Introduction
Camurati-Engelmann disease (CED, OMIM 131300) is a
rare autosomal dominant disease with variable clinical
manifestations. The main clinical and laboratory manifestations
include painful limbs, waddling gait, muscular weakness, joint
contracture, cranial nerve impingement, delayed pubertal
development and increased serum alkaline phosphatase (ALP) levels.
Fusiform thickening of the diaphyseal and metaphyseal cortex of the
long bones is the typical radiographic signature. Occasionally, a
number of patients experience blindness or deafness due to
thickened skull bones. The incidence of this disease is ~1:1
million births.
The primary causative gene in CED is the
transforming growth factor β1 (TGFβ1) gene (1), which contains seven exons. TGFβ1,
encoded by the TGFβ1 gene, is a member of the TGFβ1
signaling pathway and regulates cell proliferation, migration,
differentiation and apoptosis. TGFβ1 is particularly abundant in
the bone matrix, where it is involved in the regulation of bone
formation and resorption. The inactive form of the TGFβ1 protein
(pre-pro-TGFβ1) is composed of three subunits; the signal peptide,
the latency-associated peptide (LAP) and the mature peptide. The
activation procedure includes the cleavage of the signal peptide
followed by cleavage of the LAP from the mature peptide. Mutations
in different domains of the TGFβ1 gene lead to inherited
sclerosing bone disorder or osteoporosis (2).
To date, >40 CED families from Europe, Australia,
Israel, Japan, Korea, South America, the USA and China with ~10
mutations have been reported. The majority of these mutations are
located in the LAP region (3). The
first Chinese family with CED was reported in 2006 (4); the heterozygous missense mutation
p.Arg218His (R281H) in exon 4 of the TGFβ1 gene was detected
in the affected patient.
The present study aimed to investigate the cases of
two Chinese males diagnosed with CED, using clinical and X-ray
examinations, bone scintigraphy and TGFβ1 gene mutation
screening.
Patients and methods
Patients
Patient one (P1) was a 6-year-old male (height, 114
cm; weight, 19 kg) who had been born at full term and was the third
child born to the mother. P1 was unable to walk independently until
18 months of age and experienced waddling gait, muscular weakness
and left pelvic limb pain during the last 4 years.
Patient two (P2) was a 16-year-old male (height, 147
cm; weight, 21 kg). The parents of P2 noted that the patient was
short and underweight at the age of 6, compared with others of the
same age. P2 had a waddling gait, mild muscular weakness and no
signs of sexual development.
No fractures, auditory or visual impairments or
family history have been reported in either patient. The two
patients and their healthy non-consanguineous parents were enrolled
in this study by the Department of Osteoporosis and Bone Diseases
Outpatient Clinic (Shanghai Jiao Tong University Affiliated Sixth
People’s Hospital, Shanghai, China). The present study was approved
by the Ethics Committee of the Shanghai Jiao Tong University
Affiliated Sixth People’s Hospital. Informed consent was obtained
from the parents of the two patients. Two hundred age- and
gender-matched healthy donors were used as controls for the
mutation analysis after being recruited for a previous study
(5).
Methods
Biochemical parameters, including the complete blood
count and levels of serum calcium (Ca), phosphonium (P), ALP, blood
urea nitrogen (BUN), serum creatinine (Scr), β-isomerized
C-terminal cross-linked telopeptide of type I collagen (β-CTX),
procollagen type I N-terminal propeptide (PINP), 25-hydroxy D
[25(OH)D] and parathyroid hormone (PTH) were determined in the two
patients and their parents. X-ray radiography of the thoracic and
lumbar vertebrae, limbs, hips and skull was performed individually.
A lunar prodigy dual-energy X-ray absorptiometry (DXA) densitometer
(Lunar Corporation, Madison, WI, USA) was used to measure the bone
mineral density (BMD) values of the left proximal femur, including
the femoral neck and total hip, and the anteroposterior lumbar
spine 1–4 (L1-4). The machine was calibrated daily. Prodigy enCORE
version 6.70 software was used to analyze the data (standard-array
mode; GE Healthcare, Madison, WI, USA). DXA measurements were
obtained from triplicate measurements at L1-4 and the total hip,
femoral neck and trochanter in 15 individuals; the coefficient of
variability (CV) values for the DXA measurements were 1.39, 0.70,
2.22 and 1.41%, respectively (6).
