Introduction
Human papillomaviruses (HPVs), particularly
high-risk types, cause a variety of reproductive system lesions,
including cervical neoplasia and cancer. High-risk HPV DNA has been
detected in almost all types of cervical cancer, with HPV16 being
the most prevalent type in the general population (1). RASSF1A is a tumor suppressor gene
with a highly methylated promoter involved in tumorigenesis,
development and prognosis (2–6).
Previous studies have shown an association between HPV16 infection
and RASSF1A expression, although the precise mechanism of action
still remains to be elucidated. HPV infection and inactivation of
RASSF1A appear to be inversely correlated in several types of
cervical tumors and cell lines. The presence of HPV in cervical
carcinomas has been shown to alleviate the requirement for RASSF1A
inactivation, and no association with RASSF1A methylation has been
observed. This suggests that the two events require other
interaction mechanisms but engage the same tumorigenic pathway
(7,8).
A previous study demonstrated that one of the HPV16
oncoproteins, E5, activates the vascular endothelial growth factor
(VEGF) promoter and upregulates its expression via activation of
the epidermal growth factor receptor (EGFR) (9). HPV16-E6 and -E7 are known crucial
viral oncoproteins which have been shown to be consistently
maintained after viral integration into the host cell genome. The
probability of neoplasia is increased in HPV16 infections with E6
and E7 oncoprotein expression (10). To date, both p53-dependent and
-independent mechanisms of oncogenesis, regulated by HPV proteins,
have been described (11). E6
plays a primary role as an anti-apoptotic protein through
association with p53 via interactions with E6-associated protein,
and mediation of p53 ubiquitination and degradation that prevents
eliciting of cellular responses to stress signals, such as DNA
damage. However, underlying interaction mechanisms between HPV16
and the host factor RASSF1A still remain to be fully elucidated.
Previous experiments conducted by our group showed that p53 binds
to the RASSF1A promoter, leading to downregulation of RASSF1A
expression (12). Accordingly, it
is hypothesized that HPV16 infection, p53 and RASSF1A are closely
interrelated.
The present study aimed to investigate whether HPV16
infection regulates RASSF1A expression as well as to determine the
underlying mechanisms of action. Our results provide novel insights
into the mechanisms of cancer cell development.
Materials and methods
Cells
The human cervical carcinoma cell line, SiHa, was
obtained from the Center for Type Culture Collection (Wuhan, Hubei,
China).
Quantitative PCR (qPCR)
Total RNAs were prepared with the TRIzol kit
(Invitrogen Life Technologies, Carlsbad, CA, USA) according to the
manufacturer’s instructions. All of the RNAs were digested with
RNase-free DNase I and purified according to the protocol provided
by the manufacturer. In total, ~3 μg RNA was employed as the
template for reverse transcription using 0.5 μg oligo(dt) and 200
units of M-MLV reverse transcriptase (Promega, Madison, WI, USA).
qPCR was employed for the quantification of gene expression using
the multichannel Rotor-Gene 3000 (Corbett Research, Mortlake,
Australia) according to the manufacturer’s protocol. PCR cycling
conditions were as follows: 5 min at 94°C, followed by 40 cycles of
30 sec at 94°C, 30 sec at 60°C and 30 sec at 72°C in a 25-μl
reaction mix containing 1X SYBR-Green I. The primers used were:
5′-TGGAACAACATTAGAACAGC-3′ and 5′-CTGCAA CAAGACATACATCG-3′ for
HPV16-E6; 5′-CCTGCT GGATTACATCAAAGCACT-3′ and 5′-GTCAAGGGCA
TATCCTACAACAAA-3′ for HPRT. Simultaneous detection of the HPRT gene
was performed to normalize HPV16-E6 expression. Similar
amplification procedures were employed for HPRT and HPV16-E6. To
address robustness issues, each sample was amplified at least in
triplicate. Data were analyzed with Rotor-Gene version 6 software
and subsequently plotted in Microsoft Excel.
