Introduction
Tribbles homolog 3 (TRB3), a pseudokinase, is
essential for the catalytic activity that is performed by 10% of
the kinase superfamily members (1). TRB3 was also demonstrated to be a key
factor in complementary kinome small interfering-RNA (siRNA)
function, as a regulator of mitogen-activated protein kinase (MAPK)
signaling (2). Previous studies
have demonstrated that this occurs through the control of the
MAPK-extracellular signal-related kinase (MAPK-ERK) and
transforming growth factor β (TGFβ) pathways. Furthermore, TRB3
regulates JAG1 expression and is required for the proliferation of
breast cancer cells (3). In
addition, TRB3 is required in normal tissues during conditions of
hypoxic/endoplasmic reticulum stress or nutrient deprivation, as it
is upregulated and counteracts the effects of stress (4,5).
TRB3 is also upregulated in cancer as a response to hypoxia
(4,6) and is associated with a poor outcome
(7) as it promotes the activation
of key cancer signaling pathways (such as MAPK-ERK, TGFβ and jagged
1 protein (JAG1)/Notch). TRB3 expression and molecular function has
rarely been demonstrated in cancer cell lines, with the exception
of breast cancer cell lines (2).
The most understood function of the Notch family is
cell fate regulation. This function has been regarded to be linked
to the homeostasis of stem cell compartments (8–11)
and thus, Notch signaling has been implicated in human cancer
(10). Cell-autonomous oncogenic
activation of Notch was identified in T-cell acute lymphoblastic
leukemia/lymphoma (T-ALL). Notch 1 may be activated through
chromosomal translocations and/or mutations (10,12).
Downregulated expression of Notch-related factors, including Notch
receptors, ligands and targets, has also been observed in solid
tumors (10,13), including breast (13) and lung (14) cancer. To the best of our knowledge,
no studies with regard to the correlation between cell-autonomous
activation of Notch and TRB3 expression in lung cancer have been
published to date. Furthermore, the loss of NUMB expression in
breast cancer may contribute to increased Notch activity and
Notch-dependent proliferation (15,16).
JAG1/Notch signaling has been considered to be a
mediator of cancer progression and metastasis associated with the
basal-like subtype (17,18). The majority of lung cancer patients
have basal-like disease, and despite initial responses to systemic
cytotoxic chemotherapy, the disease follows an aggressive clinical
course with early recurrence (19). Therefore, JAG1/Notch signaling and
regulators of this pathway are attractive therapeutic targets in
this lung cancer subtype. TRB3 influences the tumor cell biology
and may be regulated by the JAG1/Notch pathways. Tumor-initiating
cells represent a small population of cells within certain types of
tumors, which possess the unique ability to self-renew and to
produce derivatives that maintain the tumor. The TRB3 target
pathways, including Notch, MAPK-ERK and TGFβ (20,21),
have been implicated in tumor-initiating cell maintenance,
suggesting that through the control of these pathways, TRB3 may
regulate the initiation of tumor formation. The metastatic
potential of epithelial tumors is likely to depend on a process
known as epithelial-to-mesenchymal (EMT) transition, where
epithelial cells acquire a migratory mesenchymal phenotype
(22). The Notch, TGFβ and
MAPK-ERK pathways interact and have a synergistic effect on the
production of factors that promote EMT and metastasis (23–25).
The Notch and TGFβ pathways have been demonstrated to facilitate
metastasis, and also have a role in determining the location of
metastatic sites (26); TGFβ
released from bone metastases induces JAG1 expression in tumor
cells, which contributes to paracrine Notch activation in
osteoblasts and preosteoclasts, and thus leads to bone invasion.
This suggests that TRB3 may potentiate the initiation of tumor
formation and the metastatic capacity of lung cancer cells through
the regulation of JAG1/Notch activation. Additionally, the
activation of these pathways and processes may result in reduced
survival associated with tumors, and elevated TRB3 levels.
