Introduction
The endoplasmic reticulum (ER) is an organelle that
folds and synthesizes transmembrane, intraorganellar and secretory
proteins. The disequilibrium of ER homeostasis, including glucose
deprivation, disturbance of the redox environment, perturbation of
calcium homeostasis and exposure to free radicals disrupts the
normal function of the ER and induces ER stress (1). Pro-apoptotic proteins are expressed
under ER stress, including CHOP and caspase-12 and -3, which
results in neuronal cell death. ER stress plays a role in the
pathogenesis of a variety of human diseases, including neuronal
degenerative diseases, ischemia/reperfusion injury, and heart
diseases (2–4).
The transient receptor potential cation channels
(TRPC) are a subfamily of nonselective cation channels permeable to
Ca2+, which are present in numerous cell types including
neurons (5,6). The TRPC6 channel is involved in the
promotion of neuronal survival following focal cerebral ischemia.
Activation of calpain leads to TRPC6 degradation and neuronal
damage in ischemia (7). Previous
studies have determined that the TRPC6 channel is essential in
promoting neuronal survival and indicates that the activation of
CREB is a key downstream effector for the neuronal protective
effect of the TRPC6 channel in vitro and in
vivo(7,8). Therefore, it may be a used as a novel
therapeutic strategy to protect against ischemic brain damage as
the inhibition of TRPC6 degradation preserves neuronal
survival.
EGCG is the predominant constituent of green tea
(9). It has been shown to promote
neuronal plasticity (10) and to
improve cognitive function and learning ability (11,12).
In addition, EGCG has also been demonstrated to reduce delayed cell
death near the hippocampus and the excitotoxic neuronal damage that
occurs in ischemic lesions following transient ischemia (2,13,14).
However, the precise mechanism of the neuroprotective activity of
EGCG remains unclear. Previous studies have not fully demonstrated
the effects of EGCG on TRPC6/CREB-mediated neuroprotection.
The aim of this study was to investigate whether the
administration of EGCG immediately following ischemia exhibits a
neuroprotective effect on ischemic neurons in a middle cerebral
artery occlusion (MCAO) rat model. This study also aimed to
determine whether EGCG inhibits ER stress (ERS) and improves the
neurological status through the inhibition of calpain-mediated
TRPC6 proteolysis and the subsequent activation of CREB via the
mitogen-activated protein kinase kinase (MEK)/extracellular
signal-regulated kinases (ERK) pathway.
Materials and methods
Middle cerebral artery occlusion (MCAO)
model
Male Sprague-Dawley rats (weight, 200–250 g) were
purchased from Hunan Weasleyg Scene of Experimental Animals Co.,
Ltd. (Hubei, China). The experiments were approved by the Committee
of Experimental Animals of Tongji Medical College (Hubei, China)
and conformed to internationally accepted ethical standards (Guide
for the care and use of laboratory animals; NIH Publication 80–23,
revised 1978). Briefly, rats were anesthetized with an
intraperitoneal (i.p.) injection of chloral hydrate (400 mg/kg) and
placed in the supine position with the limbs taped to the operation
table. A midline skin incision was performed, the right external
carotid artery was exposed and its branches were ligated. A 4-0
monofilament nylon suture (Beijing Shandong Industrial Corp.,
Beijing, China) with a rounded tip was introduced into the internal
carotid artery through the common carotid artery and advanced until
faint resistance was felt. Following 2 h of transient MCAO, blood
flow was restored by the withdrawal of the nylon thread to allow
reperfusion, which was confirmed by a laser Doppler flowmeter;
Periflux system 5000, Perimed, Stockholm, Sweden. Sham-operated
rats underwent the same procedure without the filament insertion.
Throughout the experiments, body temperature was maintained at
37±0.5°C with a homeothermic (RWD Life Science Co., Ltd., Shenzhen,
China). Regional cerebral blood flow (rCBF) was monitored by a
laser-Doppler flowmeter prior to, during and following MCAO, as
well as prior to death (Fig. 1).
Animals that did not show a CBF reduction of ≥70% and animals that
died following ischemia induction were excluded from the
experimental group. Prior to reperfusion, rats with incomplete MCAO
(~10%) were excluded from further study by a blinded observer.
