1. Introduction
Breast cancer is the most common type of cancer in
females worldwide. Statistics show that during the period between
2000 and 2005, the incidence of breast cancer in Europe was
approximately 370,000 cases per year. This type of cancer is the
most common cause of cancer-related death in women (1). Apart from the mortality caused by
tumor degradation in situ, the majority of fatal cases are
caused by local, regional or distant (bone, liver, lung, kidney,
thyroid and brain) metastasis from the primary tumor site (breast
tissues), which spreads through the bloodstream or lymphatic
channels (2).
Tumorigenesis is a multistep process that transforms
normal cells into the malignant phenotype, is always accompanied by
genetic alterations. It frequently leads to the dysregulation of
numerous signal transduction pathways, which are usually involved
in cell cycle progression, cell death and survival, angiogenesis
and apoptosis. Moreover, these pathways may present rational
targets, providing opportunities for the targeted therapeutics of
cancer (3). Advances in molecular
technology have led to a better understanding of the mechanisms
involved in the development and progression of breast cancer. Over
the past few decades, an increasing body of knowledge regarding
specific genes and proteins, such as HER2 and VEGF/VEGFR, as well
as biological molecular pathways associated with the development
and progression of breast cancer, has allowed for the development
of targeted therapeutics that ideally aim at the selective,
efficient and safe treatment of cancer. These developments have
greatly improved the survival of breast cancer patients, while both
chemotherapy and hormone therapy have also significantly prolonged
the survival of breast cancer patients (3,4).
Following its initial cloning in 2002 (5), astrocyte elevated gene-1 (AEG-1) has
been found to be overexpressed in multiple carcinomas, including
melanoma, glioma and neuroblastoma, as well as carcinomas of the
breast, prostate, liver and esophagus (6). In addition, AEG-1 has been found to
play various roles in the regulation of cancer development,
progression and metastasis.
Recent studies have shown that in normal human
breast tissues, the expression of AEG-1 is decreased or is
completely absent, while it is widely overexpressed in a number of
breast cancer cell lines and breast tumors (7–10).
Current investigations have revealed that AEG-1 plays a crucial
role in the regulation of specific development and progression of
breast cancer, indicating its potential as a specific new target in
clinical-targeted therapeutics of breast cancer.
2. Cloning and molecular structure of
AEG-1
Following human immunodeficiency virus (HIV)-1
infection or treatment with HIV-1 envelope glycoprotein (gp120) of
primary human fetal astrocytes (PHFA), AEG-1 was found to be
overexpressed by the rapid subtraction hybridization (RaSH) method
(5). In 2003, a customized
microarray approach used to detect the expression of AEG-1 in PHFA
treated with tumor necrosis factor (TNF)-α showed a concomitant
change similar to that following HIV-1 infection (11).
The AEG-1 gene consists of 12 exons and 11 introns
and is located at 8q22 via genomic blast search. Excluding the
poly-A tail, the full-length AEG-1 cDNA consists of 3,611 bp
(8). The open reading frame (ORF)
from 220 to 1,968 nt of human AEG-1 encodes a 582-amino acid
protein with a calculated molecular mass of 64 kDa and a pI of
9.33. Protein motif analysis of the AEG-1 protein and three
independent transmembrane protein prediction methods (PSORT II,
TMpred and HMMTOP) predicted that the AEG-1 protein contains a
single-transmembrane domain. Notably, PSORT II and TMpred predicted
AEG-1 as a type Ib protein (C terminal in the cytoplasmic side with
no signal peptide), whereas TMHMM and TopPred 2 obtained a reverse
localization of the C terminal, indicating that it was a type II
protein. Although the analysis of the amino acid sequence of AEG-1
revealed that it does not have any recognizable protein domains,
the presence of putative, either monopartite or bipartite, nuclear
localization signals (NLS) between amino acids 432–451 and 561–580
was confirmed, suggesting that it may enter into the nucleus to
play a significant role (12). In
another study (13), it was shown
that AEG-1 contains three NLS which may regulate its distribution
and function in cells. The final results obtained suggest that
extended NLS-1 (amino acids 78–130) regulates its nucleolar
localization, and the extended NLS-2 region (amino acids 415–486)
mediates its modification via ubiquitination almost exclusively
within the cytoplasm, whereas the COOH-terminal extended NLS-3
(amino acids 546–582) is the predominant regulator of nuclear
localization (13). Moreover, AEG-1
was found to be rich in both lysine residues, which are targets for
post-translational modification by ubiquitination, and serine
residues, required for the redistribution of AEG-1 within the cell
(13).