Weekly repeated phantom measurements were carried out; these
determined that the long-term reproducibility of the DXA data
during the study was 99.55% (7).
Standardized equipment were used to determine the body weight and
height of subjects. The body mass index (BMI) was calculated as the
weight/height2 (kg/m2). Bone scintigraphy
with Tc-99m hydroxymethylene diphosphonate (HMDP) was performed to
detect abnormal bone metabolism.
For the mutation analysis, genomic DNA was isolated
from peripheral blood leukocytes using the conventional
phenol-chloroform extraction method. The entire coding region and
adjacent splice sites of the TGFβ1 gene were amplified and
sequenced directly from the two patients, their parents and 200
healthy donors.
Results
Biochemical and bone turnover
markers
Serum levels of ALP, Ca, P, PTH, 25(OH)D, PINP and
β-CTX in P1 and P2 are shown in Table
I. Notably, levels of the bone resorption marker β-CTX were
markedly elevated 7-fold compared with the upper limit of normal
(ULN) in P1, while only a slight increase was observed in P2. The
results of two bone formation markers, ALP and PINP, were
inconsistent in the two patients. In P1, the serum PINP level
exceeded the measurement range, while the serum ALP level was only
mildly increased (normal range in childhood, 85–400 U/l). However,
in P2, serum ALP and PINP levels were within the normal range.
Furthermore, the complete blood count and renal function markers in
the two patients were normal (data not shown).
 | Table IBiochemical parameters, bone turnover
markers and bone mineral density (BMD) values of lumbar spine 1–4
(L1-4) and hip sites in patients one (P1) and two (P2). |
Table I
Biochemical parameters, bone turnover
markers and bone mineral density (BMD) values of lumbar spine 1–4
(L1-4) and hip sites in patients one (P1) and two (P2).
| Patient | Height (cm) | Weight (kg) | BMI
(kg/m2) | ALP (U/l) | Ca (mmol/l) | P (mmol/l) | PTH (ng/l) | 25(OH)D (ng/ml) | β-CTX (ng/l) | PINP (ng/ml) | L1-4
(g/cm2) | Femoral neck
(g/cm2) | Trochanter
(g/cm2) | Total hip
(g/cm2) |
|---|
| P1 | 114 | 19 | 14.62 | 435 | 2.33 | 1.57 | 56.19 | 10.66 | 3,770 | >1200 | 0.591 | 0.596 | 0.413 | NA |
| P2 | 147.5 | 21 | 9.65 | 157 | 2.16 | 1.34 | 21.42 | 12.13 | 1,590 | 70.99 | 0.512 | 0.422 | 0.407 | 0.406 |
Radiographic examinations
The X-ray examinations revealed typical fusiform
thickening of the diaphysis of the long bones among the tibias,