We hypothesized that the abnormal expression of TRB3
may participate in lung cancer development. By transfection
analysis, we demonstrated the effect of knocking down TRB3 on human
lung adenocarcinoma cells and the underlying molecular mechanism.
The aim of the current study was to investigate the therapeutic
potential of the knockdown of TRB3 in lung cancer.
Materials and methods
Reagents and antibodies
Rabbit antibodies against human TRB3 (T8076) and
Notch (SAB2101618) were purchased from Sigma-Aldrich (St. Louis,
MO, USA). Mouse antibody against human β-actin (sc-8432) and the
secondary antibodies conjugated with horseradish peroxidase against
mouse and rabbit IgG (sc-2005 and sc-2030, respectively) were
purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA).
Clinical specimens, cells, plasmids and
transfection
Clinical samples for quantitative PCR (Q-PCR) and
immunohistochemistry (IHC) were obtained from Xiangya School of
Medicine, Central South University (Changsha, Hunan, China) with
informed patient consent and approval of the institutional review
board (Xiangya School of Medicine Research Ethics Committee). Human
lung adenocarcinoma cell lines were obtained from the American Type
Culture Collection (ATCC, Manassas, VA, USA). The recombinant
expression plasmid of pcDNA3.1(t) (pc3.1) expressing TRB3 was
constructed. Briefly, the open reading frame of TRB3 (GenBank
accession: BC027484) was cloned into plasmid pcDNA3.1(t)
(Invitrogen Life Technologies, Carlsbad, CA, USA) between the
XhoI and BamHI sites to build the pc3.1-shTRB3
recombinant plasmid. The A549 cells were cultured in Dulbecco’s
modified Eagle’s medium (DMEM; Gibco-BRL, Carlsbad, CA, USA)
supplemented with 10% fetal bovine serum (FBS; Gibco-BRL) at 37°C
in an incubator with an atmosphere of 5% CO2. The cells
were transfected with pc3.1-shTRB3 using LipofectamineTM
2000 (Invitrogen Life Technologies) according to the manufacturer’s
instructions.
Semi-quantitative RT-PCR
Total RNAs were isolated using TRIzol reagent
(Invitrogen Life Technologies), according to the manufacturer’s
instructions. The first-strand complementary DNA (cDNA) was reverse
transcribed from 2 μg RNA in a final volume of 20 ml, using
SuperScript II Reverse Transcriptase (Invitrogen Life
Technologies). The primers were designed in accordance with
GenBank. The quantity of cDNA used for each PCR reaction was 20 ng
in a 50 ml reaction volume. The PCR was performed with the Applied
Biosystems 7500 Real-Time PCR system (Invitrogen Life
Technologies). The protocol was as follows: one cycle at 94°C for 4
min and 40 cycles at 94°C for 30 sec, 60°C for 30 sec and 72°C for
30 sec. The PCR products were assayed by a dissociation curve to
verify a single product generation at the end point of the
assay.
Western blot analysis
The cells were lysed in radioimmunoprecipitation
assay (RIPA) buffer on ice and centrifuged at 12,000 × g for 30 h
to obtain the supernatant. The extracted protein samples were
separated by 12% SDS-PAGE and transferred onto polyvinylidene
difluoride (PVDF) membranes (GE Healthcare, Amersham, UK). The
membranes were blocked in 5% skimmed milk for 1 h and subsequently
incubated with primary antibodies at 4°C overnight. Following
washing with PBS three times, the samples were probed by secondary
antibodies conjugated with horseradish peroxidase for 1 h at room
temperature. The signals were detected using a chemiluminescence
system SuperSignal West Pico Chemiluminescent Substrate (Pierce
Biotechnology, Inc., Rockford, IL, USA). The three independent
experiments were repeated to assess the relative protein
levels.