3. Intracellular localization of AEG-1
Intracellular localization of AEG-1 has been
examined in various cell types, using a polyclonal anti-AEG-1
antibody and immunofluorescence microscopy. In immortalized PHFA
(IM-PHFA), endogenous AEG-1 was stained. The image showed that
AEG-1 was located in the perinuclear region and in endoplasmic
reticulum (ER)-like structures, but not in the plasma membrane. It
was colocalized with the ER-specific protein calreticulin, but not
with the mitochondrial marker MitoTracker (8). Concomitantly, the observed ER
localization of AEG-1 indicates that it is a type Ib membrane
protein (8). However, following
TNF-α treatment or forced overexpression of AEG-1, localization of
the protein was detected both in the cytoplasm and nucleus in HeLa
cells (12). This phenomenon is
similar to that of the nuclear receptor coactivator SRC-3
(pCIP/ACTR/AIB-1/RAC-3/TRAM-1), which is located mainly in the
cytoplasm and translocates from the cytoplasm to the nucleus in
response to TNF-α (14), indicating
a potential direct or indirect interaction between AEG-1 and SRC-3
or other unknown factors. Although various studies (8,12) have
been performed to investigate the intracellular localization of
AEG-1, the specific localization and the relationship between its
localization and function require further clarification.
4. Molecular mechanism of AEG-1 action
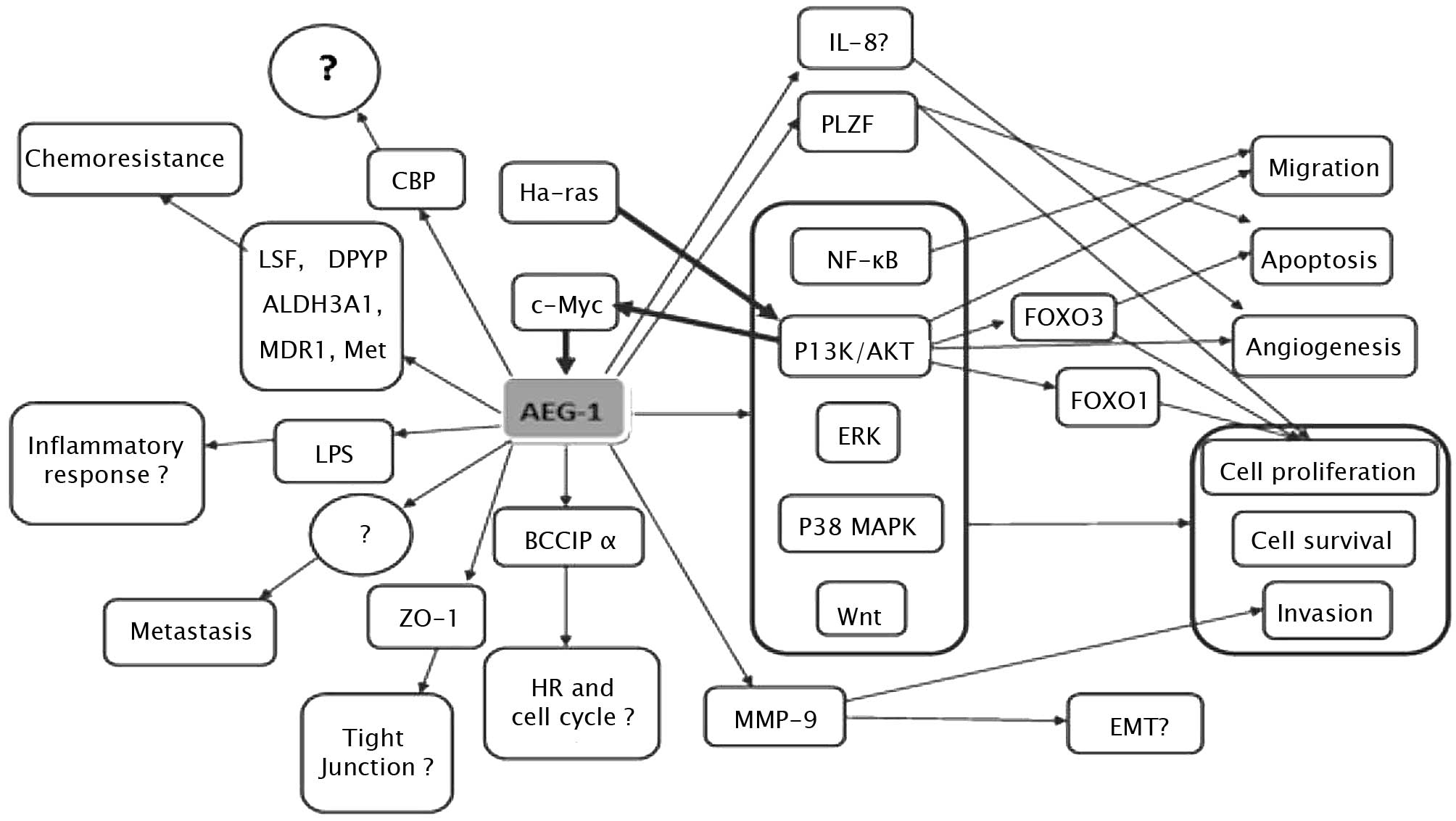
AEG-1 contributes to several hallmarks of metastatic
cancers, including proliferation, evasion of apoptosis and cell
survival under stressful conditions, such as serum deprivation and
