Introduction
Previous epidemiological studies have demonstrated
that hepatitis B virus (HBV) infection is a key risk factor for
hepatocellular carcinoma (HCC). HBV is greatly detrimental to human
health by inducing hepatitis, cirrhosis and HCC (1). The currently available diagnostic
methods, including B-mode ultrasound and α-fetal protein (AFP)
detection, lack the ability to detect liver cancer at an early
stage (2,3). In addition, therapy is currently only
partially effective against HBV. Therefore, investigation into the
pathogenesis of HBV-related HCC is of great significance. Studies
on microRNAs (miRs) may provide a valuable approach for devising
novel strategies for the diagnosis, prevention and treatment of
HCC.
miRs are non-coding, single-stranded RNA molecules
containing 19–25 nucleotides. A previous study demonstrated that
the majority of miRs regulate gene expression by imperfect
base-pairing with the 3′-untranslated region (3′-UTR) of target
mRNAs (4). miRs have been identified
as key regulators in diverse biological processes, including cell
cycle regulation, cell differentiation, proliferation and apoptosis
(5,6).
The significance of miRs in the tumorigenesis of different types of
human cancer is highlighted in the present study.
miR-34c belongs to a family of evolutionarily
conserved miRs (miR-34a, b and c) that are involved in apoptosis
and negative feedback control of the cell cycle (7,8). The
miR-34 family is downregulated in HCC and contributes to
hepatocarcinogenesis by targeting genes that include NOTCH1, MET,
E2F1-3, MYC, cyclin-dependent kinase (CDK)4, CDK6, cyclin D1,
sirtuin 1 (SIRT1) and B-cell lymphoma 2 (BCL2) (9–11). In a
preliminary study, it was revealed that miR-34c was significantly
downregulated in HBV-infected transgenic mice and HBV-related HCC
cell lines compared with their corresponding controls. Therefore,
miR-34c was selected for further investigation. Transforming growth
factor-β-induced factor homeobox 2 (TGIF2) is a transcriptional
co-repressor that interacts with Smad and negatively regulates the
transforming growth factor (TGF)-β/Smad response (12). Furthermore, it was previously reported
that TGF-β signaling is involved in cell apoptosis and
proliferation in HBV-associated HCC by upregulating Smad7 (13–15).
Therefore, the present study investigated the roles and regulatory
mechanisms of miR-34c and TGIF2 in the development of
HBV-associated HCC.
Materials and methods
Tissue samples and cell lines
Thirty pairs of tumor and adjacent non-cancerous
tissues were obtained from patients with HBV-associated HCC at the
Provincial Hospital affiliated to Shandong University (Jinan,
China) between 2008 and 2011. The tissue samples were snap-frozen
in liquid nitrogen immediately following resection and stored at
−80°C. Informed consent was obtained from all subjects (patient or
the patient's family) and the study was approved by the Provincial
Hospital Affiliated to Shandong University Ethics Committee. The
human HCC cell lines HepG2, HepG2.2.15 and BEL7402 (American Type
Culture Collection, Manassas, VA, USA) were cultured at 37°C in an
atmosphere of 5% CO2. HepG2 cells were cultured in
Dulbecco's modified Eagle's medium (Invitrogen Life Technologies,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS;
Gibco-BRL, Grand Island, NY, USA) at 37°C in a 5% CO2
incubator. HepG2.2.15 cells were cultured in minimal essential
medium (MEM; HyClone, Beijing, China) supplemented with 10% new
bovine serum (NBS; Gibco-BRL) and 380 µg/ml G418 (Promega, Madison,
WI, USA). BEL7402 cells were cultured in RPMI-1640 (Gibco-BRL)
supplemented with 10% FBS. BEL7402-HBV cells are BEL7402 cells
transfected with pcDNA3-1.1HBV.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was extracted by TRIzol® reagent
(Invitrogen Life Technologies) according to the manufacturer's
instructions. The mRNA was reverse transcribed into cDNA with the
First Strand cDNA Synthesis kit ReverTra Ace-α (Toyobo, Osaka,
Japan). The cDNA was then amplified using 2X Taq PCR Master Mix
(Tiangen Biotech, Beijing, China). The sequences of the primers for
the amplification of TGIF2 were as follows: F
5′-TCTCTGTGTTGCCTCCCTCT-3′ and R 5′-CCACCTCAGCCCAATACACT-3′. The
human β-actin gene was used as an internal control and amplified
with the following primers: F 5′-ACACTGTGCCCATCTACGAGGGG-3′ and R
5′-ATGATGGAGTTGAAGGTAGTTTCGTGGAT-3′. The PCR products were analyzed
on 1% agarose gels by electrophoresis.
Quantitative (q)PCR
qPCR was performed to detect the expression levels
of miR-34c and TGIF2. The NCode VILO miRNA cDNA Synthesis kit
(Invitrogen Life Technologies) and the all-in-one miR qPCR primer
set (GeneCopoeia, Rockville, MD, USA) were used for miR-34c
amplification. The PCR reaction was performed using the Express
SYBR® GreenER™ miRNA qRT-PCR kit (Invitrogen Life Technologies) on
a preheated qPCR instrument (ABI 7000; Applied Biosystems, Foster
City, CA, USA). All reactions were performed in triplicate. The
relative expression level of miR-34c compared with small nuclear
RNA U6 was determined using the 2−ΔΔCt method (16).
The ReverTra Ace qPCR RT kit (Toyobo) and SYBR®
Premix Ex Taq Perfect Real-Time reagent (Takara Bio, Inc., Shiga,
Japan) were used for TGIF2 amplification. The primers used for the
qPCR and RT-PCR were the same. Human β-actin RNA served as an
internal control. All qPCR reactions were performed in
triplicate.
Target prediction
The miR-34c target genes were predicted using
bioinformatics analysis, and the miR-34c target list was obtained
from a comparison of three established miR target prediction
programs, including TargetScan (http://www.targetscan.org/), PicTar (http://pictar.bio.nyu.edu/) and MiRanda (http://microrna.sanger.ac.uk/). The duplex structure
between miR-34c and the 3′-UTR of the target genes was predicted
using RNAhybrid (http://bibiserv.techfak.uni-bielefeld.de/rnahybrid/).