Cell invasion assay
The cell invasion assay was performed in 24-well
FluoroBlok cell culture inserts with 8-μm pore-size polyethylene
terephthalate (PET) membranes (BD Biosciences, Franklin Lakes, NJ,
USA). The insert was coated with 200 μl of 1 μg/μl Matrigel matrix
(BD Biosciences) at 4°C overnight. Following starvation for 6 h in
serum-free DMEM, the cells were harvested from one subconfluent 10
cm dish by cell dissociation buffer (Invitrogen Life Technologies),
centrifuged at 300 × gh for 5 min and resuspended in DMEM. Cells
(1×105, in 500 μl DMEM) were seeded onto the insert and
250 μl DMEM with 10% FBS was added into the lower chamber of the
transwells. Following incubation for 18 h at 37°C, the medium
inside the insert was removed and the insert was then placed in a
novel 24-well plate. The invaded cells on the reverse side of the
insert were labeled with a fluorescent dye, calcein acetoxymethl
ester (4 μM in PBS; BD Biosciences), for 1 h at 37°C. The
fluorescence was measured with 494/517 nm (excitation/emission
wavelength) by a DU®-8 UV-Vis spectrophotometer (Beckman
Coulter, Miami, FL, USA).
Statistical analysis
Data are presented as the mean ± standard error of
the mean of independent experiments. One-way analysis of variance
(ANOVA) was used to determine the differences among the groups. The
normality and constant variance for experimental data were tested
by the Levene’s test. Data without homogenous variance were
log-transformed to meet the necessary assumptions of the analysis
of variance. P<0.05 was considered to indicate a statistically
significant difference. When the F value exceeded the critical
value (P<0.05), the Newman-Keuls post hoc test was performed to
compare the groups.
Results
Elevated expression of TRB3 in lung
cancer
We assessed the differential expression of TRB3 in
lung cancer specimens. The mRNA levels of TRB3 in tumor lesions of
patients with non-small cell lung cancer (NSCLC) were determined by
Q-PCR. Fifty-six of the sixty tumor samples showed a higher
expression level of TRB3 compared with their respective adjacent,
normal tissues (Fig. 1A). The
upregulation of TRB3 was associated with distal metastasis and
disease recurrence (Table I).
Furthermore, the Kaplan-Meier survival curves revealed that TRB3
expression was inversely correlated with overall survival (Fig. 1B) and disease-free survival
(Fig. 1C). The IHC results
demonstrated that TRB3 was upregulated in the two types of NSCLC
investigated, particularly in adenocarcinoma; however, TRB3
upregulation was not observed in normal lung tissues (Fig. 1D). The TRB3 expression was
significantly correlated with tumor size and lymph node or distal
metastasis status in NSCLC patients (Table I). These results suggested that the
upregulation of TRB3 correlated with poor prognosis in patients
with NSCLC.
 | Table ICorrelation between TRB3 expression
and clinicopathological factors in the 60 patients with NSCLC. |
Table I
Correlation between TRB3 expression
and clinicopathological factors in the 60 patients with NSCLC.
