Introduction
Multiple osteochondroma (MO; MIM #133700 and
#133701), also known as multiple hereditary exostoses, is an
autosomal dominant skeletal disorder with characteristic multiple
cartilage-capped tumours (exostoses or osteochondromas) growing
outward from the metaphyseal region of the long tubular bones. The
estimated prevalence of MO has been recorded to range from 1/100 in
a small Guam population (1) to
1.3/100,000 in a European population (2) and at least 1/50,000 in a Western
population (3). Osteochondromas are a
consequence of the superfluous proliferation of chondrocytes and
bone growth at the juxta-epiphyseal regions of the long bones, and
are the most frequently identified benign bone tumour.
Osteochondromas are slow-growing during the first decades of life,
growing in number and size until the closure of the growth plates
upon skeletal maturation at the end of puberty. Characteristic
features of MO consist of significant inter- and intra-familial
phenotypic variability, including variability in the size and
number of the osteochondromas, the location and number of the
associated bones, and the degree of the deformity. Although benign,
osteochondromas can result in a number of secondary complications,
including compression of the blood vessels, tendons and nerves.
Individuals with osteochondroma are often of short stature, with
deformities leading to problems in functionality. The most serious
secondary complication is the malignant transformation toward a
secondary peripheral chondrosarcoma, which occurs in 0.5–5% of
cases (4).
MO is a genetically heterogeneous disease. At least
2 loci have been isolated by linkage analysis and positional
cloning: EXT1 (MIM #608177) maps to chromosome
8q24.11-q24.13, spans ~350 kb and is comprised of 11 exons
(5), while EXT2 (MIM #608210) maps to
chromosome 11p11-p11.2, spans almost 108 kb and is comprised of 16
exons (6). EXT1 and
EXT2 belong to the putative tumour-suppressor EXT