humeri, femurs, ulnas and radii (Fig.
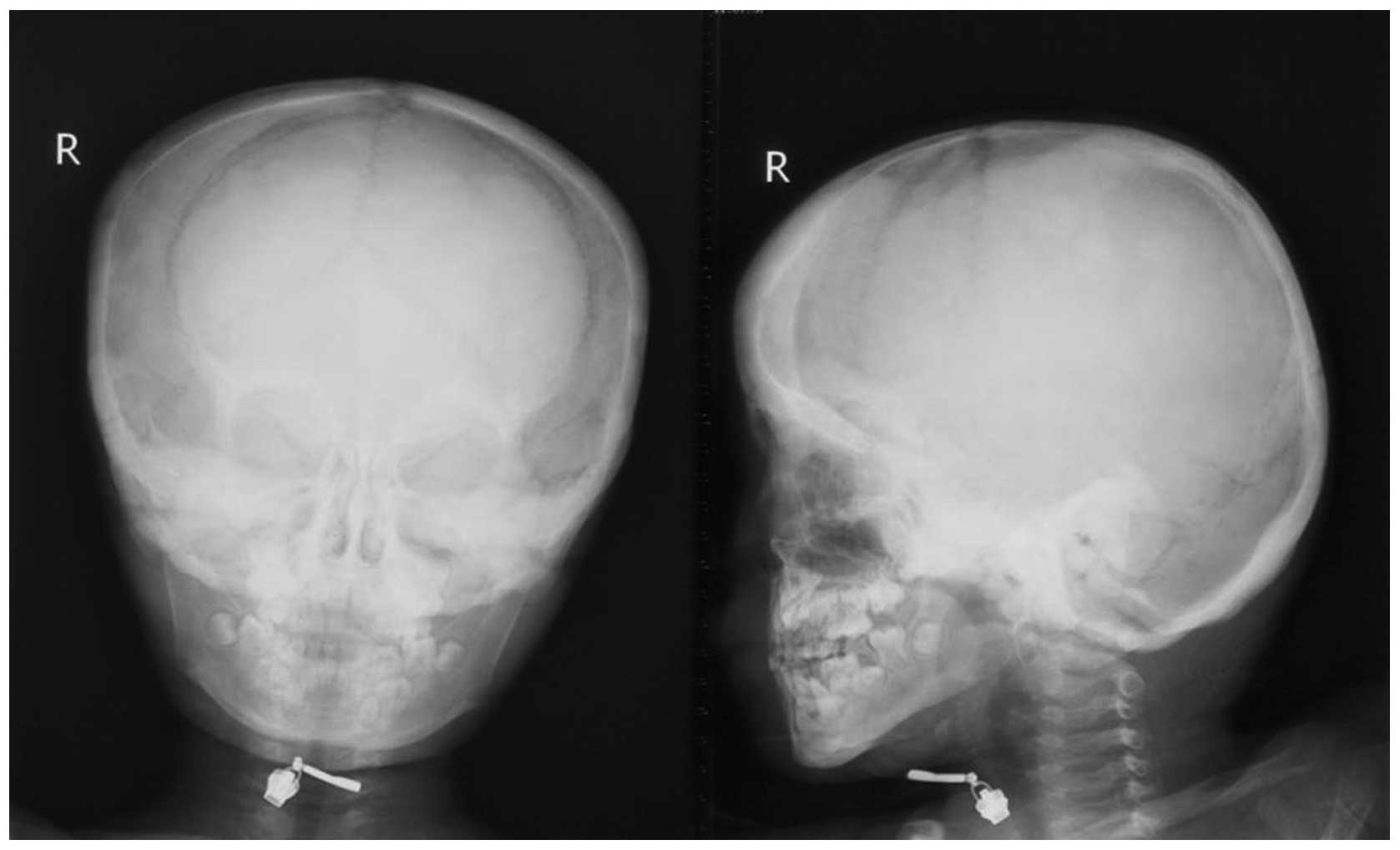
1). The skulls of the two patients exhibited cortical
thickening located at the base (Fig.
2).
Abnormal tracer uptake was observed in the skull and
both sides of upper humeri, ulnas, radii, femurs and tibias using
bone scintigraphy in the two patients (Fig. 3).
BMD
The BMD values of the two patients at L1-4 and hip
sites are shown in Table I. The Z
scores of P1 were imponderable due to the lack of reference BMD
values for Chinese children aged <10 years. The BMD values of
L1-4 and femoral neck in P1 were similar to those in a study by Wu
et al(8). For P2, the BMD
values at each site were lower compared with the age- and
gender-matched mean reference values (8,9).
TGFβ1 gene mutation
A heterozygous missense mutation p.Arg218Cys (R218C)
in exon 4 was detected in the two patients, while their parents and
the 200 healthy donors had normal wild-type genotypes (5).
Discussion
CED, also termed progressive diaphyseal dysplasia
(PDD), is a type of inherited sclerosing bone disorder
characterized by hyperostosis on the periosteal and endosteal
surface of the long bones. The age of onset varies greatly;
however, the majority of patients initially exhibit symptoms,
including pain and weakness, by adolescence. The typical
radiological characteristic is fusiform thickening of the
diaphyseal portions of the long bones. Vanhoenacker et
al(10) reported that the
radiographic manifestations were typically detected before the age
of 30 and were usually more extensive with increasing age.
Concomitant broadening of the diaphyses of long bones and the
narrowing of the medullary canal suggest that an excessive
periosteal apposition of bone and a defective resorption of bone at
the endosteal side of the long bones exist.
The TGFβ1 gene has been identified as the
causative gene of CED; numerous mutations in TGFβ1 have been
detected in CED patients worldwide. The majority of the mutations
detected in CED are missense mutations, including the arginine
residue at position 218 (R218C), R218H, H222D, C223S and C225R,
located in exon 4 at the C-terminal region of LAP, close to or
within the two cysteine residues (3). Additional mutations, including E169K
and R156C in exon 2, Y81H in exon 1 and L10-L12dup and LLL12-13ins
in the signal region, have been identified in CED family studies
(3,11–14).
