Introduction
A juxtaglomerular cell tumor (JGCT) is an extremely
rare, benign renal neoplasm of myoendocrine cell origin (1) that was first described by Kihara et
al in 1968 (2). To date, ~100
cases of JGCT have been reported (3–13),
however, few studies have systematically summarized its
characteristic manifestations on computed tomography (CT) and
magnetic resonance imaging (MRI) (4,5). JGCT is
characterized by renin production, hypokalemia and hypertension
(5). Therefore, clinical features
have assistant value for an accurate diagnosis. The diagnosis
relies on pathological identification. The present study reports
the case of a 29-year-old female who underwent a long process for
the confirmation of a JGCT. Written informed consent was obtained
from the patient.
Case report
A 29-year-old female presented to the Outpatient
Clinic of The Central Hospital of Lishui (Lishui, Zhejiang, China)
on April 15, 2013, due to headaches and hypertension.
The patient suffered from recurrent headaches and
reported a two-year history of hypertension. The patient had
previously been prescribed Norvasc, which effectively controlled
the blood pressure. Furthermore, in April 2012, the patient was
admitted to The First Affiliated Hospital of Zhejiang University
(Hangzhou, China) with hypertension and subsequently underwent
contrast-enhanced (CE)-CT, which found a left renal neoplasm.
However, due to a lack of tumor-related symptoms, no further
diagnosis was made and no treatment was provided.
Upon admission to The Central Hospital of Lishui in
April 2013, physical examinations showed no abnormal findings, but
hypokalemia was noted (potassium, 3.22 mmol/l; normal range,
3.5–5.5 mmol/l). The patient's blood pressure was 140/100 mmHg.
Renal function, urinalysis, and other chemical and hematological
profiles showed no abnormalities, with the exception of increased
peripheral plasma renin activity (32 pg/ml/h; normal range, 0.3–2.9
pg/ml/h) and a high aldosterone level (324.65 pg/ml; normal range,
10–160 pg/ml).
Abdominal US revealed a low echo solid mass
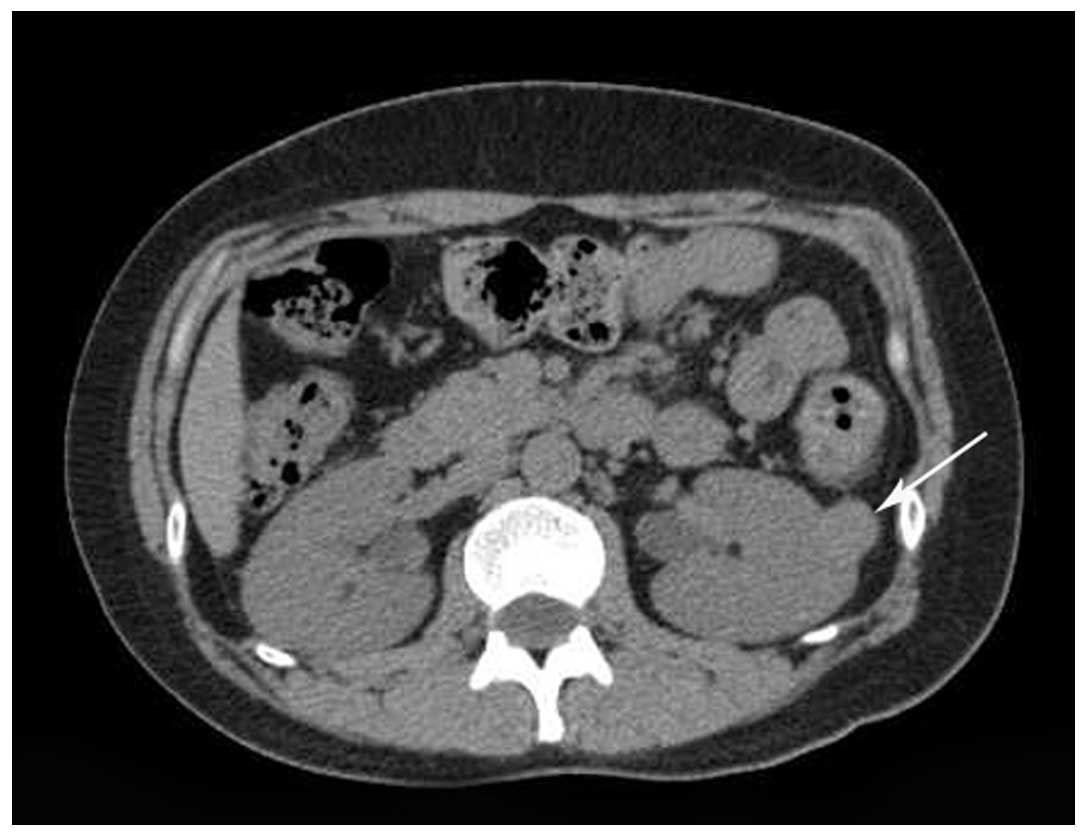
measuring 2.2×1.8 cm in the left kidney. Unenhanced CT revealed a
clearly demarcated isodensity lesion in the upper-middle region of
the left kidney (Fig. 1). CE-CT
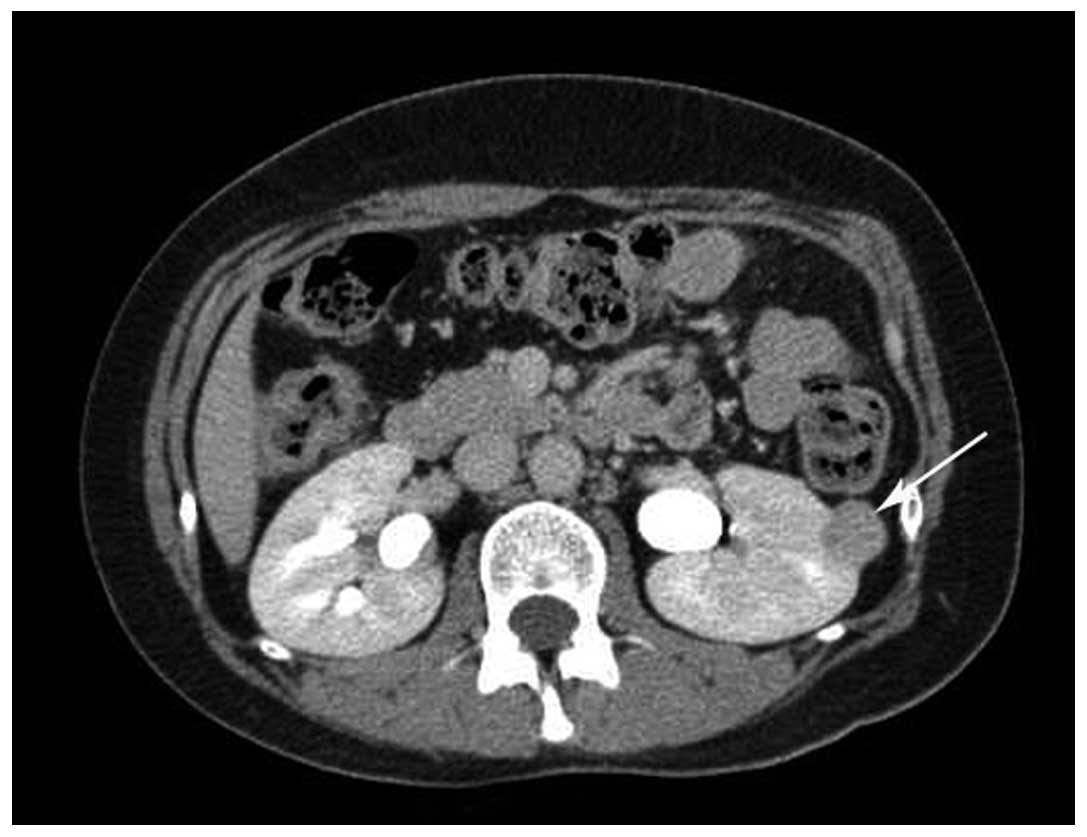
demonstrated that the tumor was not markedly enhanced in the
corticomedullary phase, but that it was further enhanced in the
parenchymal phase and that the density slightly decreased in the
excretory phase (Fig. 2). CT
angiography (CTA) revealed the mass and normal renal vessels. The
patient did not undergo MRI.
