Introduction
Prostate cancer (PC) in developed countries accounts
for ~15.3% of all male malignancies and it is the most common
non-dermatological epithelial malignant tumor in males (1).
The aggressive nature of cancer cells has been
investigated through the analysis of complex signaling pathways
that regulate important functions, including cell survival,
proliferation, invasion and migration. Cancer cell migration and
proliferation are essential factors in invasion and metastasis
(2,3). Phosphatidylinositol 3-kinases (PI3Ks)
have been shown to be involved in cell processes responsible for
malignant behaviors, including survival, proliferation,
transformation and adherence, and have been implicated in the
carcinogenic mechanisms of cancer (4,5). The
PI3K pathway has also been shown to act through its downstream AKT
(protein kinase B) molecule by regulating various cell functions,
including cell transformation, proliferation, apoptosis,
angiogenesis and tumor growth.
PIK3CA is mapped to 3q26, an area significantly
amplified in other types of human cancer, including ovarian,
breast, cervical, head and neck and urinary tract cancer (6,7).
PIK3CA mutations have been recently identified in several types of
human cancer. Moreover, PIK3CA has been found to be mutated in 25,
32, 27 and 4% of brain, colon, lung and gastric cancer,
respectively (8,9).
To elucidate the oncogenic mechanisms underlying the
development of cancer, we screened a human mammary cDNA library to
identify novel PI3K p110α-interacting proteins and to further
investigate the detailed mechanisms and functions involved in the
metastasis and proliferation of carcinoma. Among the candidate
genes, we identified receptor for activated protein kinase C1
(RACK1) as a putative target in human breast carcinoma cells.
RACK1, a homolog of the β-subunit of heterotrimeric G proteins
(10), is involved in the membrane
anchoring of multiple proteins (including protein kinase C, PKC)
and the coordination of cell adhesion, movement, growth and
division (11,12). However, the detailed mechanisms and
functions of RACK1 in the metastasis and proliferation of PC cells
have yet to be fully elucidated.
The aim of the present study was to investigate the
functions of RACK1 and its involvement in mechanisms of PC cell
proliferation, invasion and metastasis. Therefore, we investigated
the potential downstream signaling pathways following targeted
manipulation of RACK1 in the PC cell line DU145. Our data indicated
that RACK1 promotes PC cell proliferation, metastasis and invasion
in vitro and in vivo.
Materials and methods
Cells
DU145 cells stably transfected with RACK1 were
obtained from the Shanghai Medical College of Fudan University
(Shanghai, China).
Cell culture and transfection
Prostate carcinoma DU145 cells were used in the
present study. The cells were cultured in DMEM supplemented with
10% fetal bovine serum (FBS; Gibco-BRL, Grand Island, NY, USA), 50
U/ml streptomycin, 50 U/ml penicillin, 2 mmol/l glutamine and
maintained at 37°C. Transient transfection of cells was achieved
using Lipofectamine™ 2000 reagent (Invitrogen Life Technologies,
Carlsbad, CA, USA). DU145 cells were transfected 12 h after plating
(1.5×106–2×107 cells/dish) according to the
transfection protocol, and harvested for investigation within 72 h
after transfection. DU145 cells stably expressing RACK1 were
generated by transfection and selection with concentration of
geneticin selective G418 antibiotic (Gibco-BRL) for 5–6 weeks at
Shanghai Medical College of Fudan University. Surviving DU145 cells
were maintained in 1.0 mg/ml of G418. siRNAs targeting the human
RACK1 (GNB2L1) gene sequence, 50CAGATTGTCTCTGGATCTCGA, were
obtained from Qiagen (Shanghai, China).
Immunoprecipitation and
immunoblotting
Forty-eight hours after transfection, DU145 cells
were collected, washed with phosphate-buffered saline (PBS; pH
7.4), and lysed in modified RIPA buffer [50 mM Tris (pH 7.8), 5 mM
EDTA, 15 mM MgCl2, 150 mM NaCl, 1% NP-40, 1 mM DTT, 0.5%
sodium deoxycholate and 20 mM N-ethylmaleimide] supplemented with 1
tablet/50 ml of Complete Protease Inhibitor Cocktail (Roche
Molecular Biochemicals, Indianapolis, IN, USA). Lysates were
cleared by centrifugation (104 × g for 15 min at 4°C)
and incubated on ice for 2 h with 2 μg of the appropriate
antibodies (Abs; anti-Flag from Sigma-Aldrich, St. Louis, MO, USA;
anti-GFP from Roche Applied Science, Indianapolis, IN, USA;
anti-RACK1 and anti-PI3K p110α from BD Biosciences, San Jose, CA,
USA). Subsequently, 20 μl of slurry protein A/G Plus Agarose
immunoprecipitation reagent (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA) was added to each lysate, and incubation was
continued with rotation for ≥2 h at 4°C. The beads were retrieved
by centrifugation and washed (by vortex and short spin) 4 times
with RIPA buffer and once with PBS. Proteins bound to the beads
were eluted by boiling in a 29X electrophoresis sample buffer,
separated by SDS-PAGE, and electrotransferred onto PVDF membranes
(Millipore, Bedford, MA, USA). After blocking in 5% nonfat milk,
western blot analysis was performed with the indicated primary Abs
for 1 h at room temperature, followed by incubation with
horseradish peroxidase (HRP)-conjugated secondary Abs for 1 h at
room temperature. All of the blots were developed using the ECL
Plus western blot detection system (Pierce Biotechnology, Inc.,
Rockford, IL, USA) and X-ray film (Kodak, Rochester, NY, USA).