chemotherapy. AEG-1 also enhances migration, invasion, angiogenesis
and metastasis by modulating various signaling pathways (Fig. 1). It is well established that the
Ha-ras and c-Myc genes cooperate to promote
transformation, tumor development and progression, and metastasis.
However, the precise mechanism underlying this cooperation remains
to be determined. The overexpression of AEG-1 and oncogenic
Ha-Ras has been found to collaborate to enhance the soft
agar colony formation of immortalized melanocytes and astrocytes
(8). It was observed that human
AEG-1 can be induced at the transcription level by oncogenic
Ras in human fetal astrocytes, whereas AEG-1 knockdown
suppressed Ras-induced colony formation (15). Moreover, when inhibitors for various
Ras downstream signaling pathways were examined, only PI3K/AKT
inhibitors LY294002 and PTEN were able to block AEG-1
promoter activation by Ras, suggesting the involvement of the
PI3K/AKT pathway in Ha-ras-induced AEG-1 regulation (15). To confirm that the PI3K/AKT
signaling pathway is involved in oncogenic Ha-ras-induced AEG-1
expression, the phosphorylation of AKT and GSK3β was analyzed by
Western blotting, indicating a parallel overexpression of AKT and
GSK3β. Promoter mapping subsequently identified the region −356 to
−302 of the AEG-1 promoter containing two E-box elements that bind
to Ha-ras-activated transcription factors, such as c-Myc.
All of the above data indicate that Ha-ras increases
binding of c-Myc to the E-box elements in the AEG-1 promoter
through the PI3K/AKT/GSK3β/c-Myc pathway to act in Ha-ras-mediated
oncogenesis through AEG-1 (15).