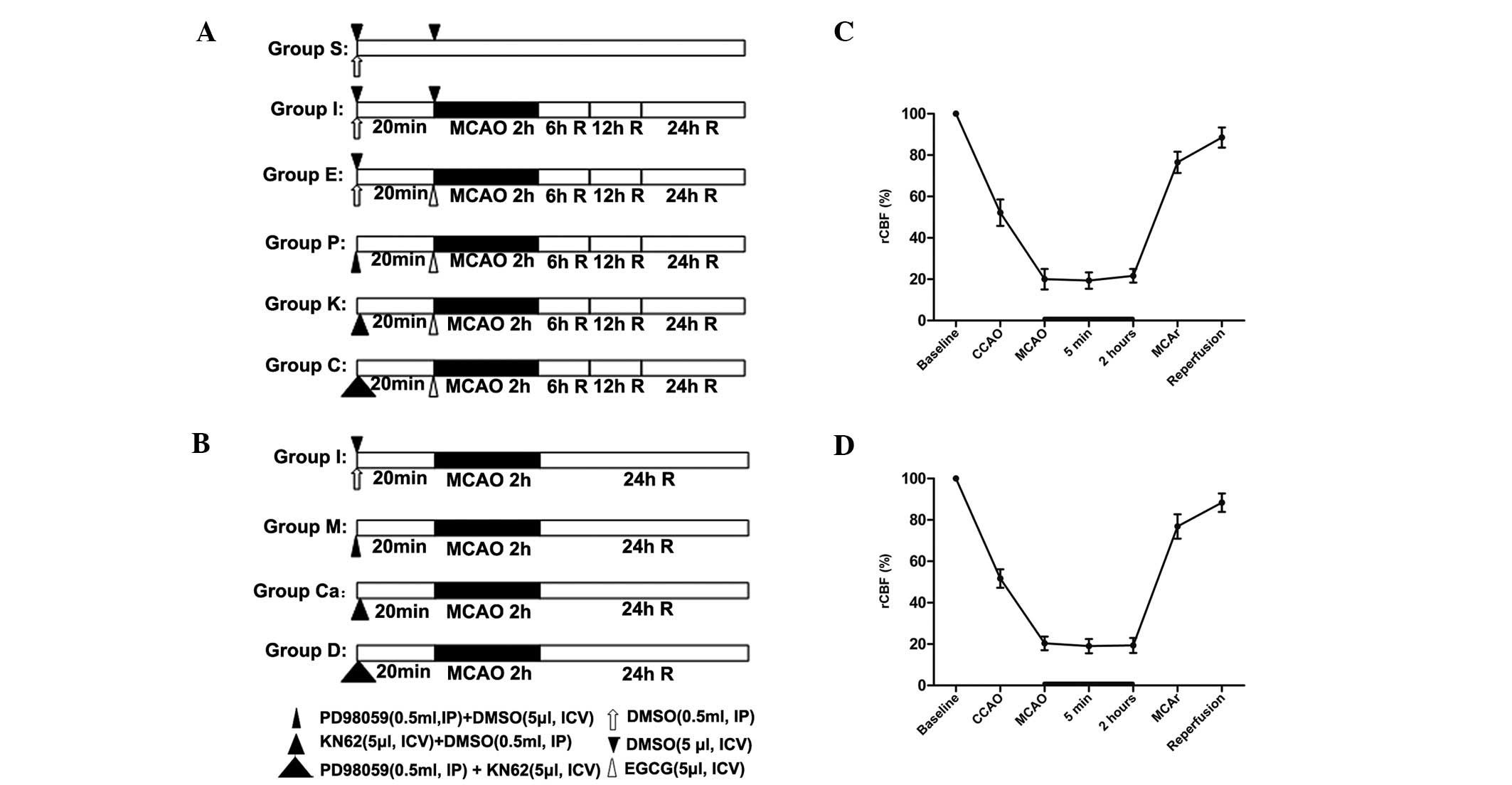 | Figure 1Experimental protocol. (A) Rats were
randomly divided into 6 groups, and each group was again divided
into 3 subgroups according to the time of reperfusion following
ischemia (6, 12 and 24 h following reperfusion) (n=12 per
subgroup). (C) Another 45 rats were randomly divided into four
groups. (B and D) Change in regional cerebral blood flow (rCBF) in
rats. Right common carotid artery (CCA) occlusion reduced CBF to
~50% of the baseline, and additional middle cerebral artery (MCA)
occlusion further decreased rCBF to ~20%. Group S (subgroup S6, S12
and S24), Sham-operation; group I (subgroup I6, I12 and I24),
middle cerebral artery occlusion (MCAO); group E (subgroup E6, E12
and E24), ischemia combined with EGCG treatment; Group P (subgroup
P6, P12 and P24), ischemia combined with EGCG plus PD98059
treatment; Group K (subgroup K6, K12 and K24), ischemia combined
with EGCG plus KN62 treatment; group C (subgroup C6, C12 and C24),
ischemia combined with EGCG plus PD98059 and KN62 treatment; group
M, ischemia combined with PD98059 treatment; Group Ca: ischemia
combined with KN62 treatment; Group D: ischemia combined with
PD98059 plus KN62 treatment. R, reperfusion; CCAO, CCA occlusion;
MCAO, MCA occlusion; MCAR, MCA remove. |
Drug treatment
Rat intracerebroventricular (ICV) injection was
performed under anesthesia using a stereotaxic instrument (RWD Life
Science Co., Ltd.) with a microsyringe pump (Shanghai Guangzheng
Medical Equipment Co., Ltd., Shanghai, China). A scalp incision was
perfomed and a burr hole was made in the right parietal skull, 1.8
mm lateral and 1.0 mm posterior to the bregma. A syringe was
inserted into the brain to a depth of 4.2 mm below the cortical
surface. EGCG or PD98059 was dissolved in dimethyl sulfoxide (DMSO)
(all obtained from Sigma-Aldrich, St. Louis, MO, USA). EGCG (1
mg/ml; 5 μl) or 1% DMSO (5 μl) was injected slowly (0.5 μl/min)
into the right ventricle immediately following ischemia. PD98059
(0.75 mg/rat, i.p.) or 1% DMSO (0.5 ml, i.p.) was administered to
rats 20 min prior to the operation.
The rats were randomly divided into four groups and
each group was again divided into three subgroups (n=12 per
subgroup) according to the time of reperfusion following ischemia.
The experimental groups and subgroups were as follows:
Sham-operation (group S; subgroup S6, S12 and S24); MCAO (group I;
subgroup I6, I12 and I24); ischemia combined with EGCG treatment
(group E; subgroup E6, E12 and E24) and ischemia combined with EGCG
plus PD98059 [a mitogen-activated protein kinase kinase (MEK)
inhibitor]treatment (group C; subgroup C6, C12 and C24). Another 27
rats were randomly divided into 3 groups (n=9 per group):
Sham-operation; MCAO (Group I) and ischemia combined with PD98059
treatment (Group P).
Measurements of infarct volume
At 24 h following reperfusion, rats were decapitated
and the brains were rapidly removed and frozen at −20°C for 10 min.
Sliced brain tissues were stained with 2%
2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich) for 30 min
at 37°C followed by overnight immersion in 4% paraformaldehyde. The
extent of ischemic infarction was traced and the integrated volume
was calculated using ImageJ 1.45 software (National Institutes of
Health, Bethesda, MD, USA). The relative infarction volume was
calculated by the following equation, giving a correction for
edema: {[Total lesion volume - (ipsilateral hemisphere volume -
contralateral hemisphere volume)] / contralateral hemisphere
volume} ×100.
