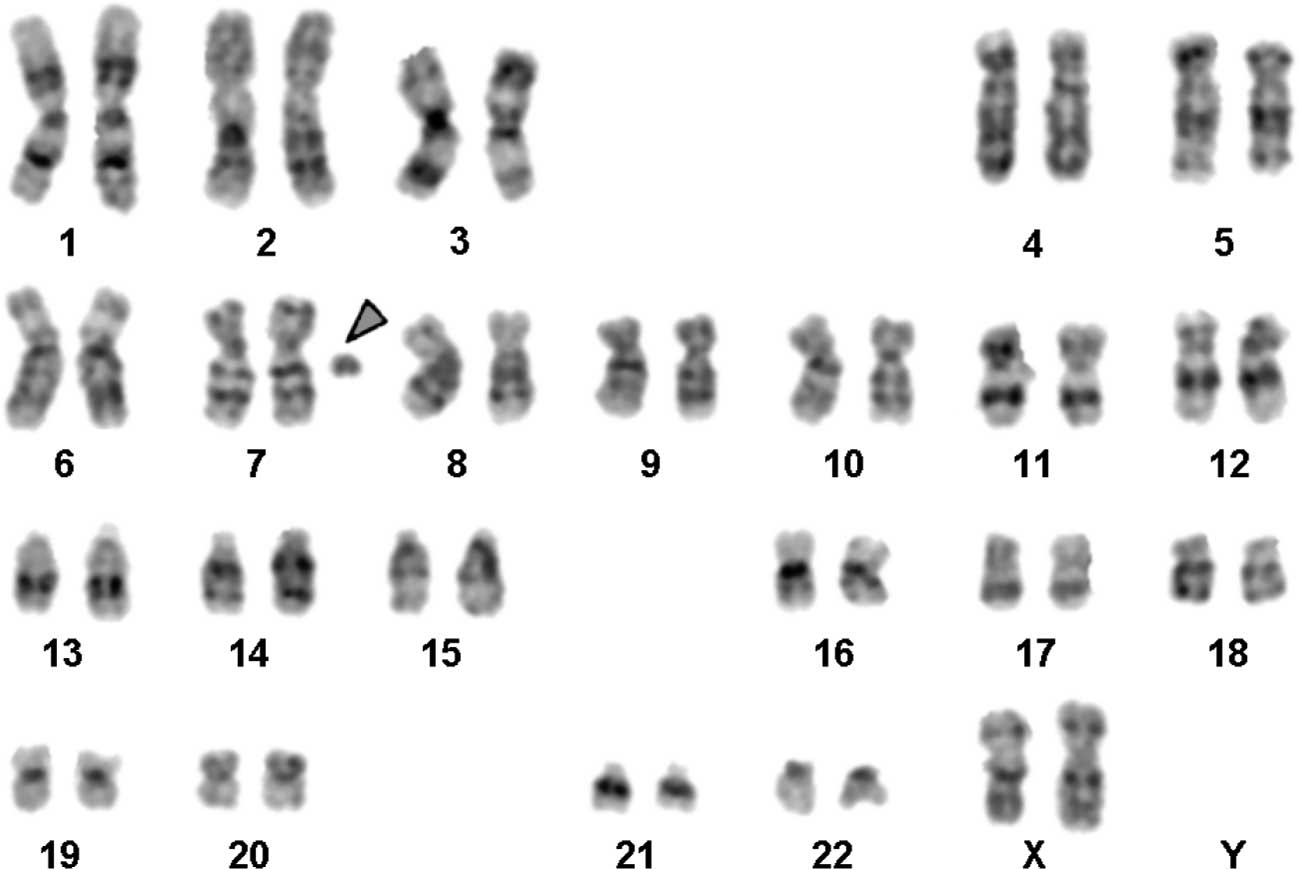
The karyotype determined by GTG-banding identified
was mos 48,XX,+mar1×2[1]/47,XX+mar1[45]/46,XX[54] (Fig. 4). The sSMC was present in 45 of 100
lymphocyte-derived metaphases. FISH excluded the presence of
SRY-specific sequences (data not shown). The sSMC was further
characterized by molecular cytogenetic studies which revealed a
centric minute-shaped chromosome 7 [min(7)(:p11.1->q11.23:)] (Fig. 5). The karyotypes of the mother and
the father were 46,XX and 46,XY, respectively.