Gel-Pro Analyzer software (version 4.0) was used for the
densitometric analysis of each film. The primary Abs used in
western blot analysis were the following: phospho-p44/42 MAPK
(Thr202/Tyr204), phospho-AKT (Ser473; Cell Signaling Technology,
Inc., Danvers, MA, USA); p21, p27, cyclin D1 and D3 (Santa Cruz
Biotechnology, Inc.); p53 (Sigma-Aldrich); and CD147 (Novocastra,
Newcastle-upon-Tyne, UK). The secondary Abs used for
immunodetection were the following: HRP-conjugated goat anti-mouse
IgG and goat anti-rabbit IgG (Amersham Biosciences, Uppsala,
Sweden). The potent inhibitor of PI3K was purchased from Cell
Signaling Technology, Inc.
Immunofluorescence
DU145 cells seeded on 6-well chamber slides were
fixed in 4% paraformaldehyde, blocked in 5% bovine serum albumin
(BSA) and permeabilized in 0.1% Triton X-100. PI3K p110α levels
were determined with anti-PI3K p110α (BD Biosciences), followed by
a 45-min incubation with Cy3-conjugated anti-mouse secondary Ab
(Amersham Biosciences). The coverslips were washed, mounted in PBS
containing 50% glycerol, and observed under a confocal laser
microscope (Leica Microsystems, Buffalo Grove, IL, USA) or an
automated microscope (Leica DMRXA2) equipped with Photometrics Cool
SnapES N&B camera driven by MetaMorph software (Universal
Imaging Corporation, Downingtown, PA, USA).
PC cell proliferation assay
PC DU145 cell proliferation was investigated using
an MTT cell proliferation kit (Roche Applied Science). DU145 cells
were cultured in 96-well plates at a density of 1.0×104
cells/well. On 1–6 days following transfection with RACK1 siRNA,
DU145 cells were incubated with 10 μl of MTT labeling reagent for 4
h, followed by the addition of 100 μl of solubilization solution
into each well. The plates were maintained in the dark overnight,
and the optical density (OD) of each sample was measured at a
wavelength of 490 nm using an ELISA multi-well spectrophotometer
(Molecular Devices, Sunnyvale, CA, USA).
In order to determine the DNA content, DU145 cells
were harvested by trypsinization 24 h after transfection, fixed
overnight at 4°C with 75% (v/v) ethanol, washed and incubated in
PBS containing 100 μg/ml RNase and 10 μg/ml propidium iodide (PI)
for 1 h at 37°C. Data were obtained by a Fluorescence-activated
cell sorter (FACS) Canto flow cytometer and analyzed using the
FACSDiva software package (BD Biosciences). A minimum of
1×104 cells were measured per sample. Gates were based
on forward and side scatter set to eliminate cell clusters and
cellular debris.
Invasion and migration assay
Scratch assay was performed with 1×106
DU145 cells seeded onto a 6-well cell plate, which were allowed to
reach full confluence. The monolayer was wounded with a 100-μl
pipette tip, followed by further culture in the appropriate media
as previously described. Images were captured at 0, 12, 24 and 48
h. A modified Boyden dual chamber assay was used to measure the
directional migration of DU145 cells. In brief, 5×104
cells were suspended in serum-free media and added to the upper
chamber of an 8-μm pore cell culture insert (Becton-Dickinson, San
Jose, CA, USA). Serum-containing medium was used as a
chemoattractant in the lower chamber. After a 24- to 72-h
incubation at 37°C in 5% CO2, the media and cells that
were still in the upper chamber were removed using a cotton swab.
The insert was fixed in ethanol and stained using hematoxylin and
eosin (H&E). DU145 cells that were attached to the lower side
of the insert were observed using an inverted microscope (Nikon,
Tokyo, Japan), and the number of invading DU145 cells was
determined by counting the cells in 5 random high-power fields and
calculating the mean number of invading cells. We then investigated
the ability of the DU145 cells to invade through growth factor
reduced Matrigel (BD Biosciences). The Rho kinase (ROCK) inhibitor
Y-27632 was purchased from Calbiochem (Darmstadt, Germany). All of
the experiments were performed in triplicate.