Among them, R218C is the most common mutation hotspot in CED
patients, accounting for >60% of the mutations (3,11).
However, to date, no CED cases caused by the TGFβ1 gene
R218C mutation have been reported in Chinese patients. Furthermore,
the correlation between polymorphisms and clinical variability has
not yet been elucidated (11).
Mutations located in the LAP region have not been
demonstrated to lead to the overproduction of TGFβ1 in functional
studies; however, they are able to increase its activity. Two
possible mechanisms may explain this; firstly, the destabilized
disulphide bridging of the LAPs leads to premature activation of
the mature peptide mediated by exon 4 mutations. Secondly,
mutations in exon 1 lead to intracellular retention of the mutant
protein, which affects secretion (15,16).
Overactive TGFβ1 proteins lead to increased bone density and
decreased body fat and muscle tissue (15,16);
this contributes to the signs and symptoms of CED.
An animal study by Tang et al(17) showed that TGFβ1 was involved in
bone resorption and formation through an SMAD signaling pathway
that mediates bone mesenchymal stem cell (BMSCs) migration. A high
level of active TGFβ1 was detected in the bone marrow of CED mice
carrying TGFβ1 gene mutations, and typical progressive
diaphyseal dysplasia manifestations were observed.
Occasionally, individuals may possess the gene
mutation that causes CED yet never develop the characteristic
features of this condition, supporting the incomplete penetrance of
CED (11). A number of individuals
with clinical manifestations of CED with no identified mutations in
the TGFβ1 gene are diagnosed with CED type II (OMIM 606631)
(18).
The two patients in the present study harbored the
most frequently detected R218C mutation in exon 4. Their clinical
manifestations, X-ray signatures and bone scintigrapy results were
consistent with previously reported phenotypes. With regard to the
6-year-old patient (P1), the ALP level was slightly increased
compared with the normal range in children aged 0–6 years.
Generally, serum ALP level is higher in childhood and adolescence
than in adults, due to the increased bone turnover associated with
growth. However, the markedly elevated levels of the bone formation
marker PINP indicated an upregulated bone formation process in CED.
Notably, the bone absorption marker β-CTX was also significantly
increased in the same patient. The 16-year-old patient (P2) also
had increased serum β-CTX levels, while the bone formation markers
ALP and PINP were within the normal ranges. To date, no studies
have reported increased β-CTX levels in CED patients. This novel
result requires further investigation in future studies.
In conclusion, the present study reported the cases
of two Chinese pediatric patients with CED caused by the
heterozygous missense mutation R218C in the TGFβ1 gene. The
results of this study suggest that abnormal bone turnover marker
levels, typical radiological findings, bone scintigraphy results
and mutations in the TGFβ1 gene are important factors for
diagnosis and appropriate genetic counseling in apparently sporadic
CED cases.
Acknowledgements
The authors would like to thank the two patients and
their family members for their cooperation. This study was
supported by the National Natural Science Foundation of China
(NSFC; nos. 30771019, 30800387 and 81070692), the Program of
Shanghai Chief Scientist (project nos. 08XD1403000 and
STCSM10DZ1950100) and the Shanghai Science and Technology
Development Fund (project nos. 08411963100 and 11ZR1427300).
References
|
1
|
de Vernejoul MC: Sclerosing bone
disorders. Best Pract Res Clin Rheumatol. 22:71–83. 2008.
|
|
2
|
Ralston SH: Genetics of osteoporosis. Ann
NY Acad Sci. 1192:181–189. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Janssens K, Vanhoenacker F, Bonduelle M,
Verbruggen L, Van Maldergem L, Ralston S, Guañabens N, Migone N,
Wientroub S, Divizia MT, Bergmann C, Bennett C, Simsek S, Melançon
S, Cundy T and Van Hul W: Camurati-Engelmann disease: review of the
clinical, radiological, and molecular data of 24 families and
implications for diagnosis and treatment. J Med Genet. 43:1–11.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liang YH, Li W, Li LY, Ye YY and Lu GX: A
mutation in TGF beta1 gene encoding the latency-associated peptide
in a Chinese patient with Camurati-Engelmann disease. Zhonghua Yi
Xue Yi Chuan Xue Za Zhi. 23:502–504. 2006.(In Chinese).
|
|
5
|
Gu JM, Zhang ZL, Zhang H, Hu WW, Wang C,
Yue H, Ke YH, He JW, Hu YQ, Li M, Liu YJ and Fu WZ: Thirteen
Chinese patients with sporadic Paget’s disease of bone: clinical
features, SQSTM1 mutation identification, and functional analysis.