Plasmid constructs
RNA interference (RNAi) clones
siRNA employed for analysis was constructed using
the Ambion online siRNA design tool (www.ambion.com/techlib/misc/siRNA_design.html; Ambion,
Austin, TX, USA). Hairpin DNA sequences were synthesized as two
complementary oligonucleotides, annealed, and ligated between the
BbsI and XbaI sites to replace the enhanced green
fluorescent protein (EGFP) coding sequence of the pmU6pro vector
(kindly provided by Dr David Turner, University of Michigan, USA)
for generating the interference vectors, HPV16-E6-RNAi and
RASSF1A-RNAi. The sequences were the following: HPV16-E6-RNAi
sense, 5′-TTTGAATGTGTGTACTGC AAGCATGGCTTGCAGTACACACATTCTTTTT-3′ and
antisense, 3′-TTACACACATGACGTTCGTACCGAACGT
CATGTGTGTAAGAAAAAGATC-5′; RASSF1A-RNAi sense,
5′-TTTGACCTCTGTGGCGACTTCAATGTGA AGTCGCCACAGAGGTCTTTTT-3′ and
antisense, 3′-TGG AGACACCGCTGAAGTTACACTTCAGCGGTGTCTCCAG
AAAAAGATC-5′.
RASSF1A-pcDNA clone
To construct the RASSF1A-pcDNA vector, full-length
RASSF1A was amplified from the vector donated by Dr Rongjia Zhou
using PCR, and subcloned into the BamHI and EcoRI
sites of the pcDNA3.0 mammalian expression vector (BD Biosciences
Clontech, Palo Alto, CA, USA). PCR cycling conditions were as
follows: 5 min at 94°C, followed by 35 cycles of 1 min at 94°C, 1
min at 65°C and 1 min at 72°C in a 20-μl reaction mix. The sense
and antisense primers used for amplification were: 5′-AACG
GATCCATGTCGGGGGAGCCTGAGC-3′ and 5′-TACGA
ATTCTCACCCAAGGGGGCAGGCG-3′, respectively.
Cell preparation and transfection
analysis
The human HPV16-positive cervical cancer cell line
SiHa, was obtained from the Center for Type Culture Collection
(Wuhan, Hubei, China). The cells were regularly maintained in
Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 20%
fetal bovine serum (FBS; HyClone, Logan, UT, USA) at 37°C with 5%
CO2. The cells were passaged every 3 days and seeded
onto 24-well plates 12 h prior to transfection. The transfection
procedure was performed with Lipofectamine™ 2000 (Invitrogen Life
Technologies) according to the manufacturer’s instructions.
Assessment of apoptosis with Annexin
V/propidium iodide (PI) staining
Apoptotic cell death was measured using the
FITC-conjugated Annexin V/PI assay (BioVision, Palo Alto, CA, USA),
followed by flow cytometry (Becton-Dickinson, Franklin Lakes, NJ,
USA). Briefly, 1×105 cells were washed with ice-cold
phosphate-buffered saline (PBS), resuspended in 0.1 ml binding
buffer, and stained with 10 μl FITC-conjugated Annexin V (10 mg/ml)
and 10 μl PI (50 mg/ml). After incubation for 15 min at room
temperature in the dark, 400 μl binding buffer was added, and the
cells were subsequently analyzed with a FACScan flow cytometer
(Annexin V excitation at 488 nm and emission at 515 nm; PI
excitation at 488 nm and emission at 580 nm).
Western blot analysis
Proteins of freshly obtained SiHa cells were
extracted with ice-cold lysis buffer and incubated on ice for 15
min. Following centrifugation for 10 min at 15,000 × g, supernatant
fractions were collected, and western blot analysis was performed
using routine protocols. Briefly, extracts were analyzed with 12%
glycine-SDS-PAGE and transferred onto PVDF membranes with a pore
size of 0.2 μm (Hybond-P; Amersham Pharmacia Biotech, Uppsala,
Sweden). Nonspecific binding of antibodies was blocked with 5%
low-fat milk powder in TBST for 1 h at room temperature. The
membranes were incubated with human anti-p53, anti-RASSF1A,
anti-HPV16-E6, anti-β-actin (1:500; Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA) or anti-caspase 3 (1:500; Epitomics, Inc.,
Burlingame, CA, USA) antibodies at 4°C overnight, followed by
incubation with horseradish peroxidase (HRP)-conjugated secondary
antibodies (1:50,000) for 1 h. Proteins were visualized with
enhanced chemiluminescence (ECL) regent.
DNA binding assay
Previous experiments conducted by our group
demonstrated that p53 binds to the RASSF1A promoter and
downregulates its expression (12). Since p53 has been suggested to be
the critical mediator linking HPV16 infection with RASSF1A
expression, we confirmed the active binding site and used a higher
dose of p53 up to 2.5 μg to investigate its regulatory effect. The
experimental method used was similar to the method reported
previously (12).