Zymographic analysis
Matrix metalloproteinase (MMP)-9 and -2 enzymatic
activity was assessed using gelatin zymography. Conditioned media
from DU145 cells (cultured in serum-free medium for 48 h) were
collected and concentrated 20-fold by a Centriprep YM-30 device
(Millipore, Bedford, MA, USA). Samples were then mixed with Laemmli
loading buffer and electrophoresed on a gelatin containing 8%
SDS-PAGE. Following electrophoresis, the gel was washed twice with
washing buffer (100 mM NaCl, 2.5% Triton X-100 and 50 mM Tris-HCl,
pH 7.5), followed by a brief rinse in washing buffer without Triton
X-100. The gel was then placed with incubation buffer (50 mM NaCl,
50 mM Tris-HCl, pH 7.5, 1 μM ZnCl2, 10 mM
CaCl2, 0.02% NaN3) at 37°C for 24 h. After
incubation, the gel was stained with Coomassie Blue R-250 and
destained with destaining solution. A clear zone of gelatin
digestion represented the MMP activity.
Establishment of the PC nude mouse
model
Male nu/nu nude mice (4–6 weeks old) were injected
with no-free DU145 control cells (group 1; parental MCF7 cells and
cells transfected with empty vector, 5 mice each), M-R4 cells
(group 2), T-47D control (group 3) or T-R2 cells (group 4). Each
group contained 10 mice. The cells were injected into the second
mammary fat pad from the flank (29×106 cells in 100 μl
of PBS). Tumor growth was externally monitored using Vernier
Calipers for 4–6 weeks. At 6 weeks after injection (when the mice
had not yet died, but some appeared to be sick), all of the mice
were sacrificed, and the tumors were removed and fixed in 10%
formalin after weighing and measuring. Tumor volume was calculated
using the formula: Tumor volume (mm3) = 0.5a ×
b2, where 0.5 is a constant to calculate the volume of
an ellipsoid, a is the longest diameter and b is the shortest
diameter. Histopathological analysis was performed according to the
procedures described below.
Immunohistochemistry
Analysis of RACK1 expression in PC tissue was
performed using the RACK1 mouse monoclonal Ab (LifeSpan
Biosciences, Inc., Seattle, WA, USA). For the analysis of Ki67,
PTEN, androgen receptor (AR) and MMP2 expression, the corresponding
mouse monoclonal Abs from Sigma-Aldrich were used. The positive
controls for RACK1 were used with sections of formalin-fixed,
paraffin-embedded human adrenal samples as indicated in the
instruction manual. The negative controls were incubated with
immunoglobulin fraction in place of the polyclonal primary Ab in
the positive tissues mentioned above. The saturation and intensity
of the immunostained cells were evaluated over 3 visual fields, at
a magnification of ×200 under a light microscope (Carl Zeiss,
Gottingen, Germany). For the statistical analysis, according to Han
et al(13) and our previous
studies (unpublished data), the total staining of RACK1 was scored
based on the intensity and percentage of cells with RACK1 cytoplasm
staining on the following scoring system: score 0, negative/weak
staining for all tumor cells or moderate staining <30%; score 1,
moderate staining in >30% but <70% of tumor cells or strong
staining within 30% of tumor cells; score 2, moderate staining in
>70%, or strong staining in >30% of all tumor cells.
Statistical analysis
All data were representative of ≥3 independent
experiments with similar results. The results are expressed as the
mean ± SEM from multiple experiments. The Student’s t-test was used
to determine significant differences (two-tailed, P<0.05). The
software SPSS 15.0 for Windows (SPSS Inc., Chicago, IL, USA) was
used for statistical analyses. P<0.05 was considered to indicate
a statistically significant difference.