| TRB3 expression |
|---|
|
|
|---|
| Characteristic | Low (0 and 1) | High (2 and 3) | P-valuea |
|---|
| Age |
| Years, mean ±
SD | 61.5 ± 5.1 | 60.87 ± 7.5 | 0.4782 |
| Gender |
| Male | 16 | 17 | 0.5712 |
| Female | 13 | 14 | |
| Smoking status |
| Smoker | 17 | 15 | 0.8575 |
| Non-smoker | 12 | 16 | |
| Histological
type |
|
Adenocarcinoma | 14 | 21 | 0.0280 |
| Squamous cell
carcinoma | 20 | 10 | |
| Large cell
carcinoma | 3 | 5 | |
| Stage |
| I and II | 18 | 10 | 0.0352 |
| III and IV | 11 | 21 | |
| Tumor status |
| T1 and T2 | 14 | 17 | 0.2782 |
| T3 and T4 | 15 | 14 | |
| Lymph node
metastasis |
| N0 | 18 | 9 | 0.0168 |
| N1-N3 | 11 | 21 | |
| Distal metastasis
status |
| M0 | 19 | 9 | 0.0316 |
| M1 | 10 | 21 | |
| Recurrence
status |
| Yes | 11 | 23 | 0.0013 |
| No | 18 | 7 | |
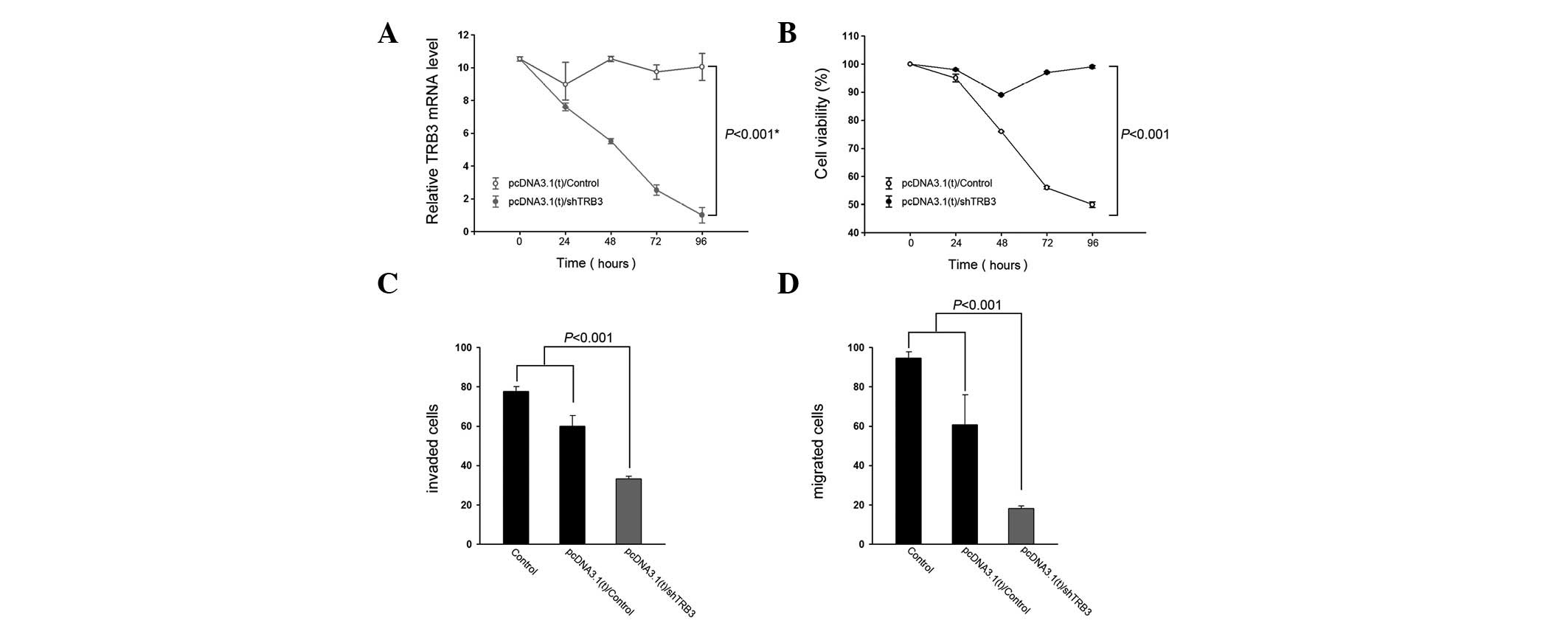
TRB3 knockdown results in apoptosis in
human lung adenocarcinoma cells
To elucidate the effect of knocking down TRB3 on
human lung adenocarcinoma cells, A549 cells were transfected with a
TRB3 interference vector and their proliferation and
characteristics were detected. The expression level of TRB3 was
significantly lower following shTRB3 transfection compared with the
control groups (Fig. 2A).