However, inhibition of the MEK pathway was found to slightly
increase Ha-ras-mediated AEG-1 promoter activation, the
significance of which remains to be determined (15). Notably, transcription of AEG-1 is
induced by c-Myc, whereas AEG-1 induces c-Myc expression (in
neuroblastoma cells it induces N-Myc expression), thereby
amplifying the tumorigenic effect (16). A recent study showed that one
mechanism by which cells with altered AEG-1 expression evade
apoptosis and increase cell growth during tumorigenesis involves
the repression of the function of promyelocytic leukemia zinc
finger (PLZF) (17). The
transcriptional repressor PLZF, which has been found to regulate
the expression of genes involved in cell growth and apoptosis,
including c-Myc, was found to interact with AEG-1 through a
yeast two-hybrid screening system. It was also noted that AEG-1
interacts with PLZF, reducing PLZF binding to promoters, thereyb
reducing PLZF-mediated repression (17).
AEG-1 activates several downstream signal
transduction pathways, including the nuclear factor κB (NF-κB),
PI3K/AKT, Wnt/β-catenin and MAPK pathways to enhance various
aspects of tumor development and progression. The first signaling
pathway identified as being activated by AEG-1 was NF-κB (12). In HeLa cells, following infection
with Ad.AEG-1 or treatment with TNF-α, AEG-1 was found to
translocate into the nucleus where it interacted with the p65
subunit of NF-κB and enhanced NF-κB-induced gene expression. AEG-1
activates NF-κB via IκBα degradation and p65 translocation
(12). A gene array analysis
revealed that Ad.AEG-1 infection resulted in a marked up-regulation
of NF-κB-responsive cell adhesion molecules (ICAM-2 and ICAM-3,
selectin E, selectin L and selectin P ligand), TLR4 and TLR5, and
cytokines, such as IL-8 (12).
Following AEG-1 overexpression or TNF-α treatment, AEG-1 was found
to interact with p65 and cyclic AMP-responsive element binding
protein-binding protein (CBP) on the IL-8 promoter, increasing IL-8
transcription, suggesting that AEG-1 acts as a bridging factor
among NF-κB, CBP and the basal transcription mechanisms (18). AEG-1-induced increase of soft agar
growth and matrigel invasion may be restrained by the inhibition of
NF-κB in HeLa cells (12).
The present review elucidates the domains of AEG-1
that are crucial for mediating its role. Functional analysis
revealed that the functions of AEG-1 in invasion, migration and
NF-κB-activating properties are mediated by NH2-terminal
71 amino acids, whereas amino acids 101 to 205 were identified as a
p65-interaction domain, indicating that p65 interaction alone is
not sufficient to mediate AEG-1 function (18). NF-κB activation by AEG-1 has also
recently been documented in prostate and liver cancer cells
(19,20). Notably, AEG-1 was found to be
induced via NF-κB activation in LPS-stimulated U937 human
promonocytic cells, and AEG-1 induced by LPS subsequently regulated
NF-κB activation in turn. Moreover, LPS-induced TNF-α and
prostaglandin E2 production were inhibited by preventing the
expression of AEG-1. Therefore, we hypothesize that AEG-1 is a
LPS-responsive gene and is involved in LPS-induced inflammatory
response (21).
PI3K/AKT is another pathway involved in the
tumorigenesis mediated by AEG-1. This pathway is not only activated
by AEG-1, but also regulates the expression of AEG-1 (15). PI3K/AKT signals modulates various
growth-regulatory transcription factors, such as the forkhead box
(FOXO) protein and NF-κB. AEG-1 knockdown was found to induce cell
apoptosis through the up-regulation of FOXO3a activity in prostate
cancer cells, and subsequently revealed that AEG-1 expression plays
a dominant role as a positive auto-feedback activator of AKT and as
a suppressor of FOXO3a to promote PC progression (20). Moreover, it was noted that AEG-1
knockdown down-regulated the constitutive activity of NF-κB and the
activator protein 1 (AP-1), while the mRNA and protein expression
levels of NF-κB and AP-1-regulated genes IL-6, IL-8 and matrix
metalloproteinase-9 (MMP-9) were significantly decreased. The
invasive properties of PC-3 and DU145 cells were also found to be
decreased (20).