Results
Promotion of proliferation, migration and
invasion by RACK1 in vitro
To investigate the importance of RACK1 in PC
tumorigenesis and progression, we first assessed RACK1 expression
in the PC cell line DU145 as mentioned in the Materials and methods
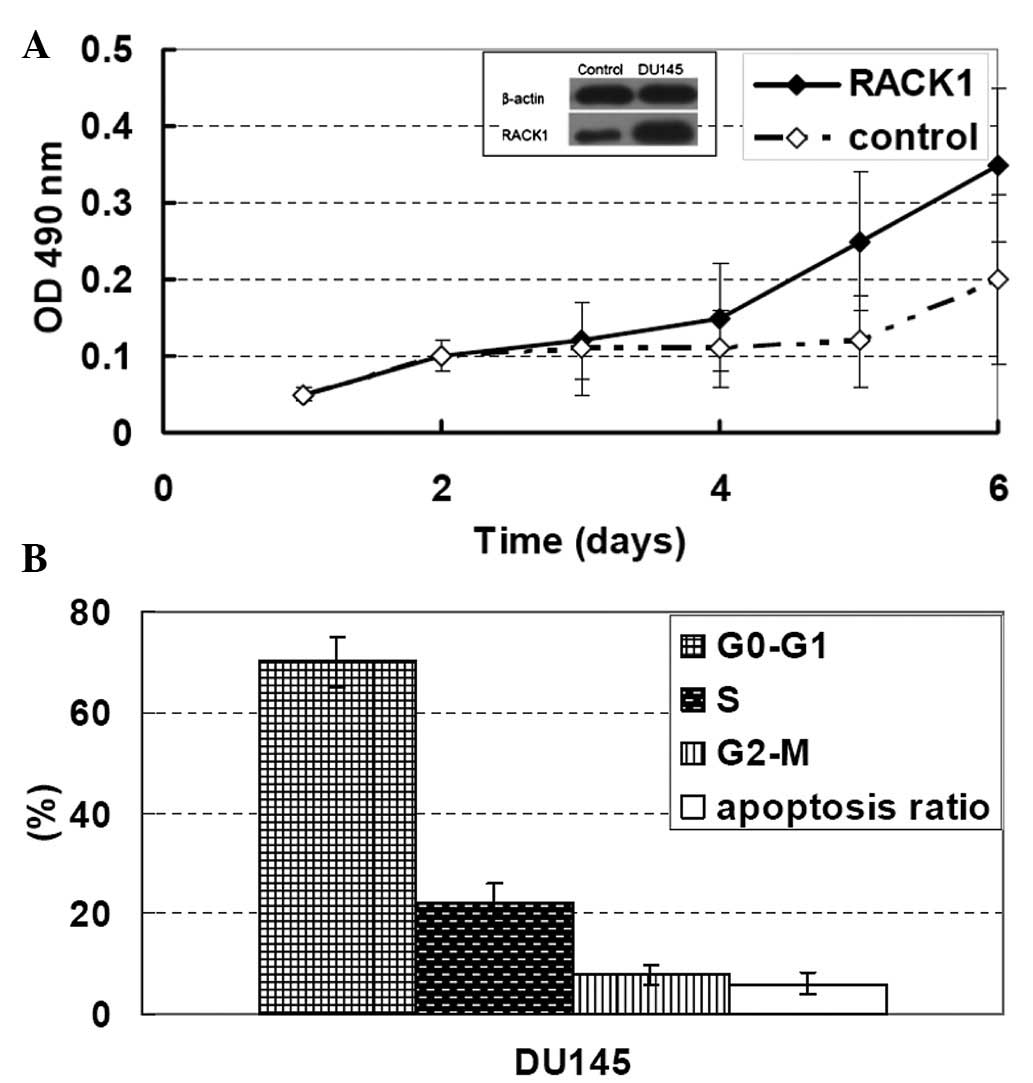
section. As shown in Fig. 1A, the
cells with a superior invasive ability had a higher RACK1
expression, while the cells with an inferior invasive ability
(non-transfected cells) had a lower RACK1 expression.
To detect the effect of RACK1, we initially
constructed two stably transfected cell lines: DU145 cells
transfected with a RACK1-expressing plasmid or an empty plasmid
(control cells) and stable cell lines were established as described
in the Materials and methods. The stably transfected clones were
obtained and further measured. The RACK1 expression level was
increased 350% in clone DU145 cells and <200% compared with the
control cells (Fig. 1A). The DU145
cell clones were stable in culture and maintained a high RACK1
level for ≥22 passages in medium containing the selection agent
G418. Similar passages of these clones were used in all of the
subsequent studies.
In order to examine the effect of RACK1 on the
tumorigenic properties of PC cells, we then investigated the in
vitro proliferation of stably transfected clones (DU145 cells).
Significantly decreased cell doubling times were observed in the
RACK1-transfected clones compared with the control cells; the mean
doubling times (95% CI) were as follows: control, 26.9 h (range,
26.4–27.4 h), P=0.07 and clone DU145 cells, 19.4 h (range,
19.1–19.7 h), P<0.001 (Fig. 1A and
B). Flow cytometric analysis of clone DU145 cells also
demonstrated the ability of RACK1 to promote PC cell proliferation
without affecting the rate of apoptosis (Fig. 1B).