Additionally, the knockdown of TRB3 exhibited a positive effect on
cell growth (Fig. 2B). The shTRB3
groups had the lowest transwell level compared with the remaining
groups.
To understand whether TRB3 is biologically
significant in the aggressiveness of lung cancer cells, A549 cells
were subjected to TRB3 knockdown and examined for their
aggressiveness in vitro. Knockdown of TRB3 was revealed to
significantly decrease the invasive and migratory abilities of the
A549 cells (Fig. 2C and D,
respectively).
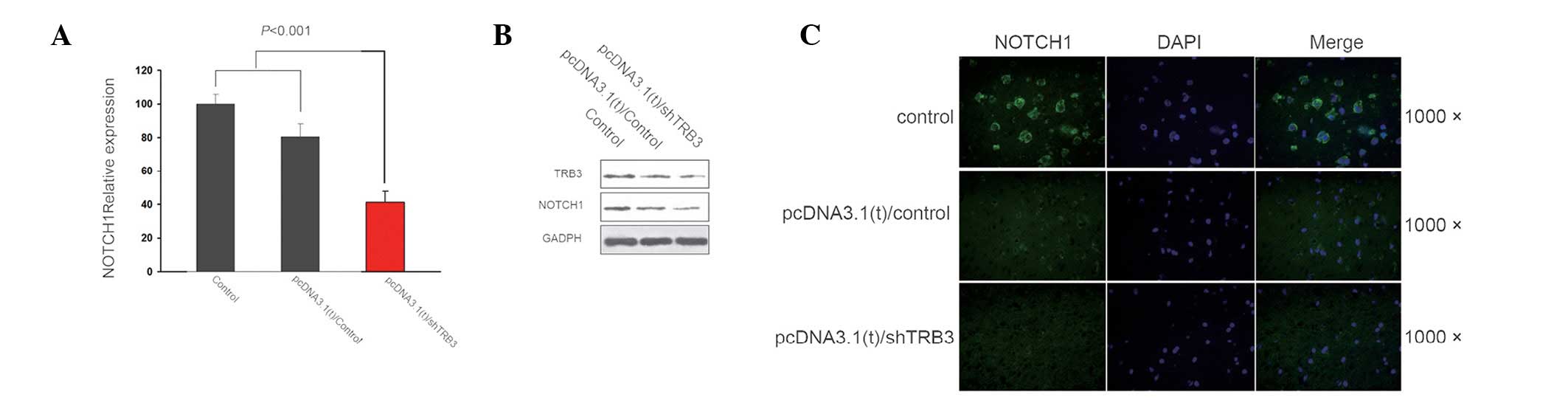
Correlation between TRB3 and Notch
expression in the lung adenocarcinoma cell lines
Following determination of the elevated expression
of TRB3 in lung cancer and the apoptotic effect of the TRB3
knockdown on the adenocarcinoma cells, the underlying mechanism of
this effect was investigated. The results demonstrated a positive
correlation between TRB3 and Notch 1 expression, at both the gene
and protein level, in the lung adenocarcinoma cell lines (Fig. 3).
Discussion
The tribbles gene family was initially identified in
Drosophila and considered as an inhibitor of mitosis that
regulates cell proliferation, migration and morphogenesis during
development (27,28). The three tribbles homologs, TRB1,
TRB2 and TRB3, are considered to be members of the pseudokinase
family, which contain a Ser/Thr protein kinase-like domain;
however, lack the ATP binding pocket and catalytic residues. TRB3
is the most widely studied member of the mammalian tribbles family.
The molecules which interact with TRB3 include transcription
factors, such as ubiquitin ligase and the BMP type II receptor,
which are members of the MAPK and PI3K signaling pathways (29,30).