AEG-1 provides protection from serum
starvation-induced apoptosis of normal cells by activating the
PI3K/AKT pathway (22). In order to
clarify the molecular mechanisms underlying AEG-1-dependent
enhanced cell growth under conditions of serum starvation, the
downstream antiapoptotic substrates of AKT were examined, revealing
that the phosphorylation-induced inactivation of GSK3β increased
c-Myc levels, inhibited serum starvation-dependent p21/mda-6
expression and phosphorylation of Bad, a proapoptotic member of the
Bcl-2 family, in Ad.AEG-1-infected PHFA (22). In less aggressive neuroblastoma
cells, the overexpression of AEG-1 enhanced proliferation and
expression of the transformed state by activating the PI3KAKT
signaling pathway (16). In
esophageal cancer cells, the up-regulation of AEG-1 reduced the
expression of p27 and induced the expression of cyclin D1 through
the AKT/FOXO3a pathway (23).
The PI3K/AKT pathway regulates AEG-1-induced
angiogenesis (24). An
immunohistochemical analysis revealed that tube formation induced
by enhanced AEG-1 expression correlates with an increased
expression of specific angiogenesis molecules, including
angiopoietin-1 (Ang1), MMP-2 and hypoxia-inducible factor 1-α
(HIF1-α) in tumor sections. To analyze the potential importance of
the PI3K/AKT signaling pathway in AEG-1-mediated angiogenesis,
Ad.DN.AKT was constructed. The result showed that AEG-1-mediated
endothelial cell tube formation was significantly inhibited by
Ad.DN.AKT, suggesting that the PI3K/AKT pathway is an integral
component of this process (24).
AEG-1 has also been associated with the
Wnt/β-catenin pathway in hepatocellular carcinoma (HCC) cells
through the activation of the Raf/MEK/MAPK branch of the Ras
signaling pathway (19). In HCC,
AEG-1 was found to activate Wnt/β-catenin signaling by activating
ERK42/44 and up-regulating lymphoid-enhancing factor 1/T-cell
factor 1 (LEF1/TCF1), the ultimate executor of the Wnt pathway
(19). Moreover, specific
inhibitors of the MAPK pathway are able to abolish the oncogenic
effect of AEG-1 on Matrigel invasion and anchorage-independent
growth, indicating that AEG-1 also plays an oncogenic role in tumor
development and progression through activation of the MAPK signal
pathway (19).
5. AEG-1 and proliferation of breast
cancer
Uncontrolled cell growth is largely associated with
alterations in genes or proteins related to the regulation of
proliferation, cell death, apoptosis and genetic stability, such as
tumor suppressor genes, oncogenes, growth factors and cell adhesion
molecules, which vary among different cancer types (25). Findings of a recent study showed
that AEG-1 promotes proliferation of breast cancer cells by
down-regulating the transcriptional activity of FOXO1 by inducing
its phosphorylation through the PI3K/AKT signaling pathway
(26). Up-regulation of AEG-1 was
found to markedly promote proliferation (detected by MTT) and
tumorigenicity (tested by anchorage-independent growth) in MCF-7
and MDA-MB-435 breast cancer cells, whereas an AEG-1-knockdown cell
model with shRNAs inhibited cell proliferation and the
colony-forming ability of cells in soft agar. Furthermore, since
these proliferative effects were significantly associated with
decreases in p27Kip1 and p21Cip1, two key
cell cycle inhibitors, overexpression of AEG-1 in breast cancer
cells may significantly impact cell cycle checkpoints, thereby
promoting the proliferation of breast cancer cells (26). In addition, BCCIP α expression was
reduced in human breast tissue and it was found that when the BCCIP
α protein was re-introduced into MCF7 breast cancer cells, the
growth of breast cancer cells was inhibited (27). Studies have shown the multifaceted
roles played by BCCIP α. It binds to p21 and enhances the
p21-mediated inhibition of Cdk2 kinase, whereas loss of BCCIP
impairs G1/S checkpoint activation following DNA damage. BCCIP also
plays a role in homologous recombination repair of DNA damage by
interacting with BRCA2 and contributes to the maintenance of
chromosome stability (27–30). Recently, it was found that AEG-1
promotes proteasomal degradation of tumor suppressor BCCIP α
(29). Therefore, whether AEG-1
promotes proliferation of breast cancer through the regulation of
the expression of BCCIP α should be confirmed. Moreover, AEG-1 was
found to promote the proliferation of various types of cancer cells
by activating the PI3K/AKT and NF-κB signaling pathways. Thus,
apart from the PI3K/AKT signaling pathway, whether the ectopic
expression of AEG-1 enhances the proliferation and
anchorage-independent growth of breast cancer cells by activating
other prosurvival signaling pathways, such as NF-κB, remains to be
determined.