Neurological scoring
Neurological scores were evaluated by a blinded
observer 24 h following reperfusion with a scoring system as
described previously (15,16).
Western blot analysis
The rats were euthanized by decapitation at 6, 12
and 24 h following reperfusion and the infarct side of the cortex
was harvested. Total protein extraction was performed according to
the manufacturer’s instructions in the kit (KGP250; Keygen Biotech,
Nanjing, China) for western blot analysis of TRPC6 and
aII-spectrin. Nuclear protein extraction was performed according to
the manufacturer’s instructions in the kit (Fisher Scientific,
Pittsburgh, PA, USA) for p-CREB. Protein levels in the extracts
were quantified using a bicinchoninic acid (BCA) assay. Equal
quantities of total or nuclear protein extracts were separated by
sodium dodecyl sulphate polyacrylamide gel electrophoresis and
transferred to polyvinylidene difluoride membranes by
electrophoresis. Membranes were incubated in Tris-buffered saline,
TBS containing 1% Tween-20 (TBST) blocking buffer and 5% non-fat
dry milk for 1 h at room temperature. Membranes were incubated
overnight at 4°C with either a mouse monoclonal anti-aII-spectrin
(dilution, 1:1000; Enzo Biochem, New York, NY, USA), rabbit
polyclonal anti-TRPC6 (dilution, 1:1000; Abcam Cambridge, MA, USA),
rabbit monoclonal anti-p-CREB (dilution, 1:1000) mouse monoclonal
anti-CHOP (dilution, 1:1000), rabbit polyclonal anti-GRP78
(dilution, 1:1000; Cell Signaling Technology Inc., Beverly, MA,
USA), rabbit polyclonal anti-caspase-12 (dilution, 1:200; Beijing
Biosynthesis Biotechnology Co., Ltd, Beijing, China), rabbit
polyclonal anti-Lamin B1 (dilution, 1:500; Bioworld Technology
Inc., Harrogate, UK) or mouse monoclonal anti-glyceraldehyde
3-phosphate dehydrogenase (GAPDH) antibody (dilution, 1:1000;
Proteintech Group, Inc., Hubei, China). This was followed by
incubation with horseradish peroxidase-labeled secondary anti-mouse
IgG or anti-rabbit IgG antibodies (dilution, 1:5000; Proteintech
Group, Inc.), respectively. Labeled proteins were detected with the
ChemiDocXRS+ chemiluminescence imaging system (Bio-Rad,
Hercules, CA, USA) and bands were quantified using lab imaging
software. The experiments were repeated in triplicate.
Quantum dot-based immunofluorescence and
immunohistochemistry
At 24 h following reperfusion, the rats were
perfused with 250 ml of 0.9% cold saline followed by 100 ml of 4%
paraformaldehyde in phosphate-buffered saline (pH 7.4). The brains
were then rapidly removed, blocked and embedded in paraffin.
Paraffin-embedded brains were cut into 4-μm thick sections
according to standard procedures. The paraffin sections (n=3 for
each group) were incubated overnight with antibodies against TRPC6
(1:100; Abcam) at 4°C subsequent to blocking with bovine serum
albumin (BSA). The samples were then incubated with a biotinylated
secondary antibody at 37°C for 30 min. Subsequent to blocking with
BSA, the paraffin sections were incubated with
streptavidin-conjugated QDs605 (dilution, 1:100; Wuhan Jiayuan
Quantum Dots Co., Ltd., Hubei, China). The cell nuclei were stained
with 4′,6-diamidino-2-phenylindole (DAPI). TRPC6-positive cells
were measured at a magnification of ×200 per visual field in the
peri-infarct region; three visual fields per section and three
brain sections per rat were analyzed. Fluorescent signals were
detected by fluorescence microscopy (BX51; Olympus, Tokyo, Japan)
and signal intensities were collected for statistical analysis.
Images were captured with a Doppler imaging system (CRi Nuance Fx;
Caliper Life Sciences, Hopkinton, MA, USA).
TdT mediated dUTP nick-end labeling
(TUNEL) assay
TUNEL staining analysis was used to detect apoptotic
cell death 24 h following reperfusion. TUNEL staining was conducted
with a kit (Roche Diagnostics GmbH, Mannheim, Germany).
TUNEL-positive nuclei with chromatin condensation and fragmented
nuclei were considered as probable apoptotic cells. The total
number of TUNEL-positive neurons in the ipsilateral hemisphere was
counted in three different fields for each section (by an
investigator who was blinded to the studies) by light microscopy at
a magnification of ×400.
Statistical analysis
GraphPad Prism software (version 5 for Windows,
GraphPad software Inc., La Jolla, CA, USA) was used for all
statistical analyses. Values are presented as the mean ± standard
error of the mean. The neurological score data comparison was
analyzed using the Kruskal-Wallis test followed by a post hoc
Dunn’s test. For all other measurements, one-way analysis of
variance followed by Newman-Keuls multiple comparison test was
used. P<0.05 was considered to indicate a statistically
significant difference.