Construction and transfection of
recombinant plasmids
Primer Premier 5.0 (Premier Biosoft, Palo Alto, CA,
USA) was used to design the miR-34c primers according to the
sequence of precursor miR-34c cDNA provided by GenBank (National
Center for Biotechnology Information, Bethesda, MD, USA). The
primer sequences were as follows: F
5′-CGCGGATCCTCTATTTGCCATCGTCTA-3′ and R
5′-CTGAAGCTTCAGGCAGCTCATTTGGAC-3′. The restriction enzyme cutting
sites of BamHI and HindIII are included in the primer sequence. The
recombinant plasmid was named pS-miR-34c and confirmed by PCR,
restriction enzyme digestion and DNA sequencing. Additionally, a
control vector containing an antisense sequence was constructed and
denoted as pS-control.
HepG2.2.15 cells were plated 24 h prior to
transfection at a density of 2×105 cells per well in
6-well plates. The cell medium (Invitrogen Life Technologies) was
replaced with serum-free Opti-MEM® (Invitrogen Life Technologies)
prior to transfection. A total of 2 µg of the constructed plasmids
were transfected into HepG2.2.15 cells using FuGENE® 6 transfection
reagent (Roche Diagnostics, Basel, Switzerland) according to the
manufacturer's instructions. Following incubation for 6 h, the
medium was replaced with fresh medium supplemented with 10% FBS.
The supernatant and cells were collected 48 h following
transfection for further studies.
Luciferase assay
The 3′-UTRs of human TGIF2 mRNA sense and antisense
strands were amplified using the following primers: Sense, F
5′-TAGGTACCCAGAAGCAGCAGGACCCA-3′; sense, R
5′-GCAGATCTTCATCACTGAGCGGAGGC-3′; antisense, F
5′-AGATCTCAGAAGCAGCAGGACCCA-3′; antisense, R
5′-GGTACCTCATCACTGAGCGGAGGC-3′. The segments were digested with the
restriction enzymes KpnI and BglII (Takara) and cloned into the
corresponding restriction sites of the pGL3-REPORT vector (Promega
Corporation) to produce the pGL3-TGIF2 and pGL3-anti-TGIF2
plasmids. HepG2 cells were seeded in 24-well culture plates at a
density of 2×105 cells per well and cultured to 90%
confluence. The cells were transfected with pGL3-TGIF2 or
pGL3-anti-TGIF2 (100 ng) and co-transfected with pRL-TK plasmid (40
ng) by FuGENE® HD transfection reagent (Roche). The cells were
harvested in 100 µl of cell lysis buffer (NP40; Invitrogen Life
Technologies) 24 h post-transfection. Firefly and Renilla
luciferase activity levels were detected using the Dual-Luciferase
Reporter assay (Promega Corporation) according to the
manufacturer's instructions. Experiments were performed in
triplicate.
Western blot analysis
Transfected cells were lysed with 1%
radioimmunoprecipitation assay lysis buffer (Beyotime
Biotechnology, Haimen, China) 72 h after transfection. The
supernatants were reserved and the concentration of protein was
determined using the Pierce BCA protein assay reagent kit (Pierce,
Rockford, IL, USA). Subsequently, equal amounts (30 µg) of protein
were separated by 10% SDS-PAGE and transferred to a polyvinylidene
difluoride membrane. The membrane was blocked with 5% milk and
incubated with a mouse monoclonal primary antibody against human
TGIF2 (dilution, 1:1,000; cat no. ab57527; Abcam, Cambridge, MA,
USA). Following incubation with goat anti-mouse horseradish
peroxidase secondary antibody, the signals were detected using
enhanced chemiluminescence (ECL Plus™; GE Healthcare, Little
Chalfont, UK).
ELISA assay for the detection of
hepatitis B virus surface antigen (HBsAg) and hepatitis B virus e
antigen (HBeAg)
The concentrations of HBsAg and HBeAg in culture
supernatants were measured by an ELISA assay using HBsAg and HBeAg
enzyme diagnostic kits (Autobio, Zhengzhou, China) according to the
manufacturer's instructions. The levels of HBsAg and HBeAg were
determined from the optical density at 450 nm using an absorbance
microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
The assays were performed in triplicate. Inhibitory rates were
calculated according to the following formula: Inhibitory rate (%)
= (Ccontrol-Ctested)/Ccontrol ×
100%, in which C is the optical density of the HBV antigen.
qPCR of HBV DNA
HBV DNA in cell culture supernatants collected
post-transfection were extracted and purified by standard methods.
qPCR was performed using the HBV PCR Fluorescence Quantitative
Detection kit (Bioer Technology Co., Ltd., Hangzhou, China)
following the manufacturer's instructions. Experiments were
performed in triplicate, and the average cycle threshold (Ct)
values were used to determine the concentration of HBV DNA.
Cell proliferation and apoptosis
assays
HepG2.2.15 cells were seeded in 96-well culture
plates (Costar®; Costar, Corning, NY, USA). As indicated, 10 µl of
Cell counting kit-8 (CCK-8) reagents were added into each well and
incubated for 1 h at 0, 24, 48 or 72 h post-transfection. The cell
proliferation rates were calculated by measuring the optical
density at 450 nm using an absorbance microplate reader (Bio-Rad
680; Bio-Rad Laboratories, Inc.).
Flow cytometry (FCM) was performed 48 h
post-transfection in 6-well plates. The cells were resuspended in
binding buffer containing Annexin V-fluorescein isothiocyanate and
propidium iodide (PI) according to the manufacturer's instructions
(KeyGen Biotech Co., Ltd., Nanjing, China). The samples were
analyzed using a Beckman Coulter Flow Cytometer (Beckman Coulter,
Fullerton, CA, USA) and experiments were performed in
triplicate.