Immunohistochemistry
Human testicular chips (no. CC23-01) were purchased
from Chao Ying Biotechnology (Xi’an, Shaanxi, China). Each chip
contained 23 samples, including normal testis, seminoma, lymphoma
and fibroma with three slices in the same area for each sample. The
expression levels of RASSF1A and p53 proteins were analyzed with
anti-RASSF1A (eBioscience, Inc., San Diego, CA, USA) and anti-p53
(Santa Cruz Biotechnology, Inc.) antibodies, respectively, using
streptavidin-biotin complex (SABC) and 3,3′-diaminobenzidine (DAB)
visualization methods according to the manufacturer’s instructions
(Boster Biological Technology, Ltd., Wuhan, China).
Results
HPV16-E6 regulates p53 and RASSF1A
levels, and suppresses apoptosis
To gain an insight into the association between
HPV16-E6 and RASSF1A expression, the HPV16-E6-RNAi vector was
cloned and transfected into SiHa cells containing endogenous HPV16,
and RASSF1A expression was detected by western blot analysis.
HPV16-E6 expression was decreased by 66% (Fig. 1) upon siRNA transfection,
subsequently leading to an increase in p53 and decrease in RASSF1A
levels (Fig. 2A). Further
examination of the biological effect of E6 RNAi revealed a 29.85%
increase in apoptosis (Fig.
2B).
p53 binds to RASSF1A promoter and
suppresses RASSF1A expression
According to a previous study conducted by our
group, the presence of a p53 binding site in the RASSF1A promoter
region was confirmed (12), and a
downregulatory effect on RASSF1A was demonstrated upon p53 binding.
To validate whether p53 binding is the key factor linking HPV16-E6
and RASSF1A expression, we repeated the experiment with 0–2.5 μg of
p53, and examined the binding (Fig.
3A) and regulatory effects (Fig.
3B) of p53. Gel-shift assay showed that His-p53 specifically
and efficiently bound to RASSF1A promoter (Fig. 3A, lane 1) which was confirmed by
adding the p53 antibody and formed a supershift band
(antibody/p53/RASSF1A; Fig. 3A,
lane 2). However, single-stranded probes and a mutant RASSF1A probe
decreased the formation of the p53/RASSF1A complex (Fig. 3A, lanes 3–7).
p53 inhibits apoptosis induction through
RASSF1A regulation
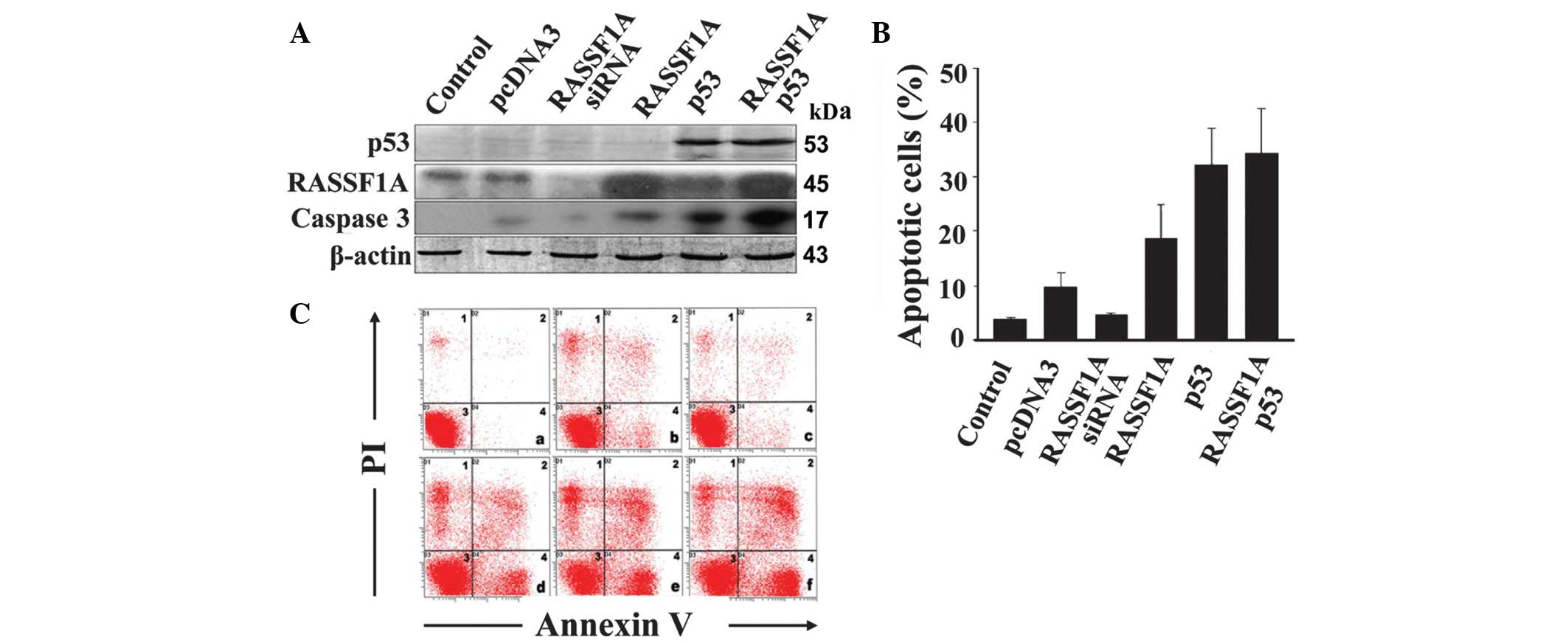
In RASSF1A-expressing SiHa cells, p53 significantly
inhibited RASSF1A expression. Consequently, we further investigated
the impact of p53 on RASSF1A-induced apoptosis. Flow cytometry
combined with Annexin V/PI staining (Fig. 4B and C) revealed that treatment of
RASSF1A-expressing SiHa cells with RASSF1A siRNA inhibits apoptosis
by 54%, compared to pcDNA. Upon overexpression of RASSF1A, the
proportion of apoptotic cells increased from 10.1 to 19.4%,
compared with pcDNA, while in the presence of p53, the proportion
of apoptotic cells increased to 33.53%. Although both RASSF1A and