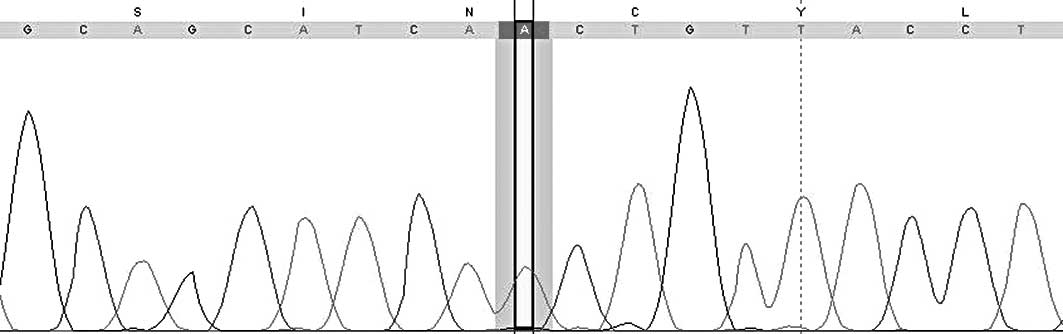
Sequencing of the CYP21A2 gene revealed the
homozygous mutation c.518T>A (p.Ile173Asn) in exon 4
(NM_000500.5; ATG=1). Both parents are heterozygous for that
mutation (Fig. 6).
Discussion
sSMC(7) is very
rare and usually small in size. They consist of the centromere and
small amounts of euchromatic material, a fact which also applies to
our patient. Only 15 patients with sSMC originating from the
proximal region of the long arm are described in the literature.
Comparing the phenotype of cases reported, delay of speech is often
reported (http://www.med.uni-jena.de/fish/sSMC/07.htm#Start07).
The phenotypic spectrum associated with duplication
of the 7q11.23 Williams-Beuren Syndrome (WBS) region has previously
been delineated. In these patients, the main clinical feature is a
moderate to severe expressed language delay (19). By contrast, subjects with deletion
of the same interval have good communication competence, with a
relative strength in verbal skill. These findings support the idea
that this region contains genes that may affect speech performance
in a dose-dependent manner. Four genes (CALN1, STX1A, LIMK1 and
CYLN), involved in brain development or function, have been
identified (20). As was observed
in our patient, facial anomalies are non-specific, but some traits
are common to both ring chromosome 7 [r(7)] carriers and patients with WBS
duplication, and thus they can be associated with 7q proximal
region triplication. Our patient shares a prominent forehead, a
triangular face, a high nasal bridge, normal eyes, thin lips, short
philtrum and normal ears (21)
with the other patients. By contrast, other aspects, such as
hirsutism, were observed in our case and other cases as well
(21). These features should be
carefully sought when a new case of r(7) is discovered, since they could
represent helpful clues to better delineate the r(7) phenotype.
CAH is found in a wide range of clinical severity
ranging from subtle hormone imbalance in adults to severe
life-threatening SW in newborns (22). Detection of the underlying
mutations in the CYP21A2 gene encoding steroid 21-hydroxylase
enzyme is helpful both for confirmation of diagnosis and management
of CAH patients (22).
Approximately 95% of the mutated alleles in patients with steroid
21-OHD are generated by transfer of DNA sequences from CYP21A1P to
CYP21A2 by gene conversion events (23). The remaining 5% of the alleles show
new/rare mutations due to random events (24). Most of these mutations are unique
to individual families, but some are population-specific (25,26).
Different kinds of mutations result in different degrees of
enzymatic impairment of P450c21, which result in varied phenotypes
of CAH patients. A large number of mutations detected in the
CYP21A2 gene have been characterized to prove their clinical
relevance and impact on the P450c21 protein. The residual enzyme
activity is then measured towards both natural substrates (17-OHP
and progesterone) and compared to the wild-type protein. The
percentage of the residual enzyme activity is correlated with the
clinical phenotype and subsequently mutations are classified as
simple virilization, SW or non-classic types of AGS (27–31),
with the change c.518T>A being a typical mutation for simple
virilization.
To our knowledge, in this study, we present the
first report of a female pseudohermaphroditism, classified as
simple virilization form of 21-OHD, with an additional
min(7(:p11.1->q11.23:) and the homozygous mutation c.518T>A
(p.Ile173Asn) in the CYP21A2 gene.
Acknowledgements
The authors thank Professor I. Othman, the Director
General of Atomic Energy Commission of SYRIA (AECS), and Dr N.