6. AEG-1 and the invasion and migration of
breast cancer
Overexpression of AEG-1 significantly enhances the
invasive ability of cells as revealed by Matrigel invasion assay in
HeLa cells, while inhibition of AEG-1-induced NF-κB activation
blocks invasion (12).
Overexpression of AEG-1 increases, whereas siRNA inhibition of
AEG-1 decreases the migration and invasion of human glioma cells,
respectively (31). AEG-1 was also
found to induce invasion in normal immortal cloned rat embryo
fibroblast (CREF) cells (24).
During the early progression of metastasis, a number of signal
transduction pathways are activated, including PI3K/AKT and NF-κB.
The two signaling pathways have been found to be linked to the
promotion of cancer cell invasion and migration through the
down-regulation of cell-cell contact protein E-cadherin and the
up-regulation of MMP-2 and MMP-9 (32–34).
It is known that AEG-1 activates the PI3K/AKT and NF-κB signaling
pathways, and AEG-1 has been found to up-regulate MMP-9 and induce
human glioma cell invasion (35).
Although experiments investigating the invasion and migration of
breast cancer have yet to be conducted, it is thought that AEG-1
promotes the invasion and migration of breast cancer by activating
the PI3K/AKT and/or NF-κB signaling pathways, or other signaling
pathways, which requires confirmation.
7. AEG-1 and metastasis of breast
cancer
Tumor metastasis is a complex multistep process in
which cancer cells detach from the primary tumor tissue and
establish metastatic foci at specific sites. Brown and Ruoslahti
(9)used a phage expression library
of cDNAs from metastatic breast carcinoma to identify protein
domains that bind to lung vasculature. These authors found an
extracellular lung-homing domain (LHD; amino acids 378–440 in mouse
or 381–443 in human) in AEG-1 to be a mediator of 4T1 mouse mammary
tumor cell adhesion to lung vasculature (9). In addition, antibodies to AEG-1 showed
a high expression of AEG-1 throughout human breast tumors and
breast tumor xenografts, while markedly lower levels of AEG-1 were
present in normal breast tissue (9). Moreover, antibodies reactive to the
lung-homing domain of AEG-1 and siRNA-mediated knockdown of AEG-1
expression inhibited experimental breast cancer lung metastasis.
Conversely, enhanced localization to lung vasculature was noted in
HEK293T cells transiently transfected with AEG-1. These results
suggest that AEG-1 plays an important role in breast cancer
metastasis (9). To further clarify
the mechanism of metastasis of breast cancer, human breast cancer
cell line MDA-MB-231 was used to investigate the roles of a number
of genes in metastasis, including UQCRB, PTDSS1, TSPYL5, AEG-1,
LAPTM4β and SDC2 (10). The data
showed that AEG-1 overexpression significantly accelerated the
development of lung metastasis and led to a modest, but
significant, increase in bone and brain metastases (10). These results suggest that AEG-1
preferentially promotes metastasis to the lung, but has a modest
impact on metastasis to other organs. As mentioned above, the
PI3K/AKT and NF-κB signaling pathways are involved in the early
progression of metastasis. AEG-1 may promote metastasis through the
interaction of the LHD, with an unknown receptor expressed in the
surface of endothelial cells, or indirectly through the activation
of signaling pathways, such as PI3K/AKT and NF-κB, which involve
adhesion molecules, such as MMPs.