gene family, which also contains three homologous EXT-like (EXTL)
genes: EXTL1, EXTL2 and EXTL3 (7–9). All
EXT gene family members share a homologous carboxyl terminus
and encode glycosyltransferases that are involved in heparin
sulfate (HS) biosynthesis (10). A
number of studies have investigated MO-causing mutations in a
variety of populations (11–13). These data are being gathered in the
Multiple Osteochondromas Mutation Database (http://medgen.ua.ac.be/LOVDv.2.0/). To date, >400
and 200 mutations have been identified in EXT1 and
EXT2, respectively. The majority of the mutations (80%) are
nonsense, frameshift and splice site mutations, which lead to the
premature termination of translation, or are involved in the
partial or total deletion of the gene (14).
The present study screened for mutations in the
EXT1/EXT2 genes using polymerase chain reaction (PCR)
and direct sequencing in three generations of a family with MO. A
novel frameshift mutation, c.119_120delCT (p.Thr40ArgfsX15), was
consequently identified in exon 2 of the EXT2 gene.
Patients and methods
Patients
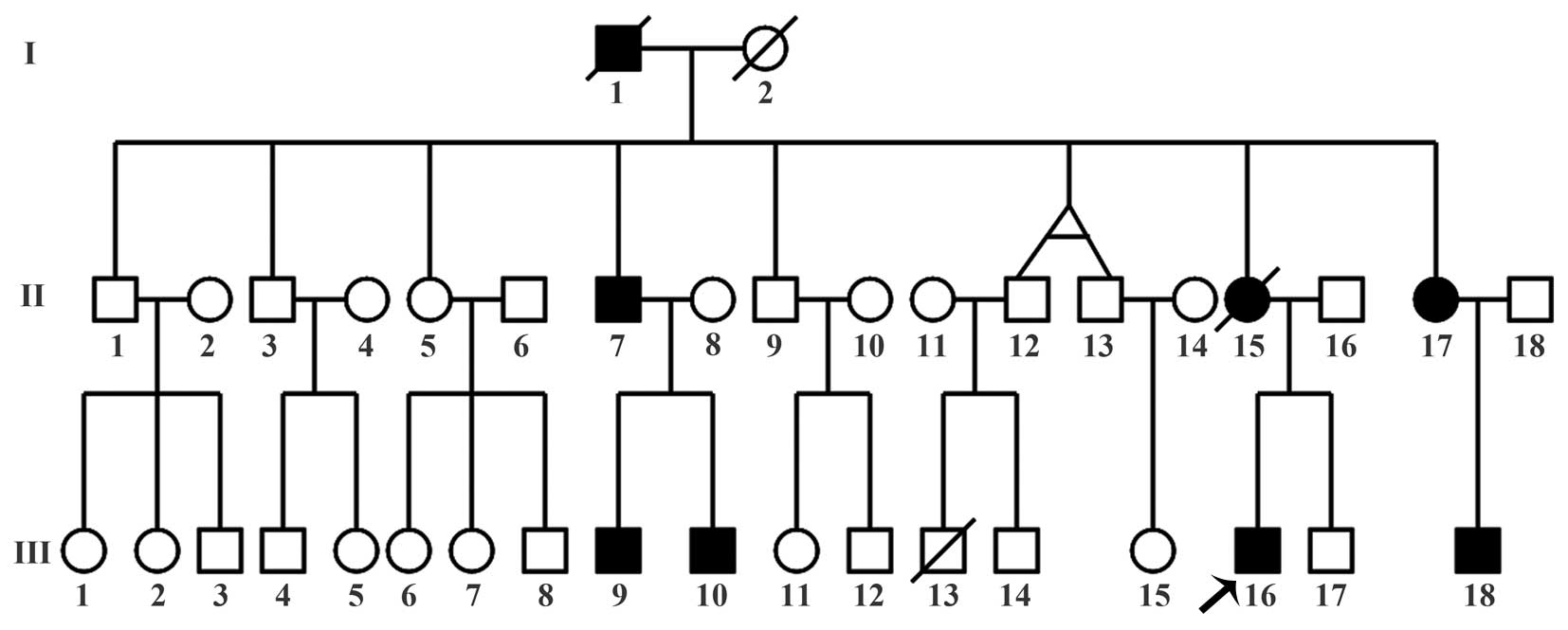
Three generations of a MO family from Jilin, China,
were investigated in the present study (Fig. 1). From the family, 8 members were
affected with MO, including 2 deceased individuals. The proband
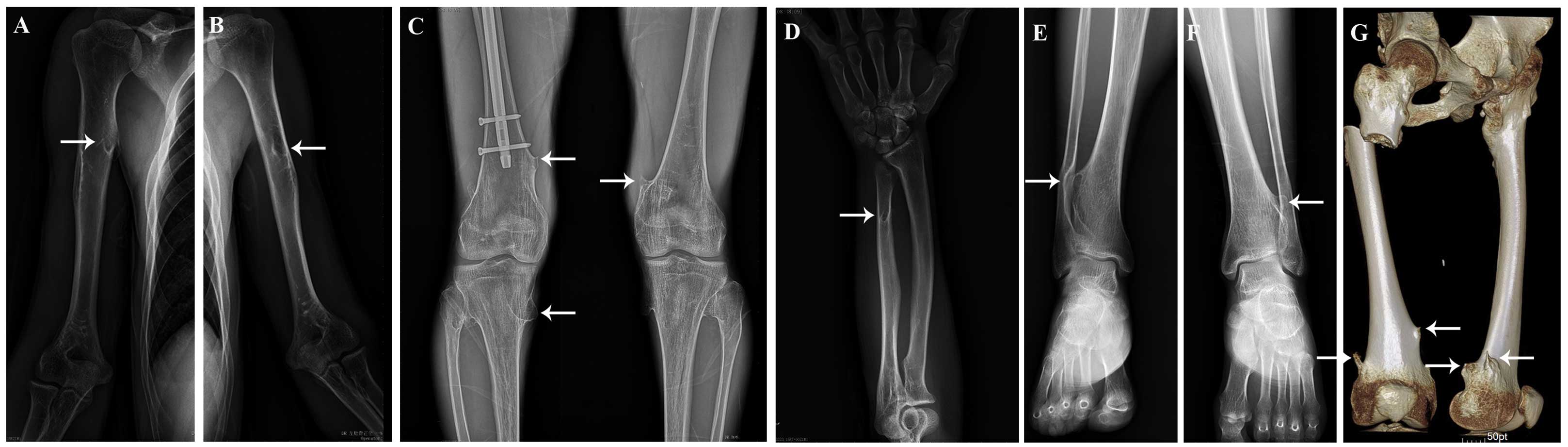
(III-16) was a 28-year-old man suffering from multiple exostoses
involving the bilateral proximal humeri, the left ulnar, the
bilateral proximal and distal tibiae, the bilateral distal femurs
and the right proximal femur (Fig.
2). On January 13, 2014, the proband underwent osteochondroma
surgery for the right femur at Country Hospital of Fusong (Fusong,
China) due to pain and functional limitations. At 15 days
post-surgery (January 28, 2014), the proband suffered a
subtrochanteric fracture of the right femur and was referred to the
Second Hospital of Jilin University (Changchun, China), where
internal fixation of the fracture site was performed.