Mirali, Head of Molecular Biology and Biotechnology Department, for
their support. This study was supported by the AECS, in parts by
the DAAD (D/07/09624).
References
|
1
|
Liehr T, Claussen U and Starke H: Small
supernumerary marker chromosomes (sSMC) in humans. Cytogenet Genome
Res. 107:55–67. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liehr T and Weise A: Frequency of small
supernumerary marker chromosomes in prenatal, newborn,
developmentally retarded and infertility diagnostics. Int J Mol
Med. 19:719–731. 2007.PubMed/NCBI
|
|
3
|
Liehr T: Small supernumerary marker
chromosomes (sSMCs): a spotlight on some nomenclature problems. J
Histochem Cytochem. 57:991–993. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Miyoshi O, Kondoh T, Taneda H, Otsuka K,
Matsumoto T and Niikawa N: 47,XX,UPD(7)mat,r(7)pat/46,XX,UPD(7)mat
mosaicism in a girl with Silver-Russell syndrome (SRS): possible
exclusion of the putative SRS gene from a 7p13-q11 region. J Med
Genet. 36:326–329. 1999.PubMed/NCBI
|
|
5
|
Liehr T: Cytogenetic contribution to
uniparental disomy (UPD). Mol Cytogenet. 3:82010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
White PC, Tusie-Luna MT, New MI and
Speiser PW: Mutations in steroid 21-hydroxylase (CYP21). Hum Mutat.
3:373–378. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Speiser PW and White PC: Congenital
adrenal hyperplasia. N Engl J Med. 349:776–788. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kawaguchi H, O’hUigin C and Klein J:
Evolutionary origin of mutations in the primate cytochrome P450c21
gene. Am J Hum Genet. 50:766–780. 1992.PubMed/NCBI
|
|
9
|
Levine LS, Zachmann M, New MI, Prader A,
Pollack MS, O’Neill GJ, Yang SY, Oberfield SE and Dupont B: Genetic
mapping of the 21-hydroxylase-deficiency gene within the HLA
linkage group. N Engl J Med. 299:911–915. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tajima T, Fujieda K and Fujii-Kuriyama Y:
De novo mutation causes steroid 21-hydroxylase deficiency in one
family of HLA-identical affected and unaffected siblings. J Clin
Endocrinol Metab. 77:86–89. 1993.PubMed/NCBI
|
|
11
|
White PC and Speiser PW: Congenital
adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev.
21:245–291. 2000.PubMed/NCBI
|
|
12
|
Jaaskelainen J, Levo A, Voutilainen R and
Partanen J: Population-wide evaluation of disease manifestation in
relation to molecular genotype in steroid 21-hydroxylase (CYP21)
deficiency: good correlation in a well defined population. J Clin
Endocrinol Metab. 82:3293–3297. 1997.PubMed/NCBI
|
|
13
|
Krone N, Braun A, Roscher AA, Knorr D and
Schwarz HP: Predicting phenotype in steroid 21-hydroxylase
deficiency? Comprehensive genotyping in 155 unrelated, well defined
patients from southern Germany. J Clin Endocrinol Metab.
85:1059–1065. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Claussen U, Michel S, Mühlig P, Westermann
M, Grummt UW, Kromeyer-Hauschild K and Liehr T: Demystifying
chromosome preparation and the implications for the concept of
chromosome condensation during mitosis. Cytogenet Genome Res.
98:136–146. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shaffer L, Slovak M and Cambell L: ISCN
(2009): An International System for Human Cytogenetic Nomenclature.
S. Karger; Basel: 2009
|
|
16
|
Nietzel A, Rocchi M, Starke H, Heller A,
Fiedler W, Wlodarska I, Loncarevic IF, Beensen V, Claussen U and
Liehr T: A new multicolor-FISH approach for the characterization of
marker chromosomes: centromerespecific multicolor-FISH (cenM-FISH).
Hum Genet. 108:199–204. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Oliver-Bonet M, Liehr T, Nietzel A, Heller
A, Starke H, Claussen U, Codina-Pascual M, Pujol A, Abad C, Egozcue
J, Navarro J and Benet J: Karyotyping of human synaptonemal
complexes by cenM-FISH. Eur J Hum Genet. 11:879–883. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Starke H, Nietzel A, Weise A, Heller A,
Mrasek K, Belitz B, Kelbova C, Volleth M, Albrecht B, Mitulla B,
Trappe R, et al: Small supernumerary marker chromosomes (SMC):
genotype-phenotype correlation and classification. Hum Genet.