A retroperitoneoscopic left nephrectomy was
performed on April 30, 2013. Grossly, a well-demarcated mass of
2.2×1.8×1.5 cm in size was located in the left kidney. Microscopic
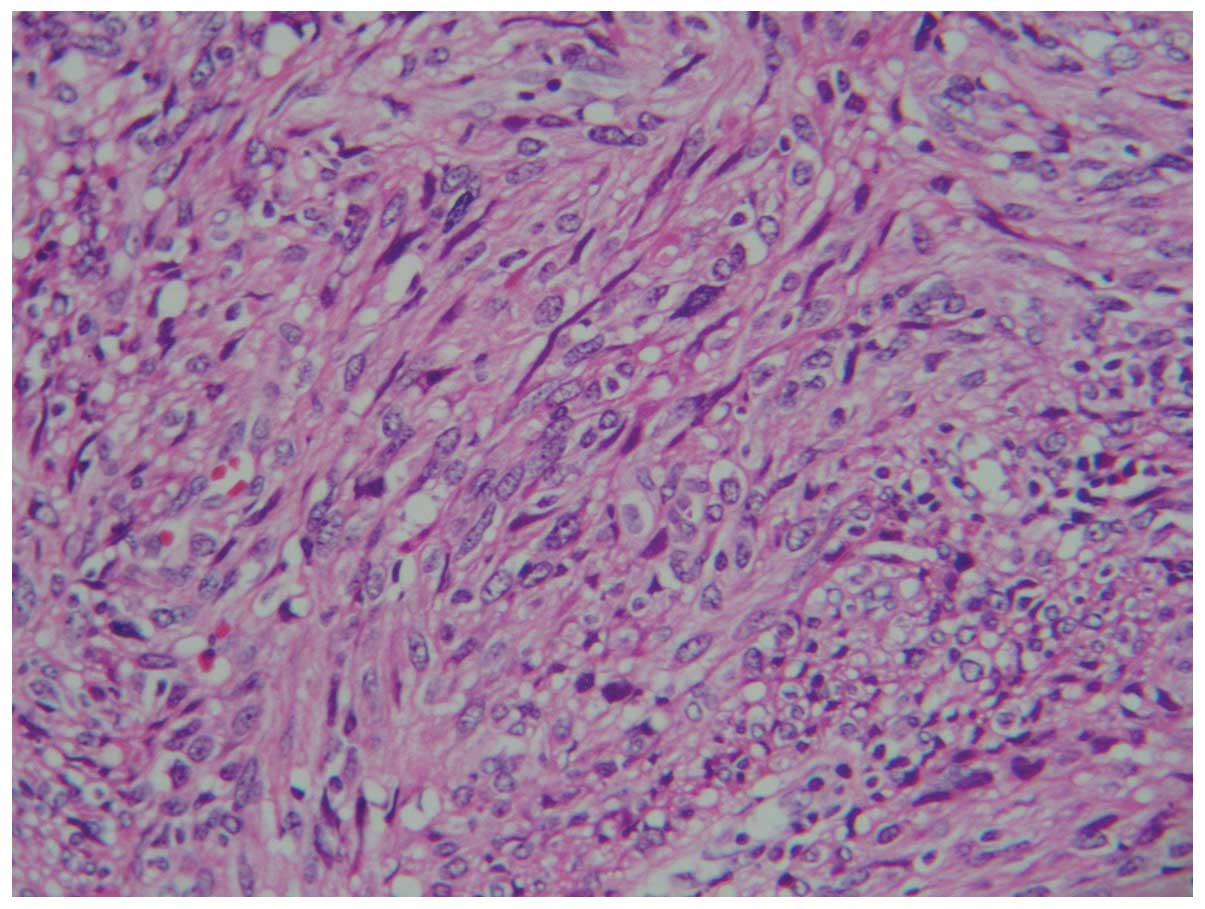
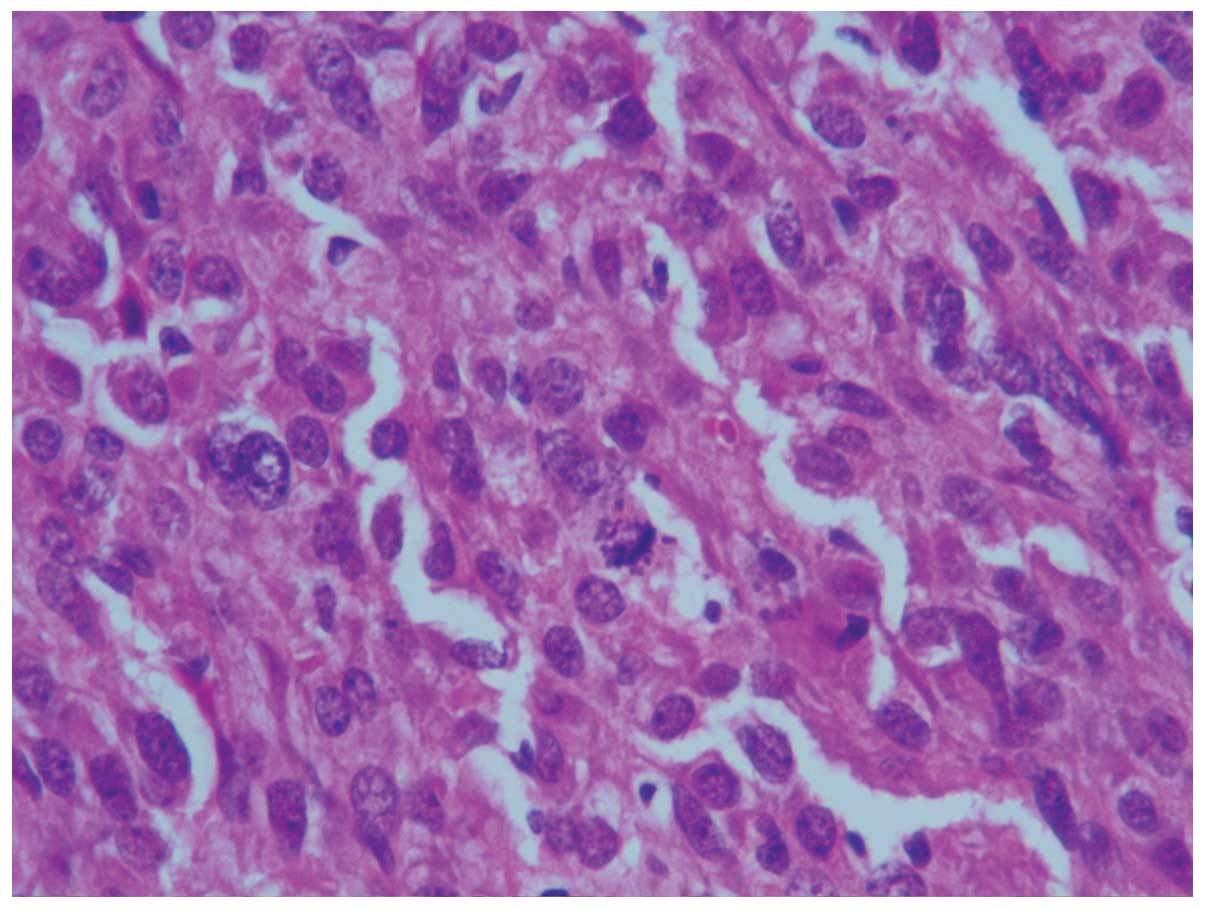
examination revealed that the tumor was composed of spindle cells
(Fig. 3) with well-defined cell