Clinical data of each subject was recorded by
nurse-administered questionnaires. Each participant underwent
careful physical and/or radiographic examinations by two or more
experienced experts at the Department of Orthopedics, The Second
Hospital of Jilin University (Changchun, Jilin, China). MO was
classified as at least 2 osteochondromas, arising from the lateral
ends of the humeri, ulnae, femurs, tibiae, fibulae or knee joints.
A group of 50 unrelated healthy subjects, matched for geographical
ancestry, were included as controls. EDTA-anticoagulated peripheral
blood (5 ml) was drawn from 10 family members (Table I) and from the 50 healthy controls.
Written informed consent was obtained from all subjects or their
respective guardians prior to their participation in the
experimental protocol. This study was approved by the Ethics Review
Committee of the Second Hospital of Jilin University.
 | Table I.Characteristics of study subjects in
the MO family. |
Table I.
Characteristics of study subjects in
the MO family.
| Subject | Age, years | Gender | MO status |
|---|
| II-5 | 62 | Female | Unaffected |
| II-7 | 61 | Male | Affected |
| II-17 | 48 | Female | Affected |
| III-9 | 37 | Male | Affected |
| III-10 | 33 | Male | Affected |
| III-11 | 30 | Female | Unaffected |
| III-14 | 29 | Male | Unaffected |
| III-16 | 28 | Male | Affected |
| III-17 | 24 | Male | Unaffected |
| III-18 | 25 | Male | Affected |
Genetic analysis
Genomic DNA was extracted from peripheral blood
leukocytes using the QIAamp DNA Blood Mini kit (Qiagen, Hilden,
Germany) according to the manufacturer's protocols. All exons and
flanking intronic regions of the EXT1 and EXT2 genes
were amplified by PCR using the primers listed in Table II. The amplifications were performed
to a final volume of 50 µl, containing 1 µl genomic DNA, 25 µl 2X
Premix Taq™ (Takara Biotechnology Co., Ltd., Dalian, China), 1 µl
of each primer and 22 µl sterilized distilled water. All PCR
programs included an initial denaturation of 5 min at 95°C,
followed by 35 cycles of 30 sec at 95°C, 45 sec at annealing
temperature (Ta) and 30 sec at 72°C. Finally, an extension at 72°C
was performed for 10 min. Ta was 60°C for all primer combinations,
with the exception of primers for the amplification of overlapping
regions of exon 1 of EXT1. For these primer combinations, Ta
was set at 57°C for ex1–2 and ex1–3. Direct sequencing of the PCR
products was performed using BigDye® Terminator v3.1 (Applied
Biosystems, Foster City, CA, USA) with the ABI 3730 automated
sequencer. Sequences generated from patients were compared with the
published EXT1 and EXT2 reference sequences from the
National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/; GenBank accession nos.
NM_000127.2 and NM_207122.1, respectively). When an
insertion/deletion mutation was found, the amplified products with
mutations were first ligated to a pMD-18T vector (Takara
Biotechnology Co., Ltd.) and transformed into Escherichia
coli. Next, sequencing was performed with plasmids extracted
from E. coli using the AxyPrep Plasmid Miniprep kit (Axygen
Biosciences, Union City, CA, USA). In addition, detected mutations
were confirmed in another 50 unrelated healthy controls.
| Table II.Primers used to amplify the exons of
the EXT1 and EXT2 genes. |
Table II.
Primers used to amplify the exons of
the EXT1 and EXT2 genes.