Results
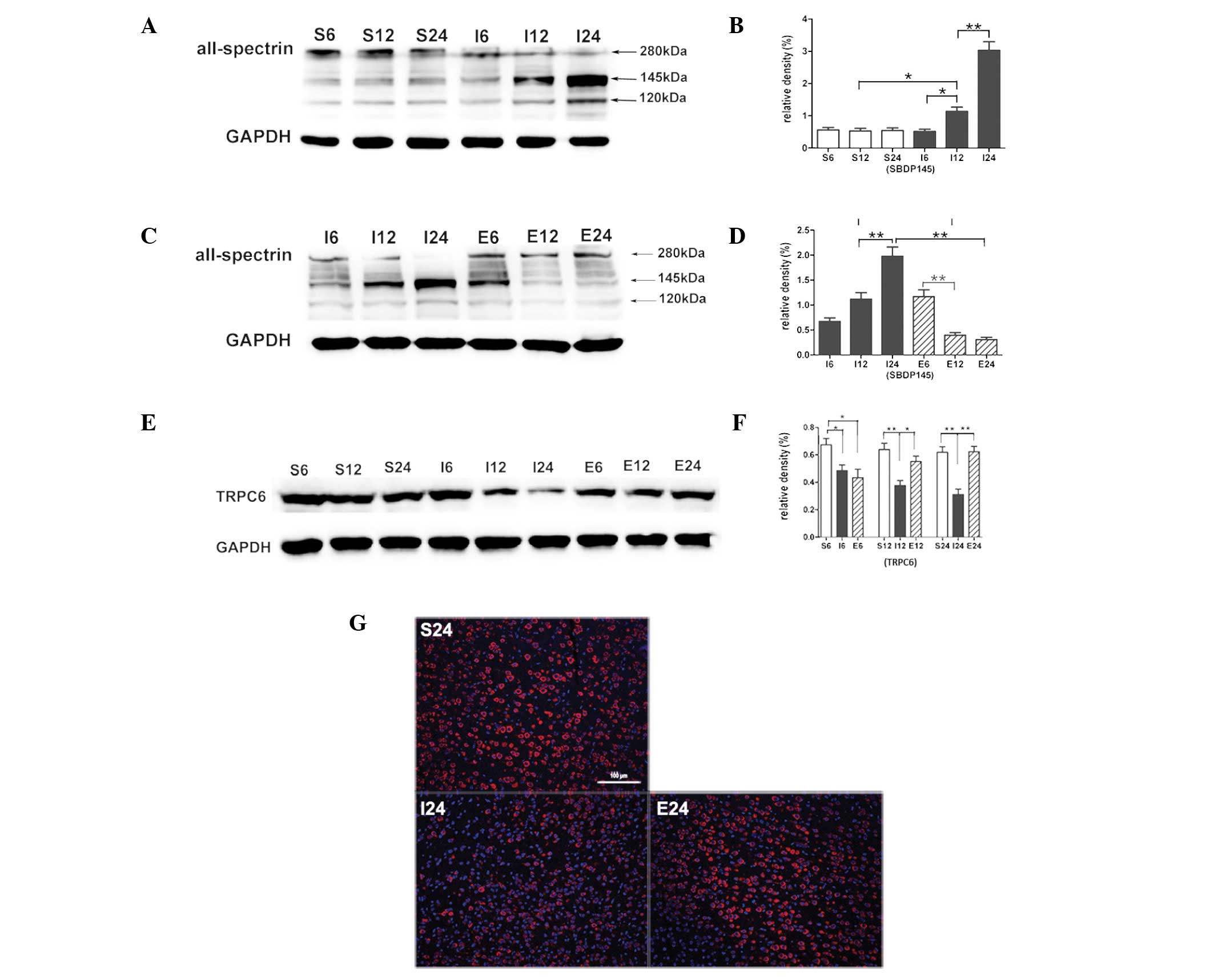
EGCG blocks calpain-specific aII-spectrin
breakdown product (SBDP145) formation
Compared with the sham-operated group the SBDP145
level in the MCAO group was significantly increased after 12 h
(P<0.05), an increase that was greatest at 24 h (P<0.01;
Fig. 2A and B). When MCAO rats
were treated with EGCG the SBDP145 level was significantly
decreased at 12 h (P<0.01) and the decrease was greatest at 24 h
(P<0.01; Fig. 2C and D).
EGCG inhibits calpain-mediated TRPC6
channel degradation
Compared with the sham-operated group, TRPC6 levels
in the MCAO group were significantly decreased at 6 (P<0.05), 12
and 24 h (P<0.01; Fig. 2E and
F). When MCAO rats were treated with EGCG, the protein level of
TRPC6 was significantly increased at 12 and 24 h (P<0.05 and
P<0.01, respectively). Immunofluorescence analysis showed a
cytomembrane staining pattern of TRPC6 in neurons of the cerebral
cortex (Fig. 2G).
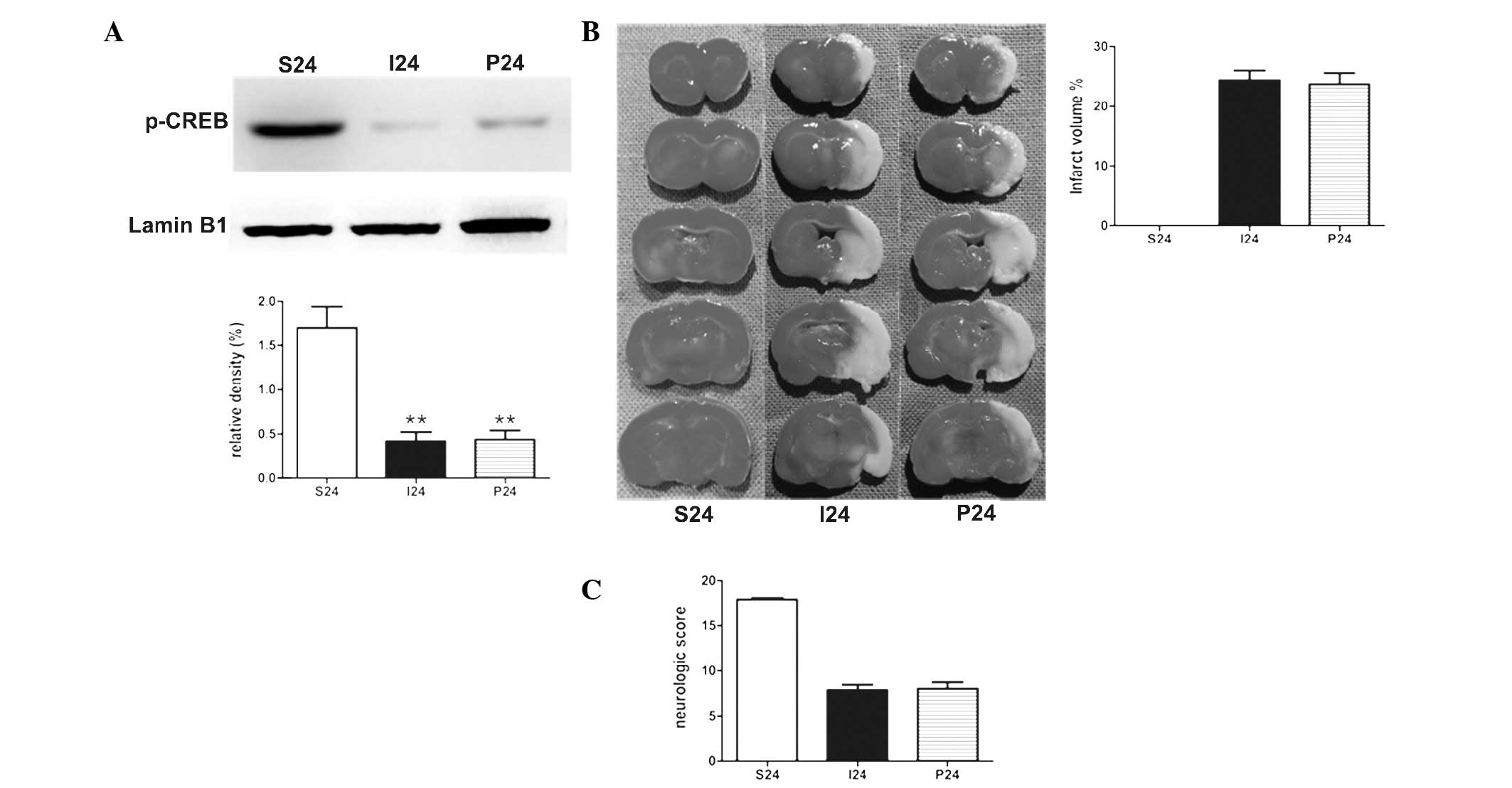
PD98059 exhibits no effect on ischemic