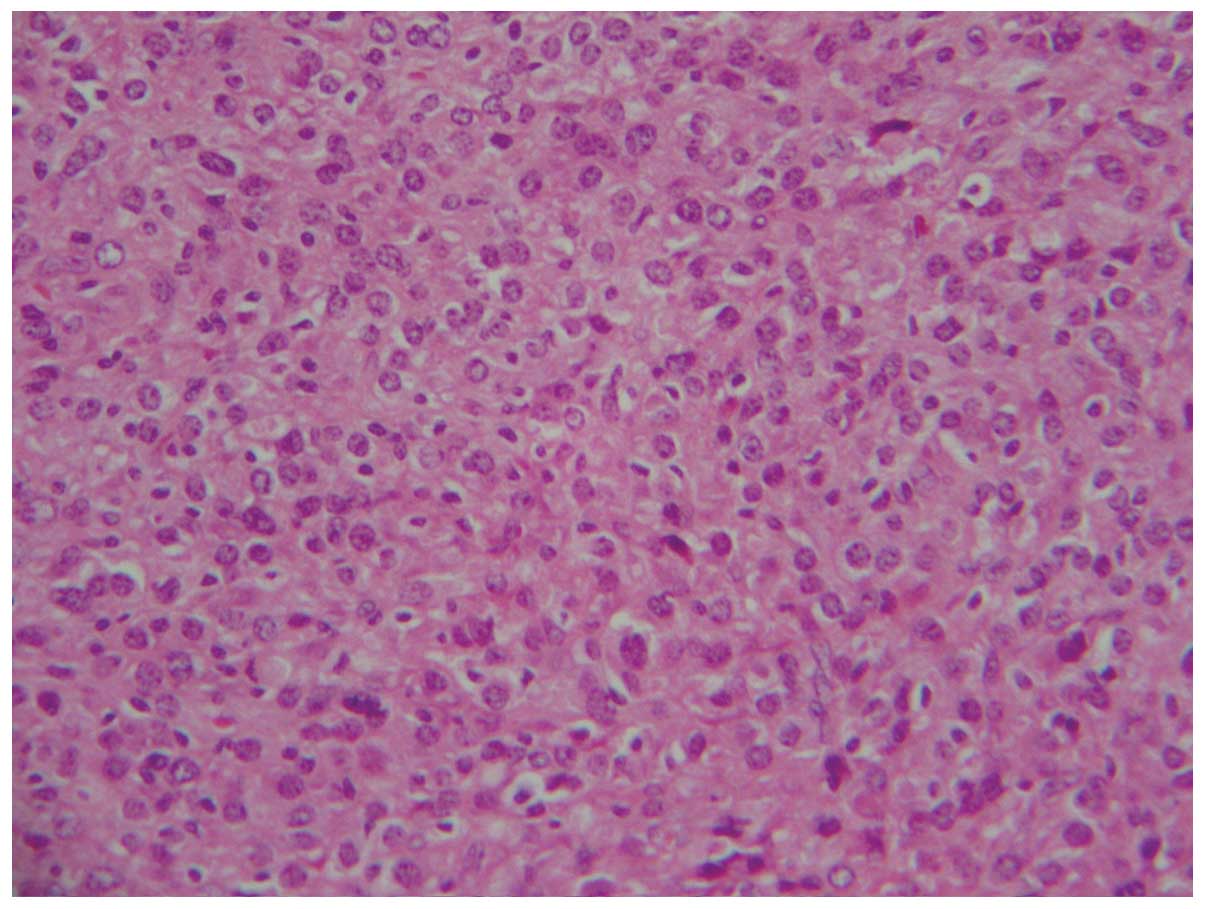
membranes. Polygonal cells (Fig. 4)
with abundant eosinophilic cytoplasm, indistinct cell borders and
nuclear atypia were observed (Fig.