| Gene | Exon | Upstream primer
(5′-3′) | Downstream primer
(5′-3′) | Amplicon length,
bp | Ta, °C |
|---|
| EXT1 | Ex1–1a |
GGAGAGTTTGAAGTCTTTACAGGC |
TGGGGTCCGAGGTGTAGAAC | 561 | 60 |
|
| Ex1–2a |
GGCTTGCAGTTTAGGGCATCGAG |
TTGTTCCACAAGTGGAGACTCTG | 485 | 57 |
|
| Ex1–3a |
CCAGTTGTCACCTCAGTATGTGC |
AACTTCACACCTGGACCAAG | 543 | 57 |
|
| Ex2 |
TGCGTAAATTCATGCACATGG |
GGAGAGGTGATAATGTTAAACC | 268 | 60 |
|
| Ex3 |
TGCCAGTCATTGAGTTTGTAC |
GGAGATTTTGTTGGAAAGTGAA | 386 | 60 |
|
| Ex4 |
CTATATGCTAGAAGCCAAATGC |
CAATATATCCAAGTACAGGAATC | 372 | 60 |
|
| Ex5 |
TCCATTACTCTCTCTGTCTTG |
ATGCAGGGTGTTAGATGGAC | 410 | 60 |
|
| Ex6 |
CCTGTCAGGACATAAGAAGC |
TGAAAAGGGTGTAACGAGGC | 426 | 60 |
|
| Ex7 |
TCTGCCGTTTTGTCTTGCTG |
ATACACACAAGGTCACAAAGC | 450 | 60 |
|
| Ex8 |
GTGAGGATGGGAGAATTGTC |
AGGAATCGGGCTGATTAAAAC | 334 | 60 |
|
| Ex9 |
GAATTAATGTTTCGCCACAGTC |
AAACACACATTTGACACATCAG | 435 | 60 |
|
| Ex10 |
CATCATCATTATCATTACCATTC |
GGAGAGCTTTACATCCTTGG | 525 | 60 |
|
| Ex11 |
TTGCTGCTTGCTCATTTGCC |
TTTGTCATTCTGCTCATCTAAG | 420 | 60 |
| EXT2 | Ex2-1a |
TGAGTGACAGAGTGAAACCC |
CAGAGCATAGATATACACCTTG | 488 | 60 |
|
| Ex2-2a |
AGCCGACAGTCCCATCCC |
AAACCAACTCAAGAGCAGAAG | 425 | 60 |
|
| Ex3 |
TGGGATTTCCAGGAGTTTGC |
CACTTCTAAATCTTCAGGAGG | 390 | 60 |
|
| Ex4 |
AGCAGAGAGGCTGTCCGTAA |
ATAGGAAGCCGTTTCAGCAA | 805 | 60 |
|
| Ex5 |
AGGGACTCAGATGTAACTAAGA |
ACGAACACAAGACACCAGAC | 792 | 60 |
|
| Ex6 |
AGTATTGCTTGGCGTCAACC |
TAGACCAGTGTACTAACTCTC | 401 | 60 |
|
| Ex7 |
AATGGAGCTGTAAGAGAACTC |
ATCTAGTGGAGGAAGTAAACC | 408 | 60 |
|
| Ex8 |
TGGCTTGAACAGCAGGGAG |
AATTATGCTGCCCTTATCAGG | 446 | 60 |
|
| Ex9 |
CACCAAGCCTGCCATGTTTG |
GGCATGCTGTCTCAGAAATG | 408 | 60 |
|
| Ex10 |
ACCTTTGGATTTGATGAGAGC |
TAACCCACACTCTTACGCAC | 450 | 60 |
|
| Ex11 |
TCTCCAGAATCCCATTATGAC |
CATATTTTCTACTATGAGCGTG | 469 | 60 |
|
| Ex12 |
GTCACTTGACCAAAAGCATTC |
GAGCTTAAAGTTTATCTAGTCC | 417 | 60 |
|
| Ex13 |
CTTGTGAGTTCTGCCGTTGG |
ACAAATTGAGTGAGTAGCATTC | 471 | 60 |
|
| Ex14 |
ACCTGTCAACCTTTTTAAGAAC |
CCAAGATCCAAGTAGGTCAAC | 445 | 60 |
In silico analysis and homology
study
When a mutation that had never been reported was
found, the functional consequences of amino acid changes were
predicted using the Mutation Taster software (http://www.mutationtaster.org). Regarding the homology
study, ClustalW2 software (http://www.ebi.ac.uk/Tools/msa/clustalw2/) was used to
compare the human EXT2 protein sequence (NP_997005.1) with the
homologs from Pan troglodytes, Macaca mulatta, Mus
musculus, Rattus norvegicus, Canis lupus
familiaris, Bos taurus, Gallus gallus, Danio
rerio and Xenopus (sequences acquired from http://www.ensembl.org/).