p53 are apoptosis inducers, overexpression of the two proteins
induced a significantly lower increase in the apoptotic cell
percentage (35.8%) compared with the expected additive effect
(52.93%; 19.4 + 33.53%), indicating that apoptosis induction by
RASSF1A is at least partially inhibited by p53. Furthermore,
caspase 3 appears to be involved in the apoptotic pathways of p53
and RASSF1A (Fig. 4A).
Mislocalization of p53 and RASSF1A
proteins in human testicular tumors
To investigate the potential association between
localization of the p53 and RASSF1A proteins and tumorigenesis, we
analyzed their expression patterns in human testicular tissue
chips. Immunohistochemical analysis using specific antibodies
showed p53 and RASSF1A signals in samples of normal testis,
spermatocytic seminoma and lymphoma, while not in fibrous tissues
(Fig. 5). RASSF1A was weakly
expressed in a number of spermatocytic seminoma samples. In
spermatocytic seminoma and lymphoma samples, signals were observed
around the nuclear membrane, and additionally in nuclei in
spermatocytic seminoma cases, compared with mainly cytosolic
signals in normal testis. Furthermore, localization of p53 and
RASSF1A was coincident in each sample. Our results indicate
co-localization of the two proteins, with altered localization
patterns in human testicular tumors.
 | Figure 5Co-localization of RASSF1A and p53
proteins observed by immunohistochemical analysis using antibodies
against RASSF1A and p53 in human testicular chips. Upper left
panel, normal testicular samples; left middle panel, fibrous tissue
samples; lower left panel, lymphoma samples; right three panels,
seminoma samples. RASSF1A and p53 signals were observed in normal,
spermatocytic seminoma and lymphoma samples, in contrast to fibrous
tissues. In spermatocytic seminoma and lymphoma samples, signals
were observed around the nuclear membrane, and also in the nuclei
in spermatocytic seminomas, in contrast to the predominant
cytosolic signals in normal testis. Furthermore, RASSF1A and p53
proteins were co-localized in each sample. |
Discussion
Both oncogenes and tumor suppressor genes contribute
to the genesis of cancer, which involves multiple genes, including
those functioning in DNA repair, signal transduction, apoptosis and
cell cycle regulation. For instance, HPV16-E6 and RASSF1A are known
oncogenic and tumor suppressor genes that are critical in apoptosis
regulation (13–16).
The results of the present study indicate a novel
function of RASSF1A in the HPV16 pathway. Treatment of cells with
HPV16-E6 siRNA led to upregulation of p53 and downregulation of
RASSF1A, indicating that RASSF1A acts as an element of the negative
autoregulatory feedback loops activated in response to p53.