5). Immunohistochemical findings: Vimentin (+), cluster of
differentiation (CD)34 (++), B-cell lymphoma 2 (−/+), CD99 (−/+),
CD117 (−/+), Dog-1 (−), human melanoma black 45 (−), Melan-A (−),
S-100 (−), cytokeratin (−), epithelial membrane antigen (−),
Synaptophysin (Syn; +), CD56 (−), smooth muscle actin (−/+) and
Desmin (−), with a Ki-67 of 6–8%. The diagnosis was of a solitary
fibrous tumor (SFT). Consultation with pathologists from other
hospitals resulted in five pathologists agreeing with this
diagnosis, while three other pathologists suggested a diagnosis of
an undifferentiated sarcoma, sarcomatoid carcinoma or mesoblastic
nephroma. Finally, the diagnosis of JGCT was formed due to the
following factors: Young female patient; hypertension, hypokalemia
and elevated plasma renin activity; histological morphology;
positivity for CD34 and Syn; and post-operative normal blood
pressure and plasma renin activity. The patient was followed up for
2 years and 6 months, with no evidence of tumor recurrence.
Discussion
JGCT mostly affects young adults. The peak age of
incidence is in the second and third decades, with a 2:1 female
preponderance (5). Although a JGCT is
generally considered to be a benign tumor, one metastatic tumor has
been reported (6). Therefore, an
early diagnosis and surgery is necessary. In the present study, a
case of a JGCT is reported and the significance of clinical
characteristics for the pathological diagnosis of a JGCT is
discussed.
The majority of JGCT patients exhibit a clinically
typical presentation, including hypertension, hyperreninemia,
hyperaldosteronism and hypokalemia (5). In the present study, hypertension was
the first and most prominent manifestation, and the main reason for
attendance at hospital, which is consistent with previously
reported cases (7). Although one case
of non-functioning JGCT has also been reported (8), the clinical features are still important
for a definite diagnosis. No correlation has been found between the
severity of symptoms and the size of the tumor (7).
Thus far, the US features of JGCT have not been
reported. The majority of JGCTs reported in the literature are low-
or isodensity, well-circumscribed, cortical tumors on plain CT
scanning (7,9), which is consistent with the findings of
the current study. Despite being hypervascular, the tumor appears
hypovascular on CE-CT and CE-MRI, possibly due to renin-induced
vasoconstriction (4). None of the
reported JGCTs were stained during the corticomedullary phase, but
all were stained moderately during the late phase following
contrast enhancement. The imaging manifestations of JGCT are
non-specific and indistinguishable from those of other solid renal
neoplasms (5).
Histologically, JGCT consists of sheets of polygonal
or spindle-shaped cells and a hemangiopericytic angioarchitecture
(3). It is occasionally difficult to
differentiate a JGCT from an SFT or MN (3,10,11). Each of these three tumors is composed
of spindle cells, and has a similar morphology and CD34 expression
pattern. However, MNs are commonly found in infants aged ≤6 months
(11), and the patient in the present
case was 29 years old. The majority of SFT patients have no
clinical symptoms (10). Ultimately,
by combining clinical symptoms, medical history and laboratory
examination results, the definitive diagnosis of JGCT was
established.
In conclusion, JGCT is a rare benign renal neoplasm.
Only with sufficient expertise and integration with clinical
characteristics can pathologists obtain evidence of a JGCT.