Results
Genetic analysis identifies a novel
frameshift mutation (c.119_120delCT) in EXT2 as the causative
mutation in the MO family
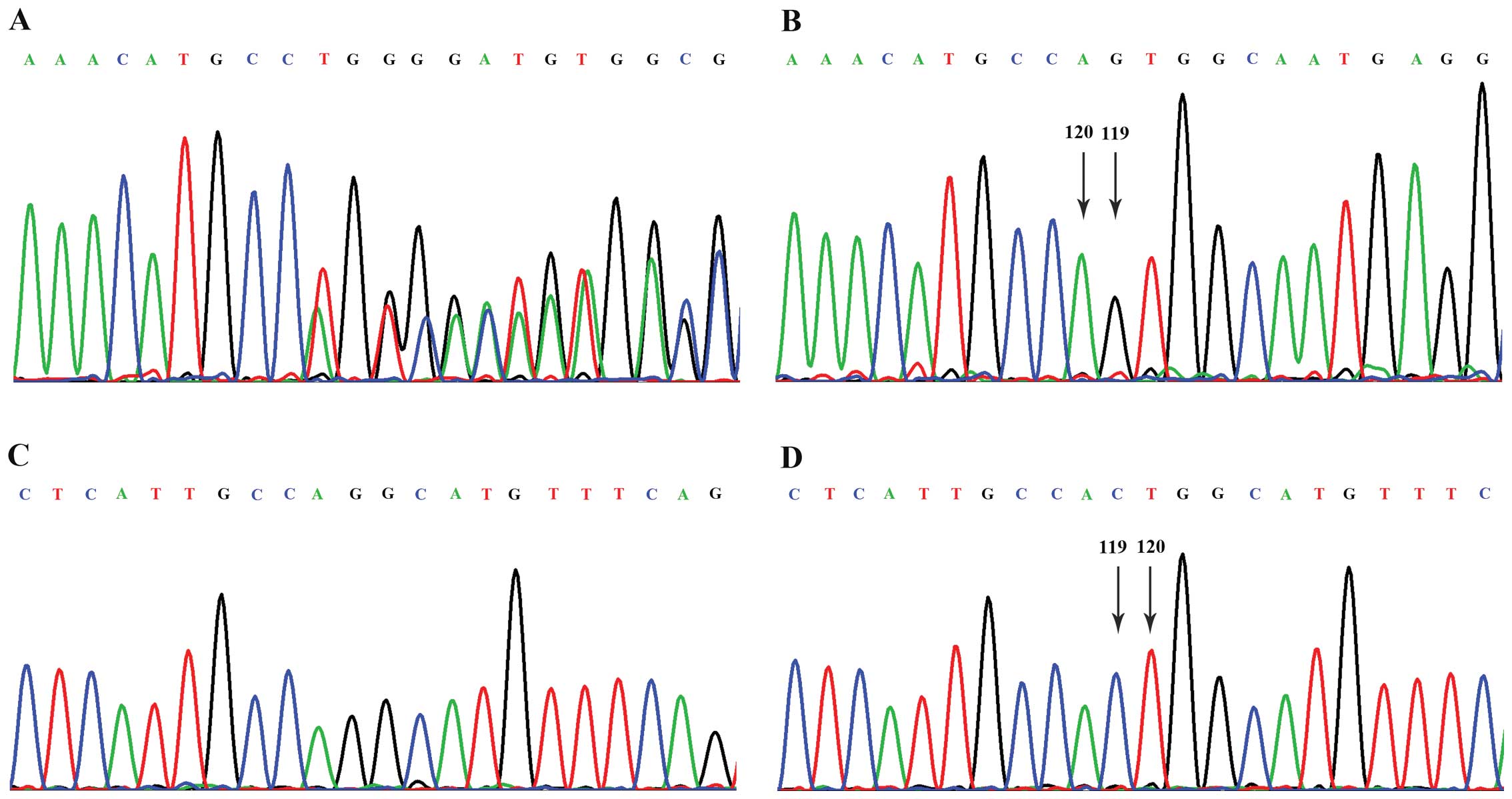
Following sequencing of all the exons and flanking
intronic regions of the EXT1 and EXT2 genes, no
mutations were detected in the EXT1 gene, but a heterozygous
deletion of two nucleotides (CT) at c.119_120 in exon 2 of the
EXT2 gene was found in the proband (Fig. 3). Subsequently, this variation was
detected in all the 5 affected individuals (II-7, II-17, III-9,
III-10 and III-18) in this pedigree by PCR and direct sequencing,
and was not detected in any of the 4 unaffected family members
(II-5, III-11, III-14 and III-17) or in the 50 healthy controls.
Thus, this finding suggested that the novel c.119_120delCT
frameshift mutation in the EXT2 gene was the causative
mutation for MO in this family.