114:51–67. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Berg JS, Brunetti-Pierri N, Peters SU,
Kang SH, Fong CT, Salamone J, Freedenberg D, Hannig VL, Prock LA,
Miller DT, Raffalli P, et al: Speech delay and autism spectrum
behaviors are frequently associated with duplication of the 7q11.23
Williams-Beuren syndrome region. Genet Med. 9:427–441. 2007.
View Article : Google Scholar
|
|
20
|
Lichtenbelt KD, Hochstenbach R, van Dam
WM, Eleveld MJ, Poot M and Beemer FA: Supernumerary ring chromosome
mosaicism: case report, investigation of the gene content, and
delineation of the phenotype. Am J Med Genet Part A. 132:93–100.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bertini V, Valetto A, Uccelli A,
Bonuccelli A, Tarantino E, Taddeucci G and Simi P: Molecular
cytogenetic characterization of a de novo mosaic supernumerary ring
chromosome 7: report of a new patient. Am J Med Genet part A.
146A:2955–2959. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dubey S, Idicula-Thomas S, Anwaruddin M,
Saravanan C, Varma RR and Maitra A: A novel 9-bp insertion detected
in steroid 21-hydroxylase gene (CYP21A2): prediction of its
structural and functional implications by computational methods. J
Biomed Sci. 16:32009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Higashi Y, Tanae A, Inoue H and
Fujii-Kuriyama Y: Evidence for frequent gene conversion in the
steroid 21-hydroxylase (P450c21) gene: implications for steroid
21-hydroxylase deficiency. Am J Hum Genet. 42:17–25.
1998.PubMed/NCBI
|
|
24
|
Wedell A, Thilén A, Ritzén EM, Stengler B
and Luthman H: Mutational spectrum of the steroid 21-hydroxylase
gene in Sweden: implications for genetic diagnosis and association
with disease manifestation. J Clin Endocrinol Metab. 78:1145–1152.
1994.PubMed/NCBI
|
|
25
|
Billerbeck AE, Bachega TA, Frazatto ET,
Nishi MY, Goldberg AC, Marin ML, Madureira G, Monte O, Arnhold IJ
and Mendonca BB: A novel missense mutation, G424S, in Brazilian
patients with 21-hydroxylase deficiency. J Clin Endocrinol Metab.
84:2870–2872. 1999.PubMed/NCBI
|
|
26
|
Barbaro M, Lajic S, Baldazzi L, Balsamo A,
Pirazzoli P, Cicognani A, Wedell A and Cacciari E: Functional
analysis of two recurrent aminoacid substitution in the CYP21 gene
from Italian patients with congenital adrenal hyperplasia. J Clin
Endocrinol Metab. 89:2402–2407. 2004. View Article : Google Scholar
|
|
27
|
Database of CYP21A2 by human Cytochrome
P450 (CYP) Allele Nomenclature Committee. [http://www.imm.ki.se/CYPalleles/cyp21.htm].
|
|
28
|
Robins T, Bellanne-Chantelot C, Barbaro M,
Cabrol S, Wedell A and Lajic S: Characterization of novel missense
mutations in CYP21 causing congenital adrenal hyperplasia. J Mol
Med. 85:247–255. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Menassa R, Tardy V, Despert F,
Bouvattier-Morel C, Brossier JP, Cartigny M and Morel Y: p.H62L, a
rare mutation of the CYP21 gene identified in two forms of
21-hydroxylase deficiency. J Clin Endocrinol Metab. 93:1901–1908.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Soardi FC, Barbaro M, Lau IF, Lemos-Marini
SHV, Baptista MTM, Guerra-Junior G, Wedell A, Lajic S and de Mello
MP: Inhibition of CYP21A2 enzyme activity caused by novel missense
mutations identified in Brazilian and Scandinavian patients. J Clin
Endocrinol Metab. 93:2416–2420. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Riepe FG, Hiort O, Grtzinger J, Sippell
WG, Krone N and Holterhus PM: Functional and structural
consequences of a novel point mutation in the CYP21A2 gene causing
congenital adrenal hyperplasia: potential relevance of helix C for
P450 oxidoreductase-21-hydroxylase interaction. J Clin Endocrinol
Metab. 93:2891–2895. 2008. View Article : Google Scholar
|