Mouse model
Male Balb/c nude mice (n=15) were purchased from the
Animal Center of Shandong University. Mice were maintained in
laminar flow rooms, with exposure to 12 h light and 12 h dark
cycles, at a temperature of 21°C and 50% humidity, with free access
to food and water. Animals were fed according to protocols approved
by the Shandong University Animal Care Committee. A total of
2×106 BEL7402-1.1-HBV cells were injected subcutaneously
into the flanks of each BALB/c nude mouse (6–8 weeks of age). The
mice were randomly assigned to 3 groups (5 mice per group). When
the tumor grew to 90 mm3 in volume, plasmids of
pS-miR-34c or pS-control (20 µg each) was injected into tumor of
each mouse every two days five times. Tumor volume was measured by
caliper and calculated as length × width × height
(mm3).
Statistical analysis
All experiments were performed in triplicate. The
results were expressed as the mean ± standard deviation and
analyzed using GraphPad Prism version 5.0 (GraphPad Software, Inc.,
La Jolla, CA, USA). Differences between experimental groups were
evaluated by analysis of variance or the Student's t-test,
and P<0.05 was considered to indicate a statistically
significant difference.
Results
Expression of miR-34c and TGIF2 in
HBV-associated HCC is inversely correlated
In the present study, qPCR revealed that miR-34c was
downregulated in HBV-related HCC tissues compared with adjacent
non-cancerous liver tissues (Fig.
1A). The levels of miR-34c expression were also detected by
qPCR in two cell lines of HBV-associated HCC (Fig. 1B). Analysis of the microarray data
demonstrated that TGIF2 was upregulated in HBV-infected transgenic
mice compared with syngeneic BALB/c mice (unpublished data). As
presented in Fig. 1C and D, TGIF2 was
also significantly upregulated in HepG2.2.15 cells compared with
HepG2 cells, and in HBV-associated HCC tissues (C1-C4) compared
with adjacent non-cancerous tissues (L1-L4).
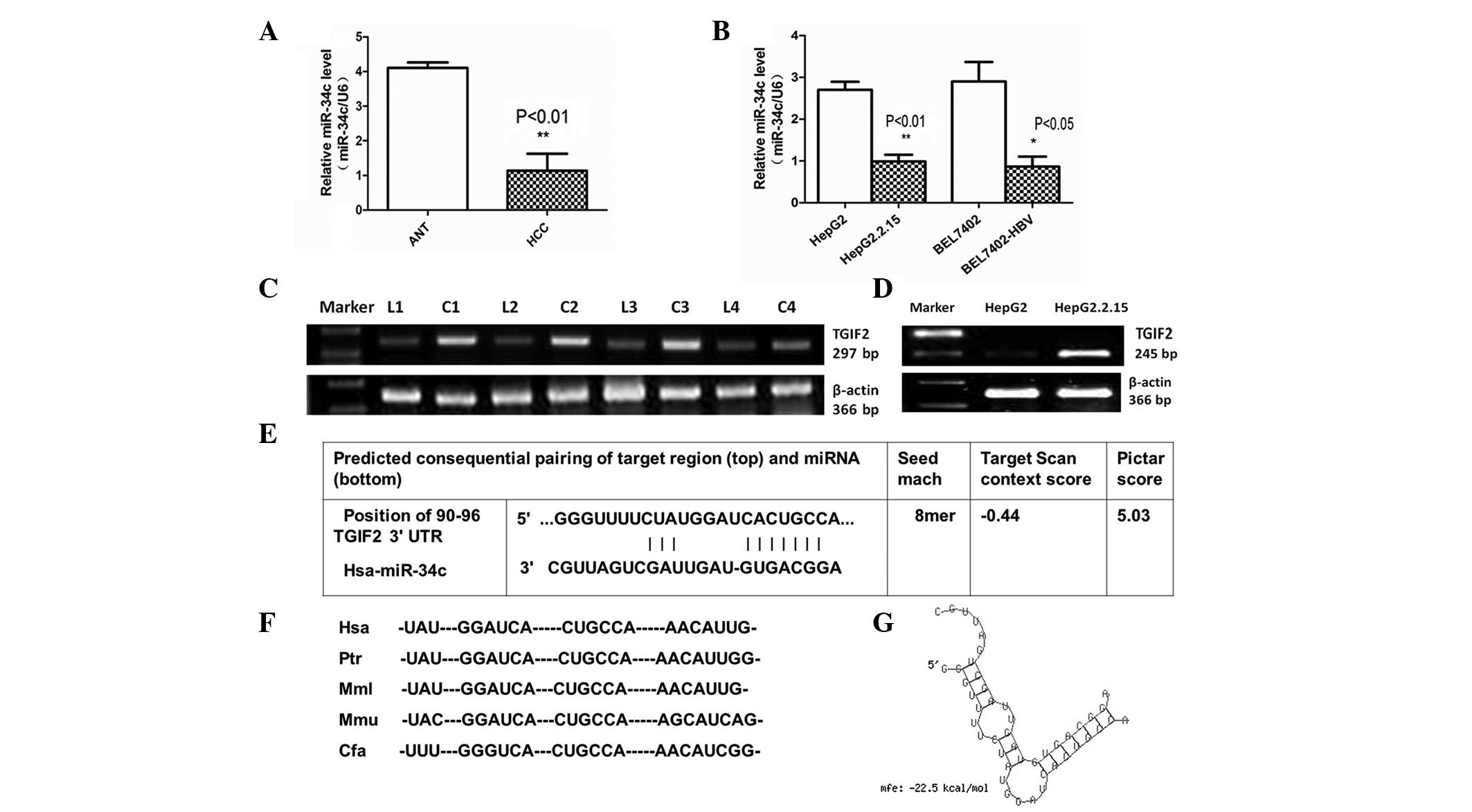 | Figure 1.miR-34c expression was downregulated
in HBV-associated HCC, and TGIF2 is a putative target of miR-34c.
(A) Expression levels of miR-34c were significantly reduced in
HBV-associated HCC clinical specimens compared with ANT
(**P<0.01). (B) Quantitative PCR results showed that the
expression levels of miR-34c were reduced in HepG2.2.15 compared
with HepG2 cells (**P<0.01), as well as in BEL7402-HBV compared
with BEL7402 cells (*P<0.05). (C) mRNA expression levels of
TGIF2 were significantly upregulated in HBV-associated HCC tissues
compared with ANT. (D) Reverse transcription-PCR results showed
that TGIF2 was upregulated in HepG2.2.15 compared with HepG2 cells.