Decreased expression of RASSF1A is known to be sufficient for
maintaining a dynamic equilibrium of cell growth and apoptosis, and
the high RASSF1A level induced by HPV16 infection could partly
counteract tumorigenesis. RASSF1A may play a pivotal role in
tumorigenesis, distinct from its earlier reported function as a
tumor suppressor. The oncoprotein E6 promotes p53 degradation whose
carcinogenic effect is suppressed by RASSF1A. In response to p53,
transcriptional networks of p53-responsive genes interact with a
number of transduction pathways and positive and negative
autoregulatory feedback loops. In the present study, RASSF1A was
identified as a novel member of the negative autoregulatory
feedback loops. While RASSF1A is a known conventional tumor
suppressor, the precise mechanisms by which it interacts with other
oncogenes and tumor suppressors remain to be elucidated.
High-risk HPV types, including types 16 and 18, have
been identified in ~2/3 of all cervical cancer patients worldwide
(17,18). HPV16-E6 binds to and degrades the
p53 tumor suppressor protein, leading to malfunction of its DNA
repair mechanism (19,20). Previous studies have shown that
RASSF1A inactivation and HPV infection are mutually exclusive, and
highlight a possible correlation between HPV infection and RASSF1A
expression, which may reflect functional interactions between
RASSF1A and viral E6 (7,21). One hypothesis is that methylation
underlies this correlation. However, in the present study, a novel
mechanism is reported where HPV16-E6 regulates RASSF1A
transcription mediated via p53 protein. Treatment of cells with
HPV16-E6-siRNA led to upregulation of p53 protein, and
subsequently, to a decrease in RASSF1A transcription. RASSF1A
induces apoptosis and cell cycle alterations via its capability to
bind and stabilize the microtubule, control mitosis and regulate
genome stability. Specific effector factors include cyclin D1,
p120E4F, Cdc20, PMCA4b, Bax, CNK1 and Raf1-MST2 (14,22–26).
Our experiments demonstrated that p53 and RASSF1A induce apoptosis
through caspase 3 activation, maintaining their reported identities
as tumor suppressors. However, overexpression of both proteins
resulted in significantly lower apoptosis compared to the expected
additive effect, indicating an additional potential role of RASSF1A
in a feedback regulatory loop to balance cell survival and
death.
In summary, our findings provide novel insights into
the cellular mechanism of tumor development that might facilitate
cancer therapy and diagnosis. Further knowledge of the molecular
mechanisms downstream of RASSF1A is required to provide a reference
for tumor gene therapy.
Acknowledgements
Funding was provided by the Fundamental Research
Funds for Central Universities (274621). The authors thank
Professor Rongjia Zhou for kindly providing RASSF1A and p53
expression plasmids.
References
|
1
|
Doorbar J: Molecular biology of human
papillomavirus infection and cervical cancer. Clin Sci (Lond).
110:525–541. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tomizawa Y, Kohno T, Kondo H, et al:
Clinicopathological significance of epigenetic inactivation of
RASSF1A at 3p21.3 in stage I lung adenocarcinoma. Clin Cancer Res.
8:2362–2368. 2002.PubMed/NCBI
|
|
3
|
Dammann R, Li C, Yoon JH, Chin PL, et al:
Epigenetic inactivation of a RAS association domain family protein
from the lung tumour suppressor locus 3p21.3. Nat Genet.
25:315–319. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lehmann U, Langer F, Feist H, et al:
Quantitative assessment of promoter hypermethylation during breast
cancer development. Am J Pathol. 160:605–612. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xing M, Cohen Y, Mambo E, et al: Early
occurrence of RASSF1A hypermethylation and its mutual exclusion
with BRAF mutation in thyroid tumorigenesis. Cancer Res.
64:1664–1668. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yegnasubramanian S, Kowalski J, Gonzalgo
ML, et al: Hypermethylation of CpG islands in primary and
metastatic human prostate cancer. Cancer Res. 64:1975–1986. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kuzmin I, Liu L, Dammann R, et al:
Inactivation of RAS association domain family 1A gene in cervical
carcinomas and the role of human papillomavirus infection. Cancer
Res. 63:1888–1893. 2003.PubMed/NCBI
|
|
8
|
Cohen Y, Singer G, Lavie O, et al: The
RASSF1A tumor suppressor gene is commonly inactivated in
adenocarcinoma of the uterine cervix. Clin Cancer Res. 9:2981–2984.