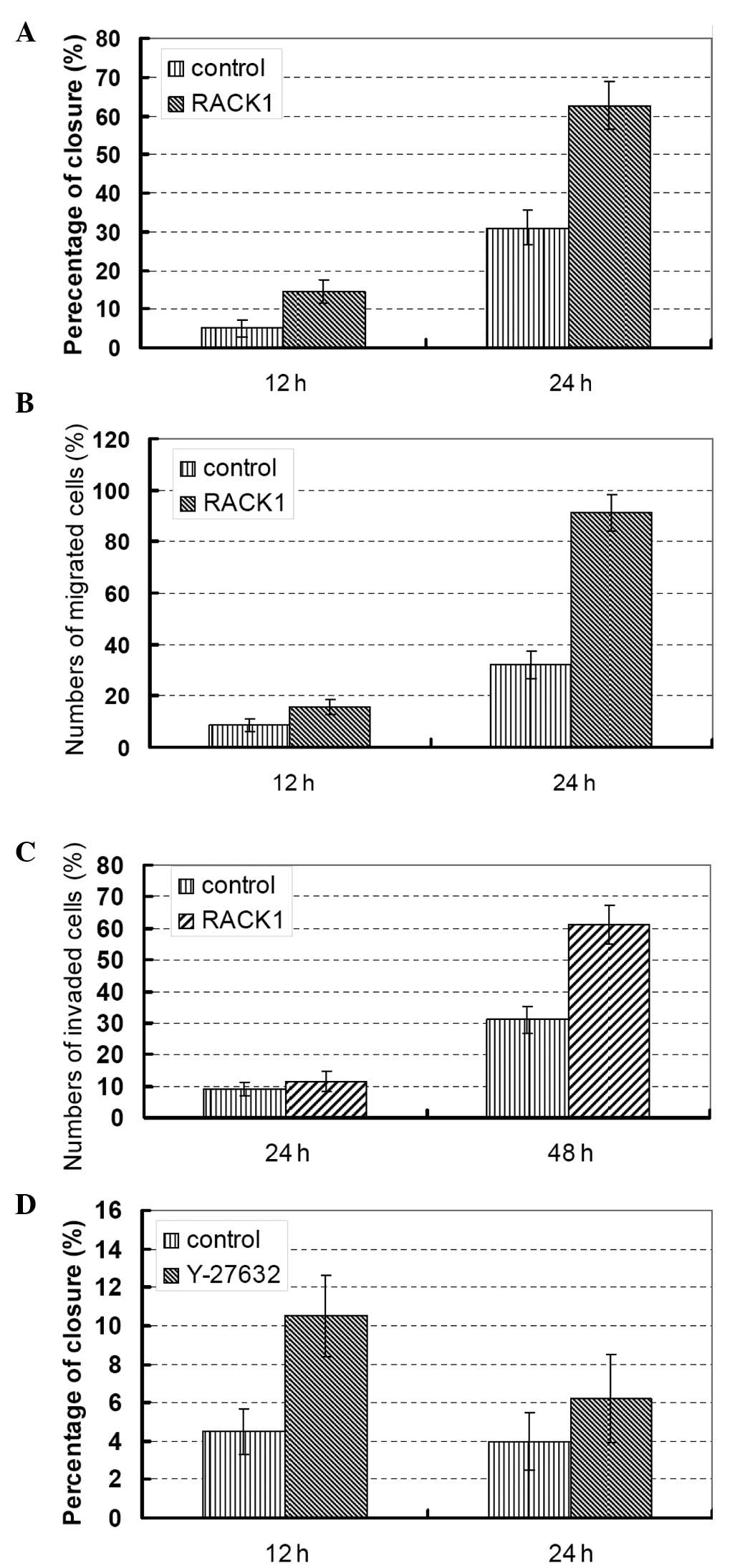
Furthermore, the effects of RACK1 on PC cell
invasion and migration were determined through scratch tests
(Fig. 2A) and Transwell assays
without (Fig. 2B) or with Matrigel
(Fig. 2C). As shown in Fig. 2, upregulation of RACK1 in clone
DU145 cells induced an increased cell migration capacity (Fig. 2B). In addition, DU145 cell invasion
abilities were significantly increased, as assessed after 48 h
using modified Boyden chamber assays. The mean number of migrated
cells/field (95% CI) was as follows: clone RACK1 siRNA-transfected
DU145 cells, 60 (range, 50–78; P<0.001; Fig. 2C).
To verify the effects of RACK1, we introduced siRNAs
specifically targeting RACK1 into the PC DU145 cells and the MTT
assay was repeated (Fig. 1B). We
next compared the migration and invasion properties of RACK1
knockdown cells compared with that of parental cells (Fig. 2A and B). These data confirmed the
promotion of cell proliferation, migration and invasion by RACK1 in
PC cells.