All of the above data were obtained from three independent
experiments. (E) TGIF2 was identified as a potential target gene of
miR-34c using TargetScan and PicTar software. (F) The TargetScan
prediction showed a common binding sequence on the 3′-UTR of TGIF2
that was conserved across vertebrate species, including human,
chimpanzee, rhesus macaque, mouse and dog. (G) The duplex structure
of miR-34c and the 3′-UTR of TGIF2 is shown. The free energy of
miR-34c bound to the target site of TGIF2 was calculated by
RNAhybrid (minimum free energy, −22.5 kcal/mol). miR-34c,
microRNA-34c; ANT, adjacent non-cancerous tissue; PCR, polymerase
chain reaction; HCC, hepatocellular carcinoma; HBV, hepatitis B
virus; 3′-UTR, 3′-untranslated region; TGIF2, TGFB-induced factor
homeobox 2. |
TGIF2 is a candidate target gene of
miR-34c
TGIF2 was identified as a candidate target gene of
miR-34c by querying MiRanda and TargetScan. The analysis of
potential miR-34c binding sites within the published 3′-UTR of
TGIF2 using the miR-target prediction programs and RNAhybrid
revealed one putative miR-34c target site (Fig. 1E). TargetScan also demonstrated that
the 3′-UTR of TGIF2 targeted by miR-34c was conserved across
vertebrate species, including human, chimpanzee, rhesus macaque,
mouse and dog (Fig. 1F). These
findings indicate that the targeting of TGIF2 by miR-34c may be
evolutionarily conserved. The free energy of miR-34c binding to the
TGIF2 targeting site was calculated (minimum free energy, −22.5
kcal/mol), and the duplex structure between miR-34c and the 3′-UTR
of TGIF2 was identified by RNAhybrid (Fig. 1G).
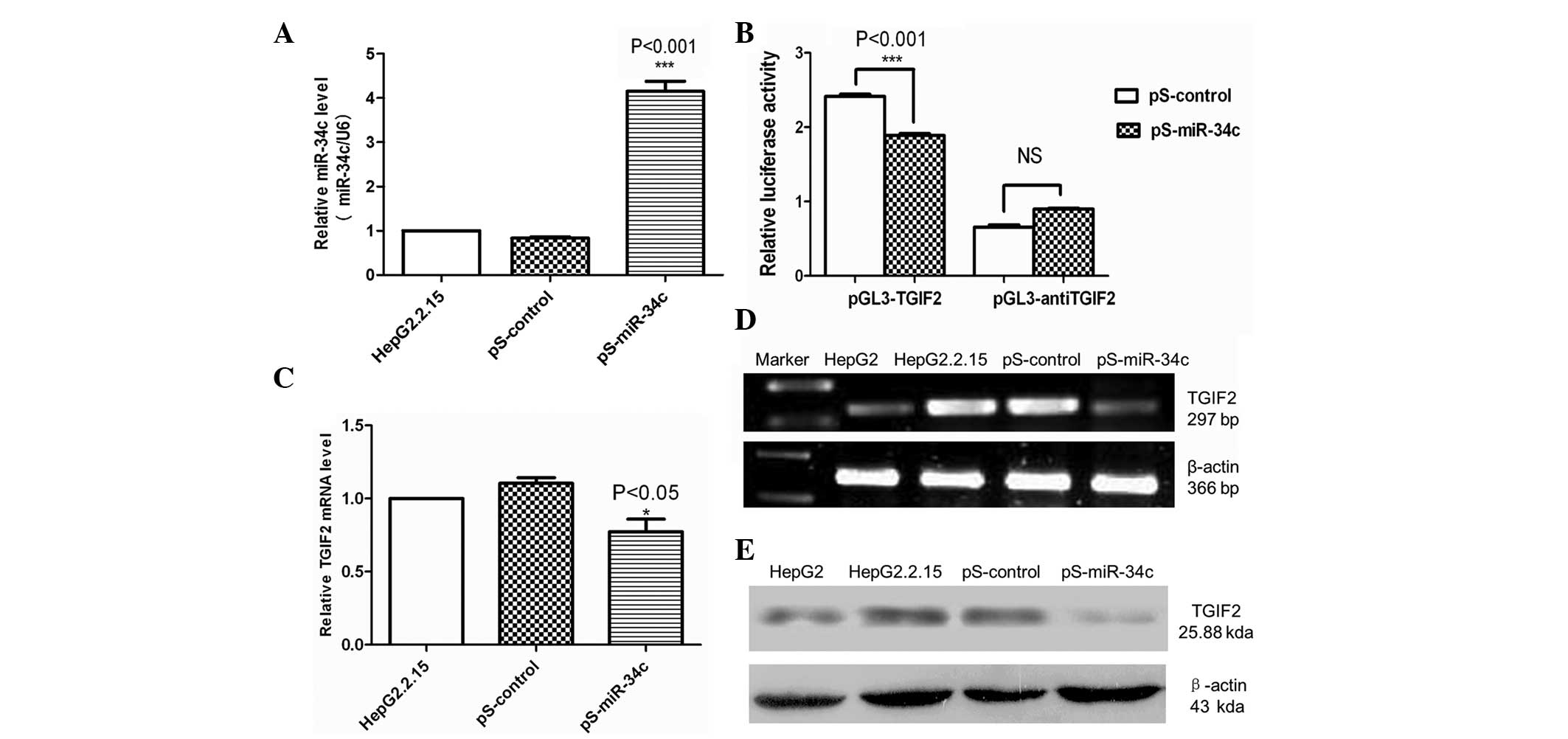
miR-34c downregulated TGIF2 expression
in HBV-associated HCC
qPCR was used to confirm that HepG2.2.15 cells
transfected with pS-miR-34c stably expressed miR-34c. The results
demonstrated that miR-34c was significantly upregulated in
HepG2.2.15 cells transfected with pS-miR-34c (P<0.001) (Fig. 2A). No difference was identified
between HepG2.2.15 cells transfected with pS-control or untreated
cells.