2003.PubMed/NCBI
|
|
9
|
Kim SH, Juhnn YS, Kang S, et al: Human
papillomavirus 16 E5 up-regulates the expression of vascular
endothelial growth factor through the activation of epidermal
growth factor receptor, MEK/ERK1,2 and PI3K/Akt. Cell Mol Life Sci.
63:930–938. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tan S, Hougardy BM, Meersma GJ, et al:
Human papilloma virus 16 E6 RNA interference enhances cisplatin and
death receptor-mediated apoptosis in human cervical carcinoma
cells. Mol Pharmacol. 81:701–709. 2012. View Article : Google Scholar
|
|
11
|
Vassallo J, Derchain SF, Pinto GA,
Martinez EZ, Syrjänen KJ and Andrade LA: High risk HPV and p53
protein expression in cervical intraepithelial neoplasia. Int J
Gynaecol Obstet. 71:45–48. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tian Y, Hou Y, Zhou X, et al: Tumor
suppressor RASSF1A promoter: p53 binding and methylation. PLoS One.
6:e170172011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Preethy CP, Padmapriya R, Periasamy VS, et
al: Antiproliferative property of n-hexane and chloroform extracts
of Anisomeles malabarica (L). R Br in HPV16-positive human
cervical cancer cells. J Pharmacol Pharmacother. 3:26–34. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Matallanas D, Romano D, Yee K, et al:
RASSF1A elicits apoptosis through an MST2 pathway directing
proapoptotic transcription by the p73 tumor suppressor protein. Mol
Cell. 27:962–975. 2007. View Article : Google Scholar
|
|
15
|
Shivakumar L, Minna J, Sakamaki T, et al:
The RASSF1A tumor suppressor blocks cell cycle progression and
inhibits cyclin D1 accumulation. Mol Cell Biol. 22:4309–4318. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Song MS, Song SJ, Ayad NG, et al: The
tumour suppressor RASSF1A regulates mitosis by inhibiting the
APC-Cdc20 complex. Nat Cell Biol. 6:129–137. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fen J, Yoshinouchi M, Nakamura K, et al:
Eradication of HPV post-surgical treatments, its correlation with
specific types, types of surgery and the physical status. Oncol
Rep. 12:375–379. 2004.PubMed/NCBI
|
|
18
|
Bosch FX, Manos MM, Muñoz N, et al:
Prevalence of human papillomavirus in cervical cancer: a worldwide
perspective. International Biological Study on Cervical Cancer
(IBSCC) Study Group. J Natl Cancer Inst. 87:796–802. 1995.
View Article : Google Scholar
|
|
19
|
Pöllänen R, Soini Y, Vähäkangas K, et al:
Aberrant p53 protein expression in cervical intra-epithelial
neoplasia. Histopathology. 23:471–474. 1993.
|
|
20
|
Boulet G, Horvath C, Vanden Broeck D, et
al: Human papilloma virus: E6 and E7 oncogenes. Int J Biochem Cell
Biol. 39:2006–2011. 2007. View Article : Google Scholar
|
|
21
|
Kuzmin I, Gillespie JW, Protopopov A, et
al: The RASSF1A tumor suppressor gene is inactivated in prostate
tumors and suppresses growth of prostate carcinoma cells. Cancer
Res. 62:3498–3502. 2002.PubMed/NCBI
|
|
22
|
Rong R, Jiang LY, Sheikh MS, et al:
Mitotic kinase Aurora-A phosphorylates RASSF1A and modulates
RASSF1A-mediated microtubule interaction and M-phase cell cycle
regulation. Oncogene. 26:7700–7708. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ahmed-Choudhury J, Agathanggelou A, Fenton
SL, et al: Transcriptional regulation of cyclin A2 by RASSF1A
through the enhanced binding of p120E4F to the cyclin A2 promoter.
Cancer Res. 65:2690–2697. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Song SJ, Song MS, Kim SJ, et al: Aurora A
regulates prome-taphase progression by inhibiting the ability of
RASSF1A to suppress APC-Cdc20 activity. Cancer Res. 69:2314–2323.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Baksh S, Tommasi S, Fenton S, et al: The
tumor suppressor RASSF1A and MAP-1 link death receptor signaling to
Bax conformational change and cell death. Mol Cell. 18:637–650.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rabizadeh S, Xavier RJ, Ishiguro K, et al:
The scaffold protein CNK1 interacts with the tumor suppressor
RASSF1A and augments RASSF1A-induced cell death. J Biol Chem.
279:29247–29254. 2004. View Article : Google Scholar : PubMed/NCBI
|