8. AEG-1 and angiogenesis of breast
cancer
Hanahan and Weinberg proposed that six essential
‘hallmarks’ or processes are required for the transformation of a
normal cell to a cancer cell. These processes include: i)
self-sufficiency in growth signals; ii) insensitivity to antigrowth
signals; iii) evasion of programmed death (apoptosis); iv) endless
replication potential; v) sustained angiogenesis; and vi) tissue
invasion and metastasis (36).
Overexpression of AEG-1 increases the expression of molecular
markers of angiogenesis, including Ang1, MMP-2 and HIF1-α (24). In vitro angiogenesis studies
further demonstrated that AEG-1 promotes tube formation in Matrigel
and increases invasion of human umbilical vein endothelial cells
via the PI3K/AKT signaling pathway (24). The angiogenesis-promoting function
of AEG-1 was also noted in human HCC (19). Numerous genes play important roles
in angiogenesis, apart from VEGF and VEGFR, which are the most
representative. IL-8 and COX2 also promote tumor angiogenesis, but
are not involved in the VEGF pathway (37–39).
Many genes are involved in the NF-κB signaling pathway, such as
IL-8 and COX2. IL-8 and COX2 are overexpressed in breast cancer
(40,41), and the expression of IL-8 can be
regulated by AEG-1 (12),
indicating that AEG-1 may also promote the angiogenesis of breast
cancer by activating the PI3K/AKT signaling pathway as well as the
NF-κB or VEGF signaling pathway. Experiments in vivo and
in vitro should be performed to clarify the mechanism.
9. AEG-1 and chemoresistance of breast
cancer
AEG-1 plays a role in chemoresistance in human HCC,
neuroblastoma cell lines and breast cancer (10,19,40).
An Affymetrix oligonucleotide microarray was performed in human HCC
to analyze the downstream genes of AEG-1. The results of the
microarray showed a cluster of genes associated with
chemoresistance, including drug-metabolizing enzymes for various
chemotherapeutic agents, such as dihydropyrimidine dehydrogenase
(DPYD), principal enzyme-inactivating 5-fluorouracil (5-FU)
cytochrome P4502B6 (CYP2B6), dihydrodiol dehydrogenase (AKR1C2) and
the ATP-binding cassette transporter ABCC11 for drug efflux
(19). AEG-1 was also found to
enhance the expression of the transcription factor LSF, which
regulates the expression of thymidylate synthase, a target of 5-FU.
In addition, AEG-1 enhanced the expression of dihydropyrimidine
dehydrogenase (DPYD), which catalyzes the initial and rate-limiting
step in the catabolism of 5-FU (42). A recent study reported a new
mechanism through which AEG-1 plays a role in chemoresistance in
human HCC (43). AEG-1 increases
the expression of multidrug resistance gene 1 (MDR1) protein,
resulting in increased efflux and decreased accumulation of
doxorubicin, thereby promoting doxorubicin resistance (43). AEG-1 increases the expression of
MDR1 by facilitating the association of MDR1 mRNA to polysomes,
resulting in increased translation. AEG-1 also inhibits
ubiquitination and subsequent proteasome-mediated degradation of
the MDR1 protein (43). In
neuroblastoma cells, a significant enhancement in chemosensitivity
to cisplatin and doxorubicin by knockdown of AEG-1 was observed
(40). In breast cancer, in
vitro and in vivo analyses of chemoresistance revealed
that AEG-1 knockdown sensitized various breast cancer cell lines
(LM2, MDA-MB-231, MCF7 and T47D) to paclitaxel, doxorubicin,
cisplatin, 4-hydroxycylcophosphamide and hydrogen peroxide
(10). AEG-1 did not affect the
uptake or retention of chemotherapy drugs in breast cancer tissue
(10). Instead, AEG-1 may increase
chemoresistance by promoting cellular survival against
antineoplastic stresses (10).
Since we confirmed that AEG-1 activates the PI3K/AKT
and NF-κB signaling pathways, chemoresistance may be mediated by
these prosurvival pathways through the interaction between AEG-1
and molecules involved in these pathways. Microarray analysis of
AEG-1-knockdown breast cancer cells revealed that two
AEG-1-down-regulated genes (TRAIL and BINP3, two cell
death-inducing genes) and a number of AEG-1-up-regulated genes
(ALDH3A1, MET, HSP90 and HMOX1) are
involved in chemoresistance (10).