To test whether miR-34c directly binds to the 3′-UTR
of TGIF2, a dual luciferase reporter assay was performed. As shown
in Fig. 2B, the luciferase activity
of the reporter containing the 3′-UTR of TGIF2 reduced in cells
expressing miR-34c compared with cells expressing pS-control
(P<0.001). This result indicated that TGIF2 was a direct target
of miR-34c.
To determine whether miR-34c overexpression affects
TGIF2, the levels of TGIF2 mRNA and protein were measured 48 and 72
h after transfection. qPCR and RT-PCR demonstrated that TGIF2 mRNA
was significantly reduced following pS-miR-34c transfection
compared with pS-control-transfected and untreated cells
(P<0.05) (Fig. 2C and D). Western
blot analysis revealed that the protein levels of TGIF2 were
reduced in HepG2.2.15 cells transfected with pS-miR-34c compared
with pS-control-transfected or untreated cells (Fig. 2E). Therefore, these results were
consistent with the changes in TGIF2 mRNA expression.
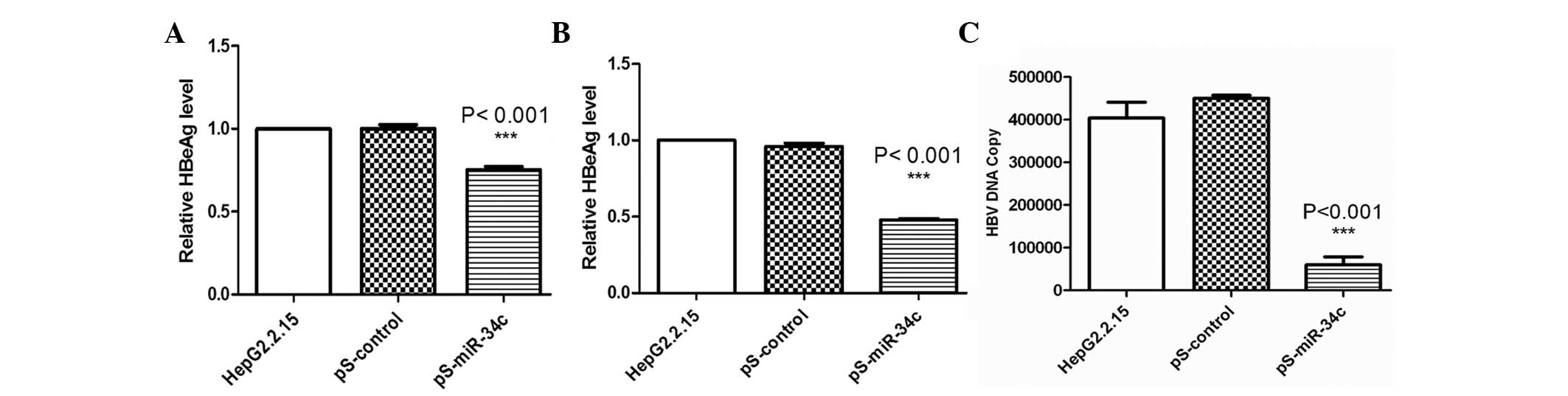
miR-34c inhibited HBV DNA replication
and viral antigen synthesis
To determine the effect of miR-34c on HBV gene
expression, HBsAg and HBeAg levels were measured 48 h after
transfection. As shown in Fig. 3A and
B, the inhibitory rates of HBsAg and HBeAg were 24.39 and
48.67%, respectively (P<0.001). No significant reduction was
observed in untreated or pS-control-transfected cells.