Hua et al demonstrated that TRB3 interacts with SMAD3 and
promotes tumor cell migration and invasion (30). The authors suggested that TRB3 is a
novel partner of SMAD3 and may be involved in retaining SMAD3 in
the nucleus by physical interaction, and maintaining the
mesenchymal status of tumor cells. These studies suggested that
TRB3 may be a potential therapeutic target for the treatment of
human tumor metastasis. In the present study, it was identified
that TRB3 exhibits an abnormally abundant expression in lung cancer
tissues in patients with NSCLC, and that the upregulation of TRB3
was correlated with an increased number of tumor metastases, a
higher recurrence of tumors and poorer survival. According to these
results, we hypothesized that TRB3 was a significant factor in
promoting the malignant progression of tumors.
Our results demonstrated that suppressing TRB3
expression significantly inhibited tumor metastasis in A549 human
lung adenocarcinoma cells. The knockdown of TRB3 affected cell
growth and metastatic ability. The cells transfected with shTRB3
remained in the G1 stage compared with those in the non-treated
group. This suggested that following the suppression of TRB3, the
cell cycle was altered to remain in the G1 stage, as opposed to
passing into the S and G2 stages. In addition, the invasive ability
of the A549 cells was significantly decreased following the
suppression of TRB3 expression. There are a limited number of
studies concerned with the effects of TRB3 on the cell cycle
regardless of the fact that studies in neuronal PC6-3 cells have
demonstrated that TRB3 is involved in neuronal apoptosis evoked by
nerve growth factor withdrawal. TRB3 is also a multi-functional
adaptor in a number of signaling pathways (31). For example, TRB3 has been
demonstrated to inhibit insulin-induced S6 kinase activation
(31). Furthermore, it has been
revealed that TRB3 binds to ATF4 and regulates its transcriptional
activity (32). The expression of
TRBs is regulated by inflammatory stimulation and is cell type
specific (33). TRB3 mRNA may be
upregulated by various stresses. Also, TRB3 is the transcriptional
target of several factors, including PPARα, ATF4-CHOP and PI-3K
(34–36). These studies indicated that TRB3
participates in multiple cellular processes and pathways.
JAG1/Notch signaling is a mediator of cancer
progression and metastasis, which is associated with the basal-like
cancer subtype (17). Therefore,
the components of the Notch 1 pathway are attractive therapeutic
targets for this cancer subtype. Whether TRB3 affects tumor cell
biology may be inferred from data concerning the pathways it
regulates. Tumor-initiating cells (TIC), a small population of
cells within certain types of tumors, are able to produce
derivatives that maintain the tumor. The pathways that TRB3s
target, such as the Notch pathway, have been implicated in TIC
maintenance. This suggests that through the control of these
pathways, TRB3 may regulate tumor initiation. The metastatic
potential of epithelial tumors likely depends on a process known as
EMT, where epithelial cells acquire a migratory mesenchymal
phenotype. The Notch 1 pathways interact with each other and have a
synergistic effect on the production of factors that promote EMT
and metastasis (24). In addition,
the locations of metastases have been shown to be influenced by the
Notch pathways (37). It has been
demonstrated that the release of TGFβ stimulates JAG1 expression in
tumor cells and enhances Notch activation in osteoblasts and
preosteoclasts, which may promote bone invasion (38). Collectively, these findings predict
that TRB3 may potentiate the initiation of tumor formation and the
metastatic capacity of cancer cells, through its regulation of
JAG1/Notch activation. However, whether the activation of these
pathways and processes are regulated by TRB3, and whether reduced
survival time is associated with tumors with an elevated level of
TRB3, have not yet been identified. In this study, we demonstrated
that Notch 1 gene expression was positively correlated with TRB3 in
A549 cells.
The current study indicated that TRB3 expression was
elevated in lung cancer tissues in patients with NSCLC. In
addition, loss of TRB3 induced an apoptotic effect in the A549 lung
adenocarcinoma cell lines. The cell cycles were held in the G1
stage and the invasive ability of the cells decreased
significantly. The results also identified a positive correlation
between TRB3 and Notch 1 in the A549 cells. Thus, this interaction
may provide potential therapeutic targets for human NSCLC.