stroke in rats at 24 h following reperfusion
To determine the effect of PD98059 in the stroke
rats, PD98059 was administered 20 min prior to the operation.
Notably, with the application of PD98059 alone, no statistical
significance was identified in the protein levels of p-CREB in MCAO
rats (Fig. 3A). There was no
significant difference between the two groups in the measurements
of the the infarct volumes and neurological scores (Fig. 3B and C).
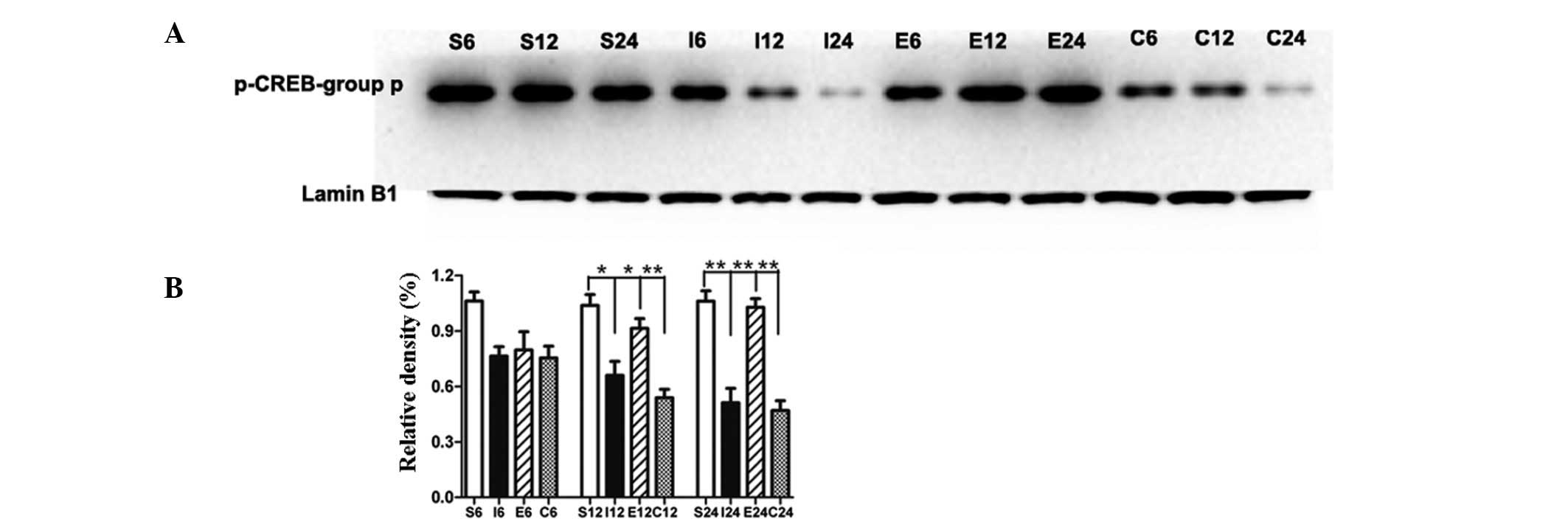
EGCG maintains phosphorylation of CREB by
blocking TRPC6 degradation
Compared with the sham-operated group, the level of
p-CREB in the MCAO group was significantly decreased at 12 and 24 h
(P<0.05 and P<0.01, respectively; Fig. 4A and B). Compared with the MCAO
group, the level of p-CREB in the EGCG-treated group was
significantly increased at 12 and 24 h (P<0.05 and P<0.01,
respectively). In the rats treated with PD98059, the level of
p-CREB was significantly decreased at 12 and 24 h (P<0.01 and
P<0.01, respectively) compared with that of the EGCG-treated
group.