qPCR was performed to determine whether transfection
with pS-miR-34c would result in a reduction in HBV DNA replication.
Quantitative assays revealed that the copy number of HBV DNA was
reduced by 83.27% in cells transfected with pS-miR-34c 48 h after
transfection (P<0.001) (Fig. 3C).
No evident changes were detected in the cells transfected with
pS-control, or untreated cells.
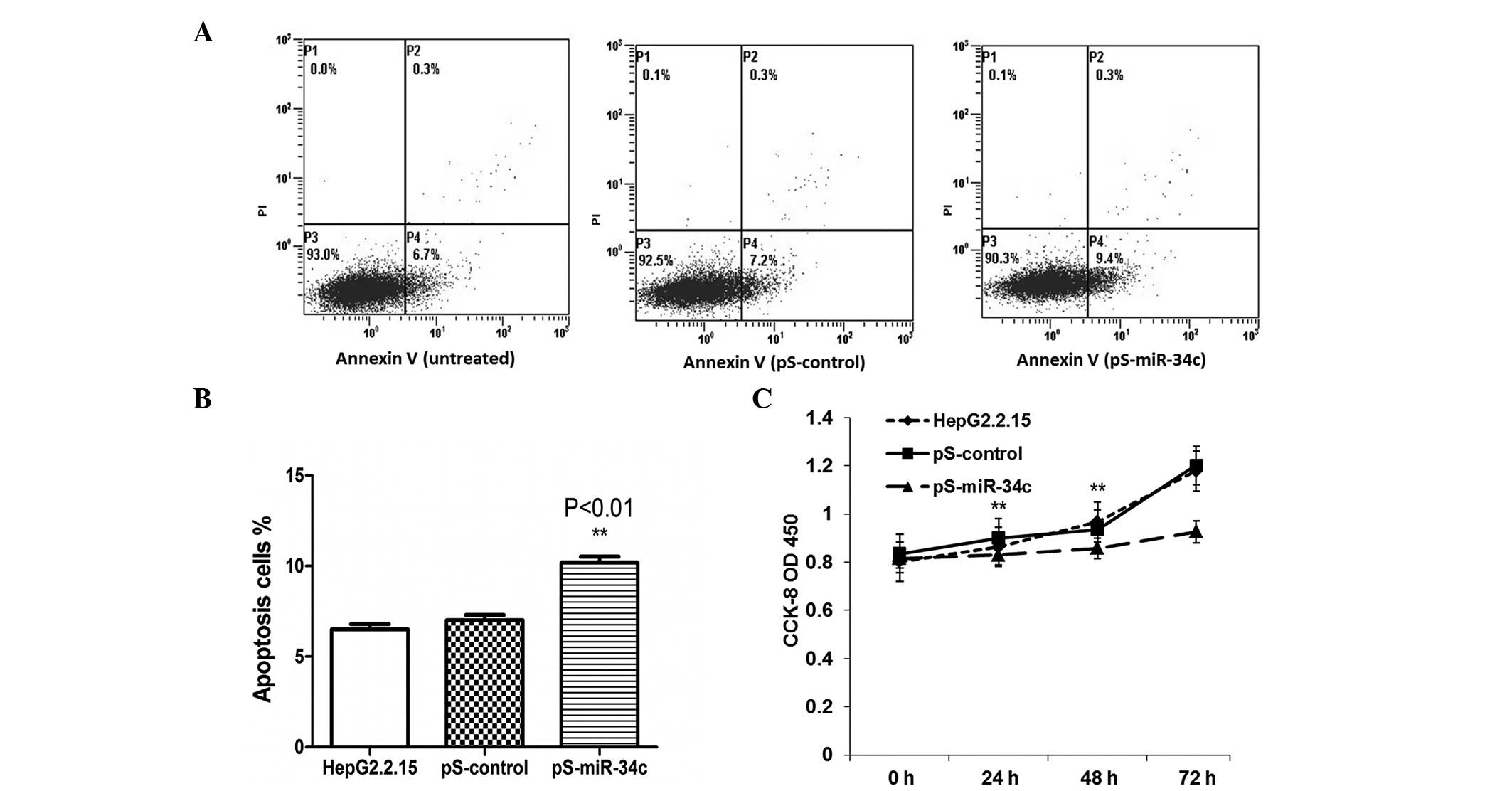
miR-34c induces apoptosis and
suppresses cell proliferation in HepG2.2.15 cells
Subsequently, the effects of miR-34c transfection on
the induction of apoptosis were examined. The data indicated that
miR-34c expression had a positive effect on apoptosis in HepG2.2.15
cells at 48 h post-transfection compared with untreated and
pS-control-transfected cells (P<0.01) (Fig. 4A and B). A CCK-8 assay was used to
observe the biological effects of miR-34c on tumor cell growth. As
shown in Fig. 4C, the overexpression
of miR-34c suppressed cell proliferation at 48 h and 72 h
post-transfection (P<0.05).
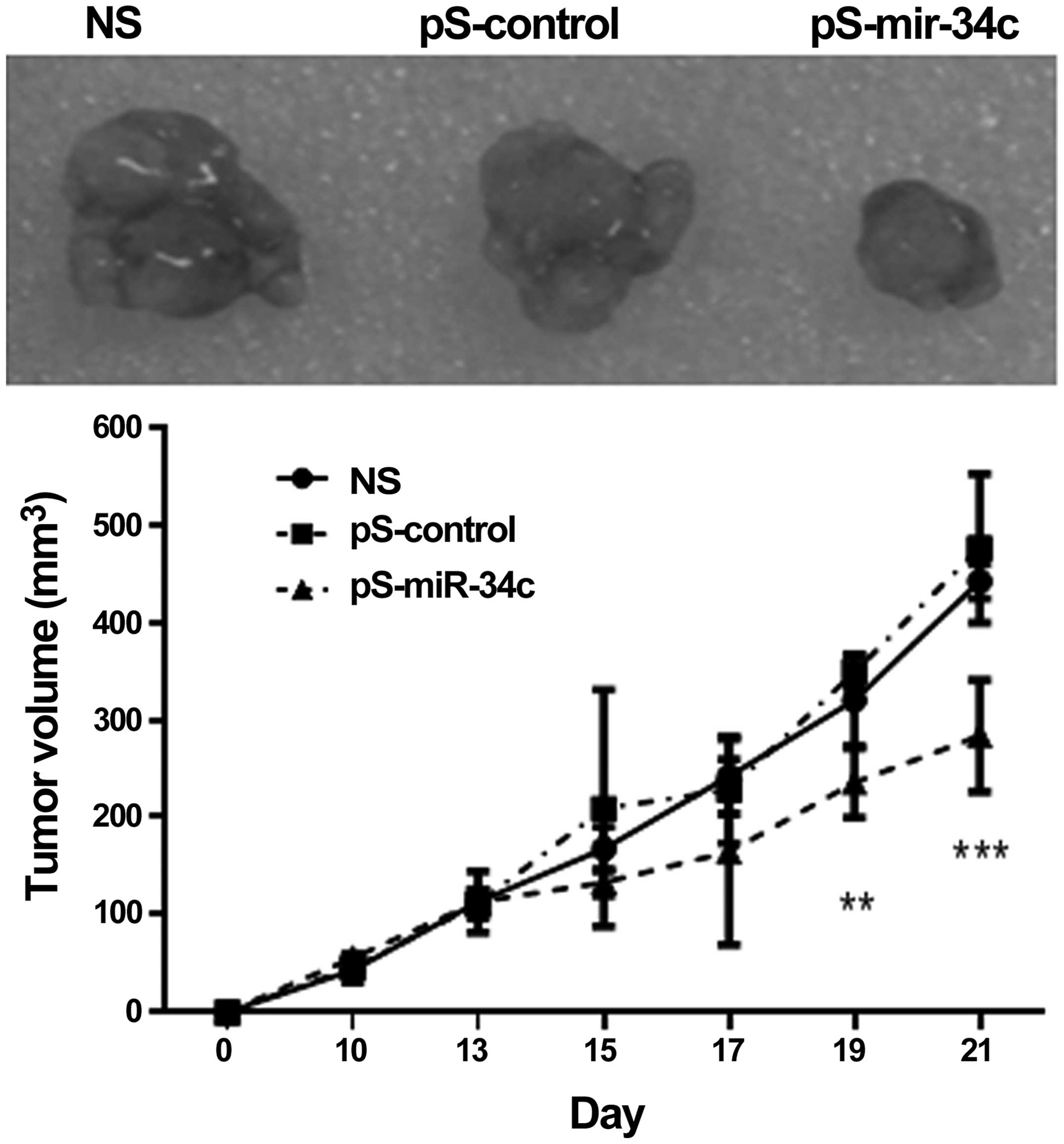
miR-34c inhibits tumor growth in
vivo
BALB/c nude mice were transplanted with
BEL7402-1.1-HBV cells and the tumors were formed in 2 weeks. As
shown in Fig. 5, tumor growth in mice
injected with miR-34c was significantly inhibited compared with the
control mice (P<0.001 at day 21).