J Bone Miner Metab. 30:525–533. 2012.
|
|
6
|
Gao G, Zhang ZL, Zhang H, Hu WW, Huang QR,
Lu JH, Hu YQ, Li M, Liu YJ, He JW, Gu JM and Yu JB: Hip axis length
changes in 10,554 males and females and the association with
femoral neck fracture. J Clin Densitom. 11:360–366. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang ZL, He JW, Qin YJ, Hu YQ, Li M,
Zhang H, Hu WW, Liu YJ and Gu JM: Association between myostatin
gene polymorphisms and peak BMD variation in Chinese nuclear
families. Osteoporos Int. 19:39–47. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu XP, Yang YH, Zhang H, Yuan LQ, Luo XH,
Cao XZ and Liao EY: Gender differences in bone density at different
skeletal sites of acquisition with age in Chinese children and
adolescents. J Bone Miner Metab. 23:253–260. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liao EY, Wu XP, Deng XG, Huang G, Zhu XP,
Long ZF, Wang WB, Tang WL and Zhang H: Age-related bone mineral
density, accumulated bone loss rate and prevalence of osteoporosis
at multiple skeletal sites in chinese women. Osteoporos Int.
13:669–676. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vanhoenacker FM, Janssens K, Van Hul W,
Gershoni-Baruch R, Brik R and De Schepper AM: Camurati-Engelmann
disease. Review of radioclinical features. Acta Radiol. 44:430–434.
2003.PubMed/NCBI
|
|
11
|
Campos-Xavier B, Saraiva JM, Savarirayan
R, Verloes A, Feingold J, Faivre L, Munnich A, Le Merrer M and
Cormier-Daire V: Phenotypic variability at the TGF-beta1 locus in
Camurati-Engelmann disease. Hum Genet. 109:653–658. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Janssens K, Gershoni-Baruch R, Guañabens
N, Migone N, Ralston S, Bonduelle M, Lissens W, Van Maldergem L,
Vanhoenacker F, Verbruggen L and Van Hul W: Mutations in the gene
encoding the latency-associated peptide of TGF-beta 1 cause
Camurati-Engelmann disease. Nat Genet. 26:273–275. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Simsek S, Janssens K, Kwee ML, Van Hul W,
Veenstra J and Netelenbos JC: Camurati-Engelmann disease
(progressive diaphyseal dysplasia) in a Moroccan family. Osteoporos
Int. 16:1167–1170. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu S, Liang S, Yan Y, Wang Y, Li F, Deng
Y, Huang W, Yuan W, Luo N, Zhu C, Wang Y, Li Y, Liu M and Wu X: A
novel mutation of TGF beta1 in a Chinese family with
Camurati-Engelmann disease. Bone. 40:1630–1634. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Saito T, Kinoshita A, Yoshiura K, Makita
Y, Wakui K, Honke K, Niikawa N and Taniguchi N: Domain-specific
mutations of a transforming growth factor (TGF)-beta 1
latency-associated peptide cause Camurati-Engelmann disease because
of the formation of a constitutively active form of TGF-beta 1. J
Biol Chem. 276:11469–11472. 2001. View Article : Google Scholar
|
|
16
|
Janssens K, ten Dijke P, Ralston SH,
Bergmann C and Van Hul W: Transforming growth factor-beta 1
mutations in Camurati-Engelmann disease lead to increased signaling
by altering either activation or secretion of the mutant protein. J
Biol Chem. 278:7718–7724. 2003. View Article : Google Scholar
|
|
17
|
Tang Y, Wu X, Lei W, Pang L, Wan C, Shi Z,
Zhao L, Nagy TR, Peng X, Hu J, Feng X, Van Hul W, Wan M and Cao X:
TGF-beta1-induced migration of bone mesenchymal stem cells couples
bone resorption with formation. Nat Med. 15:757–765. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nishimura G, Nishimura H, Tanaka Y, Makita
Y, Ikegawa S, Ghadami M, Kinoshita A and Niikawa N:
Camurati-Engelmann disease type II: progressive diaphyseal
dysplasia with striations of the bones. Am J Med Genet. 107:5–11.
2002. View Article : Google Scholar : PubMed/NCBI
|