In silico analyses indicate the
critical impact of the c.119_120delCT mutation on EXT2
function
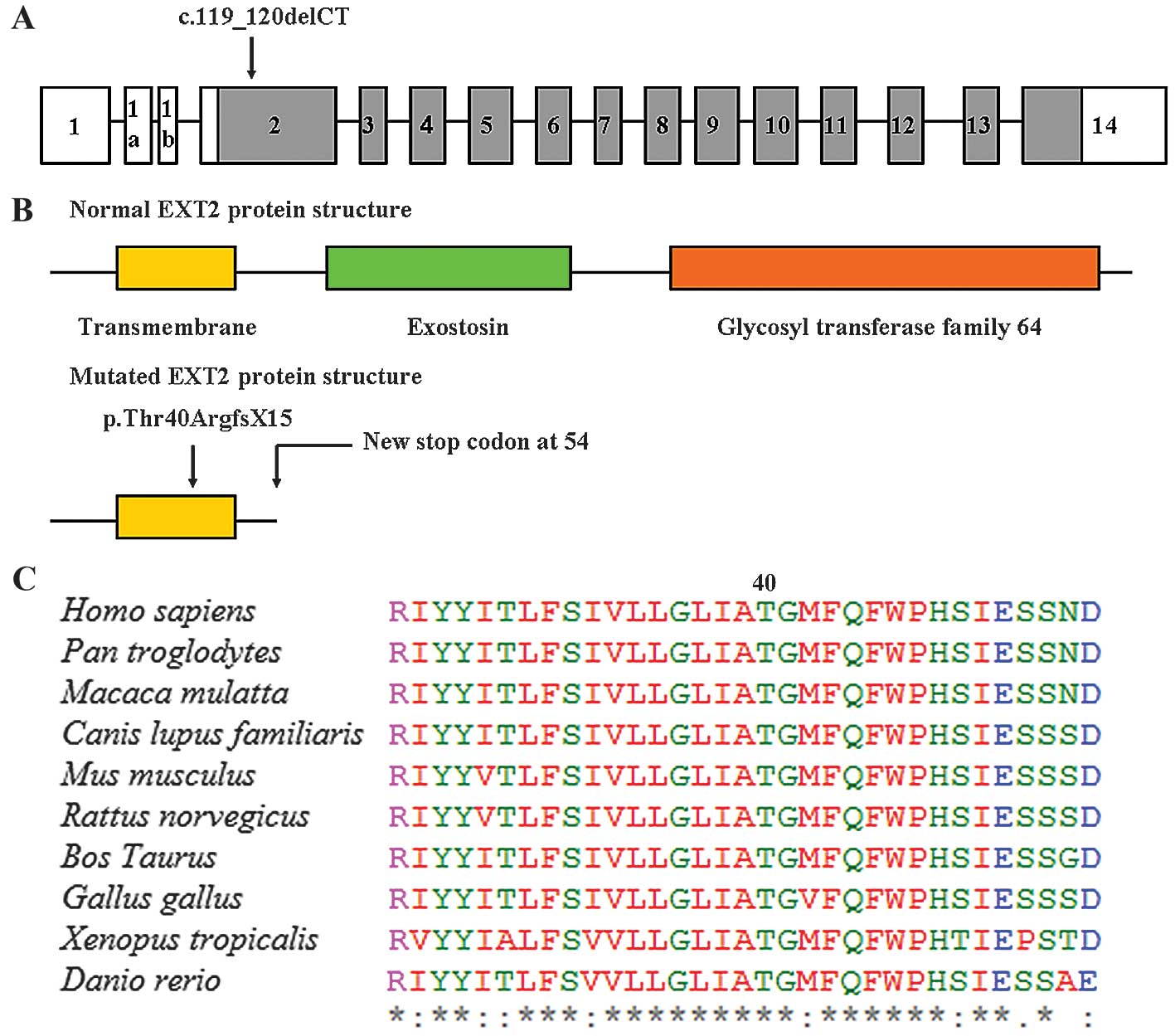
The EXT2 gene consists of 16 exons, and the
c.119_120delCT mutation was located in exon 2 (Fig. 4A). To understand the potential impact
of the c.119_120delCT frameshift mutation on EXT2 function, in
silico analyses were performed and the predicted result was
disease-causing. The EXT2 protein is composed of 718 amino acids,
with one transmembrane region (amino acids 26–46) and two critical
domains: The exostosin domain (amino acids 100–380) and the
glycosyltransferase family 64 domain (amino acids 456–701)
(Fig. 4B). The c.119_120delCT
frameshift mutation results in the substitute of amino acid
Threonine by Arginine at position 40 and creates a premature
translational termination signal at amino acid position 54 of the
EXT2 protein (Fig. 4B). Compared with
normal EXT2 protein, the mutated EXT2 protein lacks 665 amino acids
at the C-terminus, including the two critical exostosin and
glycosyltransferase family 64 domains. Thr40 is located in the
transmembrane region, which is highly conserved across various
vertebrate species (Fig. 4C),
indicating its functional importance.