Acknowledgements
This study was supported by funding from the Hunan
Provincial Department of Science and Technology (grant no.
2012SK3249) and the Hunan Provincial Department of Health (grant
no. B2012-098), China.
References
|
1
|
Boudeau J, Miranda-Saavedra D, Barton GJ
and Alessi DR: Emerging roles of pseudokinases. Trends Cell Biol.
16:443–452. 2006. View Article : Google Scholar
|
|
2
|
Izrailit J, Berman HK, Datti A, Wrana JL
and Reedijk M: High throughput kinase inhibitor screens reveal TRB3
and MAPK-ERK/TGFβ pathways as fundamental Notch regulators in
breast cancer. Proc Natl Acad Sci USA. 110:1714–1719.
2013.PubMed/NCBI
|
|
3
|
Raja E: Cross-regulation between TGFβ/BMP
Signalling and the metabolic LKB1 pathway. Doctoral thesis. Uppsala
University; 2012
|
|
4
|
Bowers AJ, Scully S and Boylan JF: SKIP3,
a novel Drosophila tribbles ortholog, is overexpressed in
human tumors and is regulated by hypoxia. Oncogene. 22:2823–2835.
2003.PubMed/NCBI
|
|
5
|
Du K, Herzig S, Kulkarni RN and Montminy
M: TRB3: a tribbles homolog that inhibits Akt/PKB activation by
insulin in liver. Science. 300:1574–1577. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wennemers M, Bussink J, Scheijen B, et al:
Tribbles homolog 3 denotes a poor prognosis in breast cancer and is
involved in hypoxia response. Breast Cancer Res. 13:R822011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Miyoshi N, Ishii H, Mimori K, et al:
Abnormal expression of TRIB3 in colorectal cancer: a novel marker
for prognosis. Br J Cancer. 101:1664–1670. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Reedijk M, Odorcic S, Chang L, et al:
High-level coexpression of JAG1 and NOTCH1 is observed in human
breast cancer and is associated with poor overall survival. Cancer
Res. 65:8530–8537. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee CW, Raskett CM, Prudovsky I and
Altieri DC: Molecular dependence of estrogen receptor-negative
breast cancer on a notch-survivin signaling axis. Cancer Res.
68:5273–5281. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Leong KG, Niessen K, Kulic I, et al:
Jagged1-mediated Notch activation induces epithelial-to-mesenchymal
transition through Slug-induced repression of E-cadherin. J Exp
Med. 204:2935–2948. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rustighi A, Tiberi L, Soldano A, et al:
The prolyl-isomerase Pin1 is a Notch1 target that enhances Notch1
activation in cancer. Nat Cell Biol. 11:133–142. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shimizu M, Cohen B, Goldvasser P, Berman
H, Virtanen C and Reedijk M: Plasminogen activator uPA is a direct
transcriptional target of the JAG1-Notch receptor signaling pathway
in breast cancer. Cancer Res. 71:277–286. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Honjo T: The shortest path from the
surface to the nucleus: RBP-J kappa/Su(H) transcription factor.
Genes Cells. 1:1–9. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Weijzen S, Rizzo P, Braid M, et al:
Activation of Notch-1 signaling maintains the neoplastic phenotype
in human Ras-transformed cells. Nat Med. 8:979–986. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pece S, Serresi M, Santolini E, et al:
Loss of negative regulation by Numb over Notch is relevant to human
breast carcinogenesis. J Cell Biol. 167:215–221. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Colaluca IN, Tosoni D, Nuciforo P, et al:
NUMB controls p53 tumour suppressor activity. Nature. 451:76–80.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee CW, Simin K, Liu Q, et al: A
functional Notch-survivin gene signature in basal breast cancer.