Discussion
miRs have been shown to serve important roles in
viral infection and carcinogenesis. Chronic infection of HBV is a
key factor in the development of HCC (17). Previously, certain mammalian miRs have
been identified that exhibit antiviral effects (9). miR-34c is involved in negative feedback
control of the cell cycle, including cell cycle arrest, senescence
and apoptosis. Previous studies have shown that miR-34c is
downregulated in different tumors and cell lines, including
laryngeal and nasopharyngeal carcinoma, and prostate, colorectal,
bronchial squamous, breast, gastric and lung cancer (18–25). The
present study extended these findings by demonstrating that miR-34c
was downregulated in HBV-associated HCC tissues as well as in
HepG2.2.15 cells that constitutively replicate HBV, compared with
HepG2 cells that do not. Previous studies have indicated that miRs
influence cell proliferation, apoptosis, migration and invasion
(26–28). The present study also demonstrated
that miR-34c inhibited cell proliferation in HepG2.2.15 cells,
which was accompanied by the induction of cell apoptosis.
Viral replication and gene expression may be
inhibited by virus-specific small interfering RNAs. For example,
previous studies have shown that human miR-125a-5p interfered with
HBsAg expression (29), miR-199a-3p
and miR-210 suppressed HBV replication (30), and miR-122 suppressed HBV replication
through the downregulation of heme oxygenase-1 and N-myc
downstream-regulated gene 3 (31,32). The
present study demonstrated that HBV DNA replication and viral
antigen secretion were significantly inhibited in HepG2.2.15 cells
by miR-34c transfection. Although further studies are required to
elucidate the mechanisms behind the antiviral effect, the present
data are important for understanding HBV-host interactions.
miR-34c is downregulated in HCC and contributes to
hepatocarcinogenesis by targeting genes, including MET, E2F1-3,
MYC, SIRT1 and BCL2. The present study combined data from
bioinformatics, gene chip analysis and a dual luciferase assay to
identify the target gene of miR-34c. TGIF2 is a transcriptional
co-repressor that interacts with Smad and represses the TGF-β
signaling pathway (12). TGF-β is a
multifunctional cell factor that inhibits cell proliferation and
induces apoptosis (33). TGIF2 may
function as an oncogene by regulating the Ras/MAPK pathway
(34). The present study identified
that TGIF2 was directly targeted and repressed by miR-34c in
HBV-associated HCC cell lines, indicating that TGIF2 participates
in HBV-associated hepatocarcinogenesis. However, TGIF2 is not the
only target gene of miR-34c that is relevant to tumorigenesis and
metastasis in HBV-associated HCC. The prediction and identification
of other target genes may be important to improve understanding of
the role of miR-34c in HBV-related HCC.
Previous studies highlight miR-34a, b and c as
direct, conserved p53 target genes that presumably mediate cell
cycle arrest, induction of apoptosis and senescence by p53
(8,9,35,36). Since miRs may regulate the expression
of hundreds of target genes, these findings add new challenges and
opportunities for studies focused on the p53 network. As reported
previously, HBV DNA may bind to the p53 protein and form
DNA-protein complexes in human hepatoma cell lines (37). Cotransfection with p53 and HBV DNA
increased the replication of HBV, chloramphenicol acetyltransferase
activity, tumor cell apoptosis and cytoplasmic p53 accumulation in
hepatoma cells (37). To further
substantiate the connection between p53 and the miR-34c target
genes, additional studies are required to determine the relevance
of miR-34c-mediated target gene downregulation for p53 tumor
suppression. Studies in gene knockout mouse models of HBV
transfection-induced hepatitis and HCC may aid in determining the
function of miR-34c genes and their relevance for tumor suppression
in vivo.
In conclusion, miR-34c may serve a significant role
as a tumor suppressor miR by targeting TGIF2 during HBV-related
HCC. The precise mechanisms that result in miR-34c downregulation
following HBV infection are not well understood, but the present
results indicate that miR-34c represses HBV replication and viral
antigen expression. HepG2.2.15 cells were influenced by miR-34c
through the inhibition of cell proliferation and induction of cell
apoptosis. Thus, miR-34c and TGIF2 represent key regulatory
factors, diagnostic markers and therapeutic targets for the
prevention and treatment of HBV-associated HCC.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 30772031 and
30801036) and the Doctorate Fund for New Teachers from the National
Education Ministry of China (grant no. 200804221065).
References
|
1
|
Lavanchy D: Hepatitis B virus
epidemiology, disease burden, treatment and current and emerging
prevention and control measures. J Viral Hepat. 11:97–107. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bruix J and Sherman M: American
Association for the Study of Liver Diseases: Management of
hepatocellular carcinoma: An update. Hepatology. 53:1020–1022.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Suzuki K, Okuda Y, Ota M, Kojima F and
Horimoto M: Diagnosis of hepatocellular carcinoma nodules in
patients with chronic liver disease using contrast-enhanced
sonography: Usefulness of the combination of arterial- and
kupffer-phase enhancement patterns. J Ultrasound Med. 34:423–433.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Carleton M, Cleary MA and Linsley PS:
MicroRNAs and cell cycle regulation. Cell Cycle. 6:2127–2132. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Skaftnesmo KO, Prestegarden L, Micklem DR
and Lorens JB: MicroRNAs in tumorigenesis. Curr Pharm Biotechnol.
8:320–325. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Paris R, Henry RE, Stephens SJ, et al:
Multiple p53-independent gene silencing mechanisms define the
cellular response to p53 activation. Cell Cycle. 7:2427–2433. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Raver-Shapira N, Marciano E, Meiri E, et
al: Transcriptional activation of miR-34a contributes to
p53-mediated apoptosis. Mol Cell. 26:731–743. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
He L, He X, Lim LP, et al: A microRNA
component of the p53 tumor suppressor network. Nature.