Discussion
EXT1 and EXT2 are tumour suppressor
genes encoding for endoplasmic reticulum-localized type II
transmembrane glycoproteins (15).
EXT1 and EXT2 form a hetero-oligomeric complex in the Golgi
apparatus that catalyses the alternative transfer of
N-acetyl-glucosamine and D-glucuronic acid residues to the
elongating HS glycosaminoglycan chain (16). This observation is consistent with the
notion that inherited mutations in either of the two EXT
genes could result in MO. HS and HS proteoglycans are known as
regulators of the distribution and receptor binding of signalling
molecule family members such as transforming growth factor-β,
fibroblast growth factor, and Indian hedgehog (IHH) (17). Previously, studies showed that
mutations in the EXT1/EXT2 genes impaired the
biosynthesis of HS and that a decreased content of HS disturbs the
IHH/parathyroid hormone-related peptide signalling pathway in MO;
this disturbance leads to abnormal endochondral ossification
resulting in exostoses formation (18,19).
EXT1 and EXT2 gene mutations have been
reported to be involved in the pathogenesis of MO and are
responsible for 70–95% of MO cases. EXT1 accounts for 56–78%
of the MO families, whereas an EXT2 mutation is detected in
21–44% of cases (20).
EXT1/EXT2 mutation analysis demonstrated that
mutations in EXT1 are more common in the Caucasian
population in North America and Europe. However, these associations
were different in the Chinese population, where mutations in
EXT2 were more common than EXT1 mutations (21). In the past 10 years, 67 Chinese
families affected with MO and 10 spontaneous cases affected with MO
have been screened for mutations in EXT1 or EXT2.
Mutations in the EXT1 gene account for 37.7% (29/77) of the
patients, whereas EXT2 mutations account for 44.2% (34/77),
which is higher than that of EXT1. Based on all the
aforementioned data, EXT2 mutations may be responsible for
more cases of MO in the Chinese population. EXT2 mutations
tend to be concentrated in the first two-thirds of the coding
region, and mutations may occur more commonly in exon 2 compared
with other exons of EXT2, whereas mutations in EXT1
are reported to be scattered throughout the complete gene.
In the present study, a large family with MO was
investigated and a sequence analysis was performed of the entire
coding regions of EXT1 and EXT2 for the proband
(III-16). A heterozygous deletion of two nucleotides (CT) was found
at c.119_120 in exon 2 of EXT2 in the proband, and this
variation was also present in all the other 5 affected patients
(II-7, II-17, III-9, III-10 and III-18) from this pedigree, but was
not detected in any of the 4 unaffected family members (II-5,
III-11, III-14 and III-17) or in the 50 healthy controls. This
mutation (c.119_120delCT; p.Thr40ArgfsX15) results in a frameshift
mutation starting from amino acid position 40 and introduces a
premature termination signal at amino acid 54, which leads to the
production of a truncated EXT2 protein with only 53 amino acids,
665 amino acids shorter than the normal EXT2 protein. Furthermore,
the c.119_120delCT mutation was located in the transmembrane region
of the EXT2 protein, the mutated EXT2 protein lacking 665 amino
acids at the C-terminus, which included the two critical exostosin
and glycosyltransferase family 64 domains.
In conclusion, the present study identified a novel
deleterious frameshift mutation, c.119_120delCT (p.Thr40ArgfsX15),
in the transmembrane region of the EXT2 gene from a large MO
family. This study is useful for extending the known mutational
spectrum of EXT2, for understanding the genetic basis of MO
in the patients studied, and for further application of mutation
screening in the genetic counseling and subsequent prenatal
diagnosis of this family.
Acknowledgements
The authors would like to thank all the study
participants for their interest and cooperation in this study.
References
|
1
|
Krooth RS, MacKlin MT and Hilbish TF:
Diaphysial aclasis (multiple exostoses) on Guam. Am J Hum Genet.
13:340–347. 1961.PubMed/NCBI
|
|
2
|
Hennekam RC: Hereditary multiple
exostoses. J Med Genet. 28:262–266. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schmale GA, Conrad EU III and Raskind WH:
The natural history of hereditary multiple exostoses. J Bone Joint
Surg Am. 76:986–992. 1994.PubMed/NCBI
|
|
4
|
Bovée JV: Multiple osteochondromas.