Promotion of tumor growth, infiltration
and metastasis by RACK1 in a nude mouse model
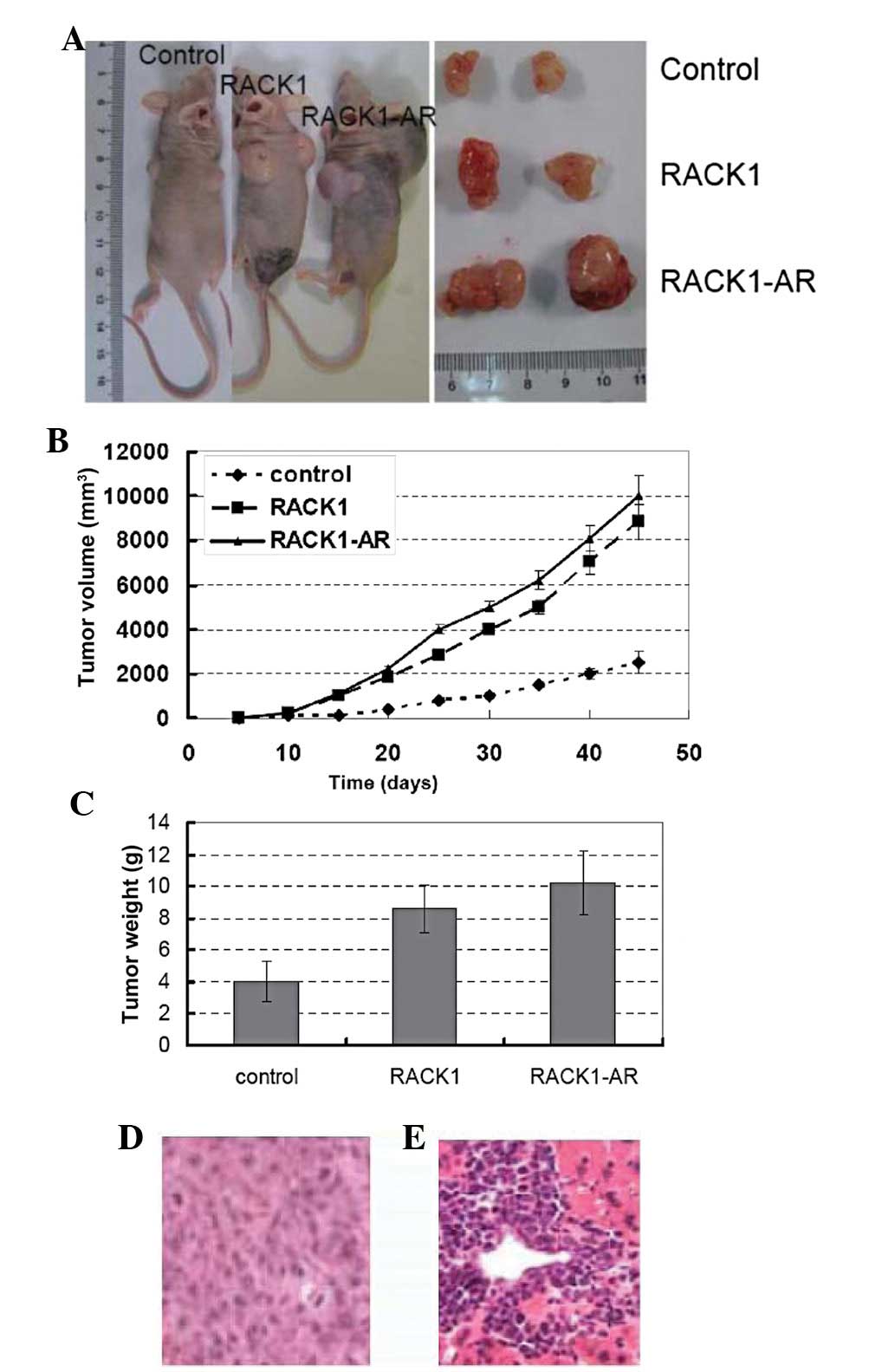
We investigated the effects of RACK1 on tumor growth
and metastasis in an orthotopic nude male primary androgen ablation
therapy mouse model. Stably transfected PC DU145 cells, DU145 cells
and AR-overexpressing endothelial cells and control cells were
implanted into the head and neck from the flank of each male nude
mouse as described in Materials and methods. Six weeks following
injection, the nude mice were sacrificed, and tumor incidence and
weight were determined (Fig. 3).
All of the mice (100%) that were orthotopically injected with
parental cells (control groups) and transfected cells developed
tumors. Mice implanted with RACK1-transfected clones or AR- and
RACK1-overexpressing cells developed significantly larger tumors
compared with the control groups (Fig.
3A–C). A mouse in Group 2 and Group 3, bearing the largest
tumor, was found to have a necrotic lower limb that may have been
caused by a tumor embolus (Fig.
3A). All of the mice (10/10) in Groups 2 and 3 were observed to
have cell infiltration into adjacent tissue (Fig. 3D) and had macroscopically visible
metastasis in the liver (one mouse) and/or microscopic metastases
in the liver (all 10 mice; Fig.