Breast Cancer Res. 10:R972008. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yamaguchi N, Oyama T, Ito E, et al: NOTCH3
signaling pathway plays crucial roles in the proliferation of
ErbB2-negative human breast cancer cells. Cancer Res. 68:1881–1888.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dent R, Trudeau M, Pritchard KI, et al:
Triple-negative breast cancer: clinical features and patterns of
recurrence. Clin Cancer Res. 13:4429–4434. 2007. View Article : Google Scholar
|
|
20
|
Farnie G and Clarke RB: Mammary stem cells
and breast cancer - role of Notch signalling. Stem Cell Rev.
3:169–175. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yin X, Wolford CC, Chang YS, et al: ATF3,
an adaptive-response gene, enhances TGF{beta} signaling and
cancer-initiating cell features in breast cancer cells. J Cell Sci.
123:3558–3565. 2010.PubMed/NCBI
|
|
22
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gavert N and Ben-Ze’ev A:
Epithelial-mesenchymal transition and the invasive potential of
tumors. Trends Mol Med. 14:199–209. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Timmerman LA, Grego-Bessa J, Raya A, et
al: Notch promotes epithelial-mesenchymal transition during cardiac
development and oncogenic transformation. Genes Dev. 18:99–115.
2004. View Article : Google Scholar
|
|
25
|
Xie L, Law BK, Chytil AM, Brown KA, Aakre
ME and Moses HL: Activation of the Erk pathway is required for
TGF-beta1-induced EMT in vitro. Neoplasia. 6:603–610. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sethi N, Dai X, Winter CG and Kang Y:
Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast
cancer by engaging notch signaling in bone cells. Cancer cell.
19:192–205. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Grosshans J and Wieschaus E: A genetic
link between morphogenesis and cell division during formation of
the ventral furrow in Drosophila. Cell. 101:523–531. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Seher TC and Leptin M: Tribbles, a
cell-cycle brake that coordinates proliferation and morphogenesis
during Drosophila gastrulation. Curr Biol. 10:623–629. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sieber C, Kopf J, Hiepen C and Knaus P:
Recent advances in BMP receptor signaling. Cytokine Growth Factor
Rev. 20:343–355. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hua F, Mu R, Liu J, et al: TRB3 interacts
with SMAD3 promoting tumor cell migration and invasion. J Cell Sci.
124:3235–3246. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Matsushima R, Harada N, Webster NJ,
Tsutsumi YM and Nakaya Y: Effect of TRB3 on insulin and
nutrient-stimulated hepatic p70 S6 kinase activity. J Biol Chem.
281:29719–29729. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Izrailit J, Berman HK, Datti A, Wrana JL
and Reedijk M: High throughput kinase inhibitor screens reveal TRB3
and MAPK-ERK/TGFβ pathways as fundamental Notch regulators in
breast cancer. Proc Natl Acad Sci USA. 110:1714–1719.
2013.PubMed/NCBI
|
|
33
|
Xu J, Lv S, Qin Y, et al: TRB3 interacts
with CtIP and is overexpressed in certain cancers. Biochim Biophys
Acta. 1770:273–278. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schwarzer R, Dames S, Tondera D, Klippel A
and Kaufmann J: TRB3 is a PI 3-kinase dependent indicator for
nutrient starvation. Cell Signal. 18:899–909. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Koo SH, Satoh H, Herzig S, et al: PGC-1
promotes insulin resistance in liver through PPAR-alpha-dependent
induction of TRB-3. Nat Med. 10:530–534. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ohoka N, Yoshii S, Hattori T, Onozaki K
and Hayashi H: TRB3, a novel ER stress-inducible gene, is induced
via ATF4-CHOP pathway and is involved in cell death. EMBO J.
24:1243–1255. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Valastyan S and Weinberg RA: Tumor
metastasis: molecular insights and evolving paradigms. Cell.
147:275–292. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Weilbaecher KN, Guise TA and McCauley LK:
Cancer to bone: a fatal attraction. Nat Rev Cancer. 11:411–425.
2011. View
Article : Google Scholar : PubMed/NCBI
|