Orphanet J Rare Dis. 3:32008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lüdecke HJ, Ahn J, Lin X, Hill A, Wagner
MJ, Schomburg L, Horsthemke B and Wells DE: Genomic organization
and promoter structure of the human EXT1 gene. Genomics.
40:351–354. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Clines GA, Ashley JA, Shah S and Lovett M:
The structure of the human multiple exostoses 2 gene and
characterization of homologs in mouse and Caenorhabditis elegans.
Genome Res. 7:359–367. 1997.PubMed/NCBI
|
|
7
|
Wise CA, Clines GA, Massa H, Trask BJ and
Lovett M: Identification and localization of the gene for EXTL, a
third member of the multiple exostoses gene family. Genome Res.
7:10–16. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wuyts W, Van Hul W, Hendrickx J, Speleman
F, Wauters J, De Boulle K, Van Roy N, Van Agtmael T, Bossuyt P and
Willems PJ: Identification and characterization of a novel member
of the EXT gene family, EXTL2. Eur J Hum Genet. 5:382–389.
1997.PubMed/NCBI
|
|
9
|
Van Hul W, Wuyts W, Hendrickx J, Speleman
F, Wauters J, De Boulle K, Van Roy N, Bossuyt P and Willems PJ:
Identification of a third EXT-like gene (EXTL3) belonging to the
EXT gene family. Genomics. 47:230–237. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Senay C, Lind T, Muguruma K, Tone Y,
Kitagawa H, Sugahara K, Lidholt K, Lindahl U and Kusche-Gullberg M:
The EXT1/EXT2 tumor suppressors: Catalytic activities and role in
heparan sulfate biosynthesis. EMBO Rep. 1:282–286. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lonie L, Porter DE, Fraser M, Cole T, Wise
C, Yates L, Wakeling E, Blair E, Morava E, Monaco AP and Ragoussis
J: Determination of the mutation spectrum of the EXT1/EXT2 genes in
British Caucasian patients with multiple osteochondromas, and
exclusion of six candidate genes in EXT negative cases. Hum Mutat.
27:11602006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ciavarella M, Coco M, Baorda F, Stanziale
P, Chetta M, Bisceglia L, Palumbo P, Bengala M, Raiteri P, Silengo
M, et al: 20 novel point mutations and one large deletion in EXT1
and EXT2 genes: Report of diagnostic screening in a large Italian
cohort of patients affected by hereditary multiple exostosis. Gene.
515:339–348. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Delgado MA, Martinez-Domenech G, Sarrión
P, Urreizti R, Zecchini L, Robledo HH, Segura F, de Kremer RD,
Balcells S, Grinberg D and Asteggiano CG: A broad spectrum of
genomic changes in latinamerican patients with EXT1/EXT2-CDG. Sci
Rep. 4:64072014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wuyts W and Van Hul W: Molecular basis of
multiple exostoses: Mutations in the EXT1 and EXT2 genes. Hum
Mutat. 15:220–227. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lind T, Tufaro F, McCormick C, Lindahl U
and Lidholt K: The putative tumor suppressors EXT1 and EXT2 are
glycosyltransferases required for the biosynthesis of heparan
sulfate. J Biol Chem. 273:26265–26268. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
McCormick C, Duncan G, Goutsos KT and
Tufaro F: The putative tumor suppressors EXT1 and EXT2 form a
stable complex that accumulates in the Golgi apparatus and
catalyzes the synthesis of heparan sulfate. Proc Natl Acad Sci USA.
97:668–673. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Esko JD and Lindahl U: Molecular diversity
of heparan sulfate. J Clin Invest. 108:169–173. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zak BM, Crawford BE and Esko JD:
Hereditary multiple exostoses and heparan sulfate polymerization.
Biochim Biophys Acta. 1573:346–355. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Koziel L, Kunath M, Kelly OG and Vortkamp
A: Ext1-dependent heparan sulfate regulates the range of Ihh
signaling during endochondral ossification. Dev Cell. 6:801–813.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jennes I, Pedrini E, Zuntini M, Mordenti
M, Balkassmi S, Asteggiano CG, Casey B, Bakker B, Sangiorgi L and
Wuyts W: Multiple osteochondromas: Mutation update and description
of the multiple osteochondromas mutation database (MOdb). Hum
Mutat. 30:1620–1627. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu L, Xia J, Jiang H, Zhou J, Li H, Wang
D, Pan Q, Long Z, Fan C and Deng HX: Mutation analysis of
hereditary multiple exostoses in the Chinese. Hum Genet. 105:45–50.
1999. View Article : Google Scholar : PubMed/NCBI
|