Among these candidate AEG-1 downstream genes, ALDH3A1
(aldehyde dehydrogenase 3 family, member A1) and MET
(hepatocyte growth factor receptor) collectively contribute to its
role in broad-spectrum chemoresistance (10). Whether AEG-1 regulates MDR1
expression in breast cancer and whether AEG-1 increases
chemoresistance by promoting cellular survival or regulating
downstream genes that directly modulate chemoresistance require
elucidation. Moreover, the specific mechanism of chemoresistance of
AEG-1 in breast cancers remains to be clarified.
10. AEG-1 and prognosis of breast
cancer
Two independent research groups analyzed AEG-1
expression and clinical associations, respectively, using breast
tumor samples and obtained similar results (7,10).
AEG-1 was found to be abundantly expressed in breast tumors with a
percentage of 47 and 44.4%, respectively, in these two groups, and
was significantly correlated with clinical staging, tumor size,
lymph node spread, distant metastasis and poor survival (7,10).
Overexpression of AEG-1 was not linked to any specific breast tumor
subtype in terms of HER2 status, triple marker status (ER/PR/HER2),
or basal epithelial cell marker CK5/6 status (10). Findings of the multivariate analysis
suggested that AEG-1 expression may be an independent prognostic
indicator independent of other clinicopathological factors for the
survival of patients with breast cancer (7,10).
Further evidence points to the fact that AEG-1 is located at
chromosome 8q22, a region frequently amplified in many cancer types
and associated with poor prognosis (44–47).
These findings suggest the clinical and prognostic significance of
AEG-1 in breast cancer.
11. Conclusion and future perspectives
Although in vitro and in vivo studies
have confirmed that AEG-1 plays a key role in the process of cancer
development and progression in multiple organs, including breast,
further investigations should be conducted to clarify the specific
mechanisms involved in the mediation of AEG-1 functions. Moreover,
additional unknown functions of AEG-1 should be further studied
(48).
Since we know that a correlation exists between
AEG-1 and tumor progression, whether AEG-1 regulates other
functions related to tumor progression, such as immortalization and
transformation, as suggested by numerous studies in other types of
cancers, and whether using immortal normal cells in breast cancer
is viable should be investigated. For example, local invasion is
considered to be a crucial first step in metastatic dissemination,
and epithelial-mesenchymal transition (EMT) and epithelial
plasticity are hypothesized to contribute to tumor progression
(49). In breast cancer, features
of EMT have been observed (50) and
it was found that developmental EMT regulators, including
Snail/Slug, Twist, Six1 and Cripto, along with developmental
signaling pathways, including TGF-β and Wnt/β-catenin, are
aberrantly expressed in breast cancer (49). MMPs also induce EMT in breast cancer
(51). Since we found that AEG-1
regulates the expression of MMP-9 and activates the Wnt/β-catenin
signaling pathway, whether AEG-1 is involved in the regulation of
EMT in breast cancer through the modulation of regulators, such as
MMP-9 or through the activation of signaling pathways, such as
Wnt/β-catenin, requires clarification. In addition, data show that
AEG-1 colocalizes with tight junction proteins ZO-1 and occludin in
polarized epithelial cells (52),
suggesting that AEG-1 is related to the loss of cell polarity known
to occur with increased epithelial tumorigenicity. Previous
findings have shown that tight junctions are comprised of a number
of types of membrane proteins, cytoskeletal proteins and signaling
molecules, many of which play roles in the development of the
mammary gland. Moreover, several of these proteins are regarded as
potential ‘tumor suppressors’ during the development and
progression of breast cancer (53).
EMT is associated with the disintegration of tight junctions
(54). These findings indicate
potential new roles of AEG-1 in tight junctions and EMT.
Acknowledgements
This study was partly supported by the Program for
New Century Excellent Talents in University, Key Project of Chinese
Ministry of Education (no. 108080), the National Natural Science