Acknowledgements
The authors would like to thank the associated staff
from the First Affiliated Hospital of Zhejiang University
(Hangzhou, Zhejiang, China), the Zhejiang Provincial People's
Hospital (Hangzhou, Zhejiang, China), the Indiana University School
of Medicine (Indianapolis, IN, USA) and the University of
Massachusetts Medical School (Worcester, MA, USA) for their
assistance with the pathological diagnosis.
References
|
1
|
Martin SA, Mynderse LA, Lager DJ and
Cheville JC: Juxtaglomerular cell tumor, A clinicopathologic study
of four cases and review of the literature. Am J Clin Pathol.
116:854–863. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kihara I, Kitamura S, Hoshino T, Seida H
and Watanabe T: A hitherto unreported vascular tumor of the kidney,
A proposal of ‘juxtaglomerular cell tumor’. Acta Pathol Jpn.
18:197–206. 1968.PubMed/NCBI
|
|
3
|
Kuroda N, Maris S, Monzon FA, Tan PH,
Thomas A, Petersson FB, Gatalica Z, Ghazalpour A, Bender RP,
Grossmann P, et al: Juxtaglomerular cell tumor: A morphological,
immunohistochemical and genetic study of six cases. Hum Pathol.
44:47–54. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tanabe A, Naruse M, Ogawa T, Ito F, Takagi
S, Takano K, Ohashi H, Tsuchiya K, Sone M, Nihei H and Toma H:
Dynamic computer tomography is useful in the differential diagnosis
of juxtaglomerular cell tumor and renal cell carcinoma. Hypertens
Res. 24:331–336. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Prasad SR, Surabhi VR, Menias CO, Raut AA
and Chintapalli KN: Benign renal neoplasms in adults,
Cross-sectional imaging findings. AJR Am J Roentgenol. 190:158–164.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Duan X, Bruneval P, Hammadeh R, Fresco R,
Eble JN, Clark JI, Vigneswaran WT, Flanigan RC and Picken MM:
Metastatic juxtaglomerular cell tumor in a 52-year-old man. Am J
Surg Pathol. 28:1098–1102. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qiang J, Gao W, Chen D, Nie Z, Guan W, Li
Y and Yu W: CT manifestations of renal juxtaglomerular cell tumor.
Zhong Hua Fang She Xue Za Zhi. 44:885–886. 2010.(In Chinese).
|
|
8
|
Sakata R, Shimoyamada H, Yanagisawa M,
Murakami T, Makiyama K, Nakaigawa N, Inayama Y, Ohashi K, Nagashima
Y, Yao M and Kubota Y: Nonfunctioning juxtaglomerular cell tumor.
Case Rep Pathol. 2013:9738652013.PubMed/NCBI
|
|
9
|
Prasad SR, Narra VR, Shah R, Humphrey PA,
Jagirdar J, Catena JR, Dalrymple NC and Siegel CL: Segmental
disorders of the nephron, Histopathological and imaging
perspective. Br J Radiol. 80:593–602. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang Z, Li K, Dong K, Liu G, Xiao X and
Zheng S: Eight cases of congenital mesoblastic nephroma, Clinical
analysis and literature review. Zhong Hua Xiao Er Wai Ke Za Zhi.
34:754–756. 2013.(In Chinese).
|
|
11
|
Lingying ZH, Enyu W, Suming CH and Linghui
X: Solitary fibrous tumor of the kidney with duodenal stromal
tumor. A case report. Zhong Hua Fang She Xue Za Zhi. 41:7782007.(In
Chinese).
|
|
12
|
Elouazzani H, Jahid A, Bernoussi Z and
Mahassini N: Juxtaglomerular cell tumor, A distinct mesenchymal
tumor of kidney. J Clin Imaging Sci. 4:332014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ren GP, Yu XR, Li YX, Wang LJ, Wang JQ,
Shi HQ and Ye HH: Juxtaglomerular cell tumor of the kidney, A
clinicopathologic analysis of five cases. Zhonghua Bing Li Xue Za
Zhi. 32:511–515. 2003.PubMed/NCBI
|