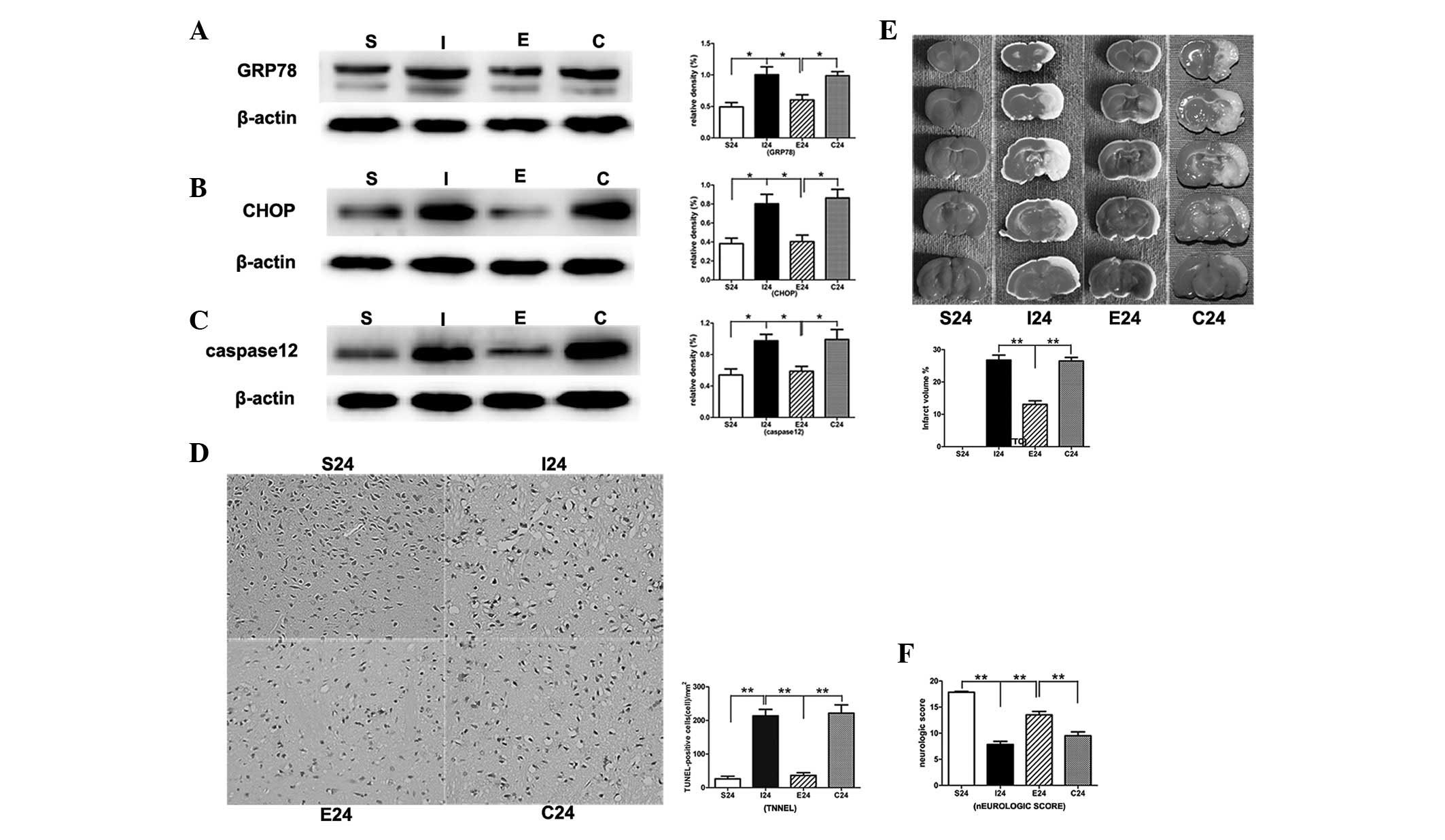
EGCG inhibits ERS and apoptosis 24 h
following reperfusion
In the MCAO group, the protein levels of ERS-related
markers GRP78, CHOP and caspase-12 were significantly increased
compared with that in the sham-operation group (P<0.01,
P<0.01 and P<0.01, respectively; Fig. 5Aa–c). When MCAO rats were treated
with EGCG, the protein levels of CHOP, GRP78 and caspase-12 were
significantly decreased (P<0.01, P<0.01 and P<0.01,
respectively). Subsequent to the administration of PD98059, the
protein levels were significantly increased compared with that of
the EGCG-treated group.
In the TUNEL assay, EGCG significantly reduced
apoptotic cell death in the right cortex compared with that in the
MCAO group (P<0.01; Fig. 5D and
E). Following treatment with PD98059, the apoptotic cell death
was significantly increased compared with that of the EGCG-treated
group (P<0.01).
EGCG significantly reduces infarct
volumes and promotes functional recovery in ipsilateral ischemic
hemispheres 24 h following reperfusion
Following ischemia/reperfusion injury, a
white-stained infarct area was observed in the MCAO group. By
contrast, treatment with EGCG significantly reduced infarct volumes
compared with that of the MCAO group 24 h following reperfusion
(P<0.01). Following the application of PD98059, the infarct
volumes were significantly increased compared with that of the
EGCG-treated group (P<0.01; Fig.
5E).
There was also significant improvement in the
neurological score 24 h following reperfusion with EGCG treatment
(P<0.01). When treated with PD98059, the neurological scores
were significantly decreased compared with that of the EGCG-treated
group (P<0.01; Fig. 5F).
Discussion
The results clearly demonstrated that ICV injection
of EGCG at low doses immediately following ischemia improved the
outcome as measured by TTC staining and neurological scoring. EGCG
treatment also significantly inhibited ERS and apoptosis. EGCG
improved the neurological status and inhibited ERS, which was
correlated with elevated TRPC6 and p-CREB activity and decreased
SBDP145 activity. When MEK activity was inhibited, the
neuroprotective effect of EGCG was attenuated and a correlated
decrease in CREB activity was observed. These results demonstrated
that EGCG, is important in the prevention of cerebral ischemic
injury (17), and may be used as a
therapeutic intervention for stroke during the acute or subacute
period similar to the drug edaravone (18).