447:1130–1134. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kong YW, Cannell IG, de Moor CH, et al:
The mechanism of microRNA-mediated translation repression is
determined by the promoter of the target gene. Proc Natl Acad Sci
USA. 105:8866–8871. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Migliore C, Petrelli A, Ghiso E, et al:
MicroRNAs impair MET mediated invasive growth. Cancer Res.
68:10128–10136. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wotton D, Lo RS, Lee S and Massagué J: A
Smad transcriptional corepressor. Cell. 97:29–39. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang P, Li Q-J, Feng Y, Zhang Y, Markowitz
GJ, Ning S, Deng Y, Zhao J, Jiang S, Yuan Y, et al:
TGF-β-miR-34a-CCL22 signaling-induced Treg cell recruitment
promotes venous metastases of HBV-positive hepatocellular
carcinoma. Cancer Cell. 22:291–303. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Karimi-Googheri M, Daneshvar H,
Nosratabadi R, Zare-Bidaki M, Hassanshahi G, Ebrahim M, Arababadi
MK and Kennedy D: Important roles played by TGF-β in hepatitis B
infection. J Med Virol. 86:102–108. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu N, Jiao T, Huang Y, Liu W, Li Z and Ye
X: Hepatitis B virus regulates apoptosis and tumorigenesis through
the microRNA-15a-Smad7-transforming growth factor beta pathway. J
Virol. 89:2739–2749. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
McGlynn KA and London WT: Epidemiology and
natural history of hepatocellular carcinoma. Best Pract Res Clin
Gastroenterol. 19:3–23. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cai KM, Bao XL, Kong XH, et al:
Hsa-miR-34c suppresses growth and invasion of human laryngeal
carcinoma cells via targeting c-Met. Int J Mol Med. 25:565–571.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tarasov V, Jung P, Verdoodt B, et al:
Differential regulation of microRNAs by p53 revealed by massively
parallel sequencing: miR-34a is a p53 target that induces apoptosis
and G1-arrest. Cell Cycle. 6:1586–1593. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li T, Chen JX, Fu XP, Yang S, Zhang Z,
Chen KH and Li Y: microRNA expression profiling of nasopharyngeal
carcinoma. Oncol Rep. 25:1353–1363. 2011.PubMed/NCBI
|
|
21
|
Toyota M, Suzuki H, Sasaki Y, Maruyama R,
Imai K, Shinomura Y and Tokino T: Epigenetic silencing of
microRNA-34b/c and B-cell translocation gene 4 is associated with
CpG island methylation in colorectal cancer. Cancer Res.
68:4123–4132. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mascaux C, Laes JF, Anthoine G, Haller A,
Ninane V, Burny A and Sculier JP: Evolution of microRNA expression
during human bronchial squamous carcinogenesis. Eur Respir J.
33:352–359. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu F, Jiao Y, Zhu Y, Wang Y, Zhu J, Cui X,
Liu Y, He Y, Park EY, Zhang H, Lv X, et al: MicroRNA 34c gene
down-regulation via DNA methylation promotes self-renewal and
epithelial-mesenchymal transition in breast tumor-initiating cells.
J Biol Chem. 87:465–473. 2012. View Article : Google Scholar
|
|
24
|
Suzuki H, Yamamoto E, Nojima M, et al:
Methylation-associated silencing of microRNA-34b/c in gastric
cancer and its involvement in an epigenetic field defect.
Carcinogenesis. 31:2066–2073. 2012. View Article : Google Scholar
|
|
25
|
Wang Z, Chen Z, Gao Y, Li N, Li B, Tan F,
Tan X, Lu N, Sun Y, Sun J, Sun N and He J: DNA hypermethylation of
microRNA-34b/c has prognostic value for stage I non-small cell lung
cancer. Cancer Biol Ther. 11:490–496. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Garzon R, Marcucci G and Croce CM:
Targeting microRNAs in cancer: Rationale, strategies and
challenges. Nat Rev Drug Discov. 9:775–789. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shenouda SK and Alahari SK: MicroRNA
function in cancer: Oncogene or a tumor suppressor? Cancer
Metastasis Rev. 28:369–378. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Potenza N, Papa U, Mosca N, Zerbini F,
Nobile V and Russo A: Human microRNA hsa-miR-125a-5p interferes
with expression of hepatitis B virus surface antigen. Nucl Acids
Res. 39:5157–5163. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang GL, Li YX, Zheng SQ, Liu M, Li X and
Tang H: Suppression of hepatitis B virus replication by
microRNA-199a-3p and microRNA-210. Antiviral Res. 88:169–175. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qiu L, Fan H, Jin W, Zhao B, Wang Y, Ju Y,
Chen L, Chen Y, Duan Z and Meng S: miR-122-induced down-regulation
of HO-1 negatively affects miR-122-mediated suppression of HBV.
Biochem Biophys Res Commun. 398:771–777. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fan CG, Wang CM, Tian C, Wang Y, Li L, Sun
WS, Li RF and Liu YG: miR-122 inhibits viral replication and cell
proliferation in hepatitis B virus-related hepatocellular carcinoma
and targets NDRG3. Oncol Rep. 26:1281–1286. 2011.PubMed/NCBI
|
|
33
|
Yamada SD, Baldwin RL and Karlan BY:
Ovarian carcinoma cell cultures are resistant to TGF-beta1-mediated
growth inhibition despite expression of functional receptors.
Gynecol Oncol. 75:72–77. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lo RS, Wotton D and Massagué J: Epidermal
growth factor signaling via Ras controls the Smad transcriptional
co-repressor TGIF. EMBO J. 20:128–136. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Corney DC, Flesken-Nikitin A, Godwin AK,
Wang W and Nikitin AY: MicroRNA-34b and microRNA-34c are targets of
p53 and cooperate in control of cell proliferation and
adhesion-independent growth. Cancer Res. 67:8433–8438. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yamakuchi M, Ferlito M and Lowenstein CJ:
miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci
USA. 105:13421–13426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Qu J, Lin J, Zhang S, Zhu Z, Ni C, Zhang
S, Gao H and Zhu M: HBV DNA can bind to P53 protein and influence
p53 transactivation in hepatoma cells. Biochem Biophys Res Commun.
386:504–509. 2009. View Article : Google Scholar : PubMed/NCBI
|