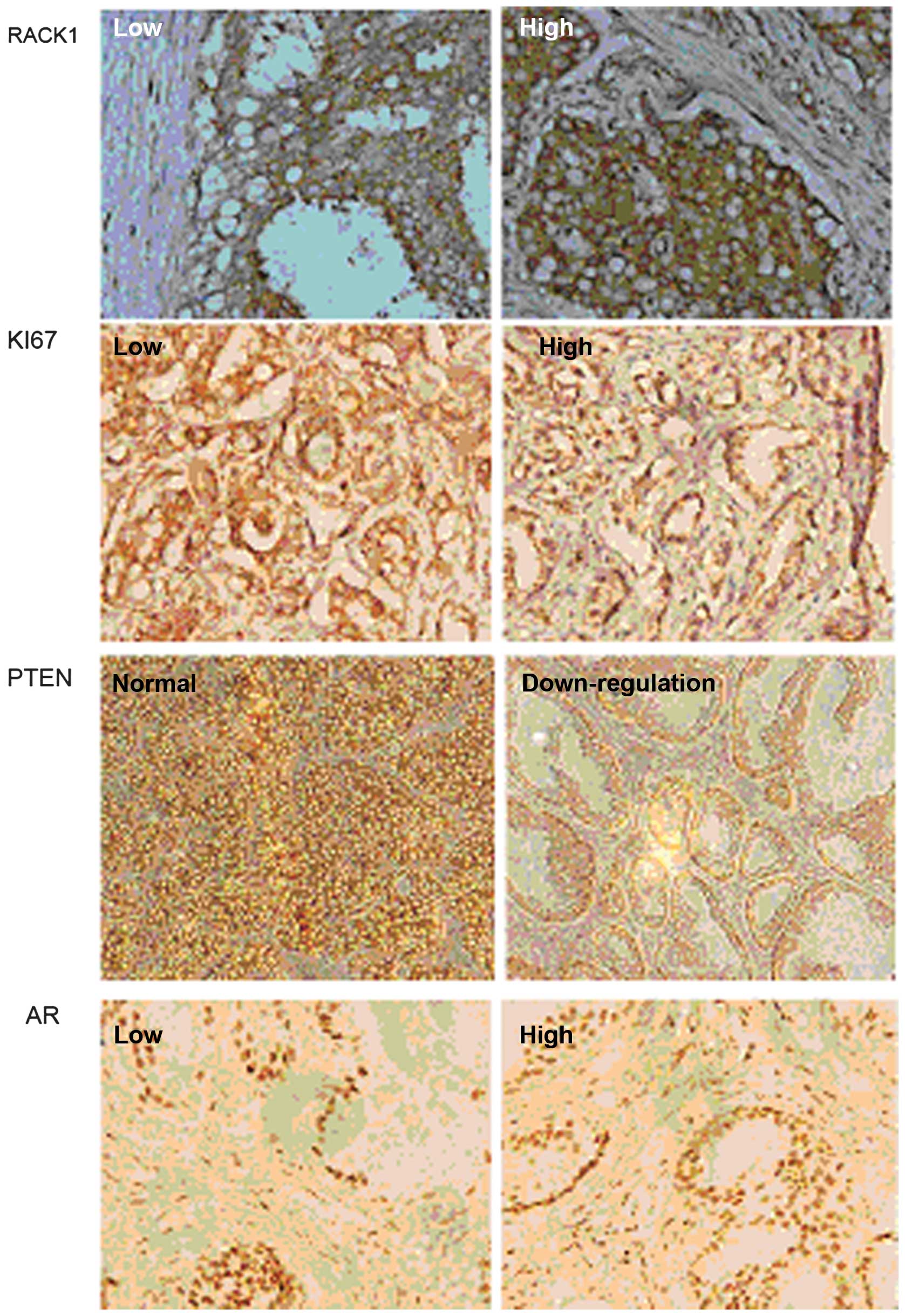
3E). The expression of Ki67, PTEN and AR was also evaluated in
tumor tissue removed from the nude mice (Fig. 4) and suggested an important role
for RACK1 in PC cell growth and metastasis in vivo.
Discussion
PI3K has been shown to be involved in cell survival,
proliferation, cytoskeletal reorganization, metabolism and membrane
trafficking (3,4), which are highly implicated in
mechanisms of prostate carcinogenesis. In the present study, a
human PC cDNA library was initially screened with PI3K p110α as
bait, to identify new mechanisms involved in PC cell growth and
progression. Of the proteins identified, RACK1 was selected for
detailed analysis. For confirmation, we investigated the PI3K
p110α/RACK1 interaction using in vivo binding experiments.
Furthermore, the physiological interaction between PI3K p110α and
RACK1 was confirmed by immunoprecipitation and immunofluorescence
analysis of DU145 cells, where RACK1 was identified as a novel
binding partner of PI3K p110α. A series of in vitro and
in vivo experiments were performed to investigate the
characteristics of RACK1 and to elucidate its role in prostate
tumorigenesis. The mechanisms underlying the effect of RACK1 on
prostate tumorigenesis were also investigated. The upregulation of
RACK1 was shown to significantly promote prostate tumor growth
in vitro, using the DU145 cell line by MTT assay and flow
cytometry, and in vivo using an orthotopic nude mouse model.