Calpains are intracellular calcium-dependent
cysteine endopeptidases that are activated by cytosolic
Ca2+ overload (19).
The most studied target of calpain is aII-spectrin, a 280-kDa
neuronal protein that localizes to axons and functions in cortical
cytoskeleton matrix support. The aII-spectrin breakdown product,
SBDP145, results from the sequential calpain cleavage of
aII-spectrin which generates SBDP150 followed by cleavage to remove
the additional 5 kDa (20,21). In the present study, MCAO rats
exhibited elevated levels of SBDP145 in the cortical regions of the
ipsilateral hemisphere in the first 24 h following ischemic injury.
EGCG treatment significantly reduced SBDP145 formation at 12 and 24
h. The results clearly demonstrated that EGCG, when applied
immediately following ischemia, inhibited calpain activation and
induced resistance to ischemia/reperfusion injuries.
TRPC channels are non-selective cation channels that
are expressed in numerous multicellular organisms with different
functions (5). TRPC6 is involved
in brain-derived neurotrophic factor (BDNF)-mediated growth cone
turning, neuron survival and spine formation (8,22).
TRPC6 was specifically degraded in transient ischemia and this
degradation occurred prior to and during neuronal cell death. In
addition, TRPC6 protein in neurons in ischemia was specifically
downregulated by calpain proteolysis. Inhibition of calpain
proteolysis of TRPC6 protected animals from ischemic brain damage.
In the present study, the protein levels of TRPC6 were markedly
decreased at 6 h and the reduction in TRPC6 protein levels remained
at 12 and 24 h in the MCAO group, these results were consistent
with a previous study (7). EGCG
treatment significantly enhanced the protein levels of TRPC6 at 12
and 24 h. In addition, EGCG-treated rats exhibited significantly
lower infarct volumes and also increased functional recovery
compared with that of MCAO rats at 24 h. Therefore, the results
indicated that EGCG treatment protected rats from ischemic brain
damage through the inhibition of calpain proteolysis of TRPC6.
A modest level of Ca2+ influx through
TRPC6 channels leads to the activation of ERK, which activates CREB
to promote neuronal survival (8).
Inhibition of TRPC6 channel degradation maintained the
phosphorylation of CREB and prevented ischemic brain damage
(7). CREB activation is a critical
event in the neuroprotection from ischemic injury (23,24).
In the present study, the protein levels of p-CREB were
significantly increased in the EGCG-treated group at 12 and 24 h.
When MEK activity was inhibited, the neuroprotective effect of EGCG
was attenuated and correlated with decreased CREB activity levels.
The results demonstrated that EGCG, when administered immediately
following ischemia, stimulated the MEK/ERK pathway that ultimately
induced CREB activation and contributed to neuroprotection at 24 h.
In addition, EGCG significantly reduced TRPC6 degradation induced
by ischemia at 24 h. Therefore, the results suggested that EGCG
blocked calpain-mediated TRPC6 channel degradation which activated
CREB through the MEK/ERK pathway and contributed to
neuroprotection.
The ER is an organelle that is important in the
maintenance of intracellular calcium homeostasis and proper folding
of newly synthesized secretory and membranous proteins (25). ER functions are disturbed by
different insults, such as the accumulation of unfolded proteins
and the disruption of intracellular calcium homeostasis (26,27),
which result in ER stress. However, if ER stress is too severe, the
unfolded protein response initiates the apoptotic pathway (28). Increased expression of GRP78 is a
marker of ER stress (29,30). In addition, CHOP participates in
apoptosis signaling pathways and serves as a hallmark of ER stress
(31,32). ER stress-induced neuronal cell
death is important in stroke pathophysiology (33) and involves the activation of
caspase-12 (34,35), which is specific to apoptosis
mediated by ER stress (9). In the
present study, EGCG treatment significantly decreased the infarct
volumes and improved functional recovery at 24 h. In addition, the
ER stress was enhanced in the brain following ischemia/reperfusion
as demonstrated by the significant elevation of the ER
stress-related markers GRP78, CHOP and caspase-12 in the cortex and
EGCG was demonstrated to inhibit this. However, the effect of EGCG
was attenuated by the MEK inhibitor PD98059. In addition, when
treated with PD98059, the apoptotic cell death was also
significantly increased. Thus, this study has demonstrated that ICV
injection of EGCG inhibited ER stress and apoptosis through the
MEK/ERK/CREB pathway, which contributed to its neuroprotective
effects. The results indicated that EGCG exerted its
neuroprotective effects by activating ERK/CREB pathways and the
subsequent inhibition of ERS.
In conclusion, the present study has demonstrated
that administration of EGCG immediately following ischemia
inhibited ER stress and improved the neurological status through
the inhibition of calpain proteolysis of TRPC6 and the subsequent
activation of CREB via the MEK/ERK pathway.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 30901984).
References
|
1
|
Kaufman RJ: Stress signaling from the
lumen of the endoplasmic reticulum: coordination of gene
transcriptional and translational controls. Genes Dev.
13:1211–1233. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang B, Rusciano D and Osborne NN: Orally
administered epigallocatechin gallate attenuates retinal neuronal
death in vivo and light-induced apoptosis in vitro. Brain Res.
1198:141–152. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vivien D, Gauberti M, Montagne A, Defer G
and Touzé E: Impact of tissue plasminogen activator on the
neurovascular unit: from clinical data to experimental evidence. J
Cereb Blood Flow Metab. 31:2119–2134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Martinou JC, Dubois-Dauphin M, Staple JK,
et al: Overexpression of BCL-2 in transgenic mice protects neurons
from naturally occurring cell death and experimental ischemia.
Neuron. 13:1017–1030. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Montell C, Birnbaumer L and Flockerzi V:
The TRP channels, a remarkably functional family. Cell.
108:595–598. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Harteneck C, Plant TD and Schultz G: From
worm to man: three subfamilies of TRP channels. Trends Neurosci.
23:159–166. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Du W, Huang J, Yao H, Zhou K, Duan B and
Wang Y: Inhibition of TRPC6 degradation suppresses ischemic brain
damage in rats. J Clin Invest. 120:3480–3492. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jia Y, Zhou J, Tai Y and Wang Y: TRPC
channels promote cerebellar granule neuron survival. Nat Neurosci.
10:559–567. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Graham HN: Green tea composition,
consumption, and polyphenol chemistry. Prev Med. 21:334–350. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xie W, Ramakrishna N, Wieraszko A and
Hwang YW: Promotion of neuronal plasticity by
(−)-epigallocatechin-3-gallate. Neurochem Res. 33:776–783.
2008.
|
|
11
|
Haque AM, Hashimoto M, Katakura M, Tanabe
Y, Hara Y and Shido O: Long-term administration of green tea
catechins improves spatial cognition learning ability in rats. J
Nutr. 136:1043–1047. 2006.PubMed/NCBI
|
|
12
|
van Praag H, Lucero MJ, Yeo GW, et al:
Plant-derived flavanol (−)epicatechin enhances angiogenesis and
retention of spatial memory in mice. J Neurosci. 27:5869–5878.
2007.
|
|
13
|
Nagai K, Jiang MH, Hada J, et al:
(−)-Epigallocatechin gallate protects against NO stress-induced
neuronal damage after ischemia by acting as an anti-oxidant. Brain
Res. 956:319–322. 2002.
|
|
14
|
Sutherland BA, Shaw OM, Clarkson AN,
Jackson DN, Sammut IA and Appleton I: Neuroprotective effects of
(−)-epigallocatechin gallate following hypoxia-ischemia-induced
brain damage: novel mechanisms of action. FASEB J. 19:258–260.
2005.
|
|
15
|
Garcia JH, Wagner S, Liu KF and Hu XJ:
Neurological deficit and extent of neuronal necrosis attributable
to middle cerebral artery occlusion in rats. Statistical
validation. Stroke. 26:627–635. 1995. View Article : Google Scholar
|
|
16
|
Tsubokawa T, Jadhav V, Solaroglu I,
Shiokawa Y, Konishi Y and Zhang JH: Lecithinized superoxide
dismutase improves outcomes and attenuates focal cerebral ischemic
injury via antiapoptotic mechanisms in rats. Stroke. 38:1057–1062.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Arab L, Liu W and Elashoff D: Green and
black tea consumption and risk of stroke: a meta-analysis. Stroke.
40:1786–1792. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang N, Komine-Kobayashi M, Tanaka R, Liu
M, Mizuno Y and Urabe T: Edaravone reduces early accumulation of
oxidative products and sequential inflammatory responses after
transient focal ischemia in mice brain. Stroke. 36:2220–2225. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Goll DE, Thompson VF, Li H, Wei W and Cong
J: The calpain system. Physiol Rev. 83:731–801. 2003.
|
|
20
|
Nath R, Raser KJ, Stafford D, et al:
Non-erythroid alpha-spectrin breakdown by calpain and interleukin 1
beta-converting-enzyme-like protease(s) in apoptotic cells:
contributory roles of both protease families in neuronal apoptosis.
Biochem J. 319:683–690. 1996.
|
|
21
|
Wang KK: Calpain and caspase: can you tell
the difference?, by kevin KW Wang Vol 23, pp 20–26. Trends
Neurosci. 23:592000.
|
|
22
|
Li Y, Jia YC, Cui K, et al: Essential role
of TRPC channels in the guidance of nerve growth cones by
brain-derived neurotrophic factor. Nature. 434:894–898. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Walton MR and Dragunow I: Is CREB a key to
neuronal survival? Trends Neurosci. 23:48–53. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Finkbeiner S: CREB couples neurotrophin
signals to survival messages. Neuron. 25:11–14. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yin XM, Oltvai ZN and Korsmeyer SJ: BH1
and BH2 domains of Bcl-2 are required for inhibition of apoptosis
and heterodimerization with Bax. Nature. 369:321–323. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
He B: Viruses, endoplasmic reticulum
stress, and interferon responses. Cell Death Differ. 13:393–403.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ginsberg MD: Neuroprotection for ischemic
stroke: past, present and future. Neuropharmacology. 55:363–389.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Love S: Apoptosis and brain ischaemia.
Prog Neuropsychopharmacol Biol Psychiatry. 27:267–282. 2003.
View Article : Google Scholar
|
|
29
|
Yoshida H, Matsui T, Yamamoto A, Okada T
and Mori K: XBP1 mRNA is induced by ATF6 and spliced by IRE1 in
response to ER stress to produce a highly active transcription
factor. Cell. 107:881–891. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Groenendyk J, Sreenivasaiah PK, Kim do H,
Agellon LB and Michalak M: Biology of endoplasmic reticulum stress
in the heart. Circ Res. 107:1185–1197. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
DeGracia DJ and Montie HL: Cerebral
ischemia and the unfolded protein response. J Neurochem. 91:1–8.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tajiri S, Oyadomari S, Yano S, et al:
Ischemia-induced neuronal cell death is mediated by the endoplasmic
reticulum stress pathway involving CHOP. Cell Death Differ.
11:403–415. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu DY, Lau L, Liu SH, Wei JS and Lu YM:
Activation of cAMP-response-element-binding protein (CREB) after
focal cerebral ischemia stimulates neurogenesis in the adult
dentate gyrus. Proc Natl Acad Sci USA. 101:9453–9457. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zinkstok SM, Vergouwen MD, Engelter ST, et
al: Safety and functional outcome of thrombolysis in
dissection-related ischemic stroke: a meta-analysis of individual
patient data. Stroke. 42:2515–2520. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Paschen W and Mengesdorf T: Cellular
abnormalities linked to endoplasmic reticulum dysfunction in
cerebrovascular disease - therapeutic potential. Pharmacol Ther.
108:362–375. 2005. View Article : Google Scholar : PubMed/NCBI
|