Notably, our results are in agreement with those obtained by Wang
et al(14), where
overexpression of RACK1 was found to be an important predictor of
oral squamous cell carcinoma. In the present study, RACK1 staining
was found to be closely associated to Ki67 staining (Fig. 4), which is one of the most widely
known proliferative predictors in malignant diseases, such as
breast carcinoma. Our data indicated that increased RACK1
expression is not only closely related to in vitro cell
proliferation, migration and invasion, but also linked to PC cell
growth and metastasis in vivo.
The RACK1 pathway is suggested to act through PI3K.
The PI3K pathway has also been shown to act through its downstream
AKT molecule by regulating various cell functions, including cell
transformation, proliferation, apoptosis, angiogenesis and tumor
growth. Phosphatase and tensin homolog deleted on chromosome ten
(PTEN) is a PI3K inhibitor. PTEN loss or mutation is common in
human PC. Inhibition of the PI3K signaling pathway by PTEN inhibits
tumor angiogenesis and growth. We have previously demonstrated that
AKT is the downstream target of PI3K in controlling angiogenesis
and tumor growth, and that PTEN inhibits angiogenesis by regulating
the expression of HIF-1 and VEGF expression through AKT activation
in PC-3 cells.
AR and PI3K signaling are two of the most important
pathways involved in PC. Previous work has shown that there is
crosstalk between these two pathways (15). AR and PI3K pathways crosstalk in PC
cells in vitro as well as in vivo. The PI3K pathway
is dominant over AR signaling in PC cells (16–19)
(Fig. 4). This should be taken
into consideration in future studies on the development of novel
therapeutic strategies for PC treatment.
References
|
1
|
Grönberg H: Prostate cancer epidemiology.
Lancet. 361:859–864. 2003.PubMed/NCBI
|
|
2
|
Harbeck N, Dettmar P, Thomssen C, et al:
Risk-group discrimination in node-negative breast cancer using
invasion and proliferation markers: 6-year median follow-up. Br J
Cancer. 80:419–426. 1999.PubMed/NCBI
|
|
3
|
Leevers SJ, Vanhaesebroeck B and
Waterfield MD: Signalling through phosphoinositide 3-kinases: the
lipids take centre stage. Curr Opin Cell Biol. 11:219–225. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fang J, Ding M and Yang L: PI3K/PTEN/AKT
signaling regulates prostate tumor angiogenesis. Cell Signal.
19:2487–2497. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Samuels Y, Wang Z, Bardelli A, et al: High
frequency of mutations of the PIK3CA gene in human cancers.
Science. 304:5542004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Broderick DK, Di C, Parrett TJ, et al:
Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade
astrocytomas, and medulloblastomas. Cancer Res. 64:5048–5050. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Campbell IG, Russell SE, Choong DY, et al:
Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer
Res. 64:7678–7681. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao JJ, Gjoerup OV, Subramanian RR, et
al: Human mammary epithelial cell transformation through the
activation of phosphatidylinositol 3-kinase. Cancer Cell.
3:483–495. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mao X, Kikani CK, Riojas RA, et al: APPL1
binds to adiponectin receptors and mediates adiponectin signalling
and function. Nat Cell Biol. 8:516–523. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ron D, Chen CH, Caldwell J, et al: Cloning
of an intracellular receptor for protein kinase C: a homolog of the
beta subunit of G proteins. Proc Natl Acad Sci USA. 91:839–843.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Berns H, Humar R, Hengerer B, et al: RACK1
is up-regulated in angiogenesis and human carcinomas. FASEB J.
14:2549–2558. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Han HJ, Russo J, Kohwi Y and
Kohwi-Shigematsu T: SATB1 reprogrammes gene expression to promote
breast tumor growth and metastasis. Nature. 452:187–193. 2008.
View Article : Google Scholar
|
|
14
|
Wang Z, Jiang L, Huang C, et al:
Comparative proteomics approach to screening of potential
diagnostic and therapeutic targets for oral squamous cell
carcinoma. Mol Cell Proteomics. 7:1639–1650. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cao XX, Xu JD, Xu JW, et al: RACK1
promotes breast carcinoma proliferation and invasion/metastasis in
vitro and in vivo. Breast Cancer Res Treat. 123:375–86. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mulholland DJ, Tran LM, Li Y, et al: Cell
autonomous role of PTEN in regulating castration-resistant prostate
cancer growth. Cancer Cell. 19:792–804. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Serrels B, Sandilands E, Serrels A, et al:
A complex between FAK, RACK1, and PDE4D5 controls spreading
initiation and cancer cell polarity. Curr Biol. 20:1086–1092. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang F, Osawa T, Tsuchida R, et al:
Downregulation of receptor for activated C-kinase 1 (RACK1)
suppresses tumor growth by inhibiting tumor cell proliferation and
tumor-associated angiogenesis. Cancer Sci. 102:2007–2013. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shi Y, Han JJ, Tennakoon JB, et al:
Androgens promote prostate cancer cell growth through induction of
autophagy. Mol Endocrinol. 27:280–295. 2013. View Article : Google Scholar : PubMed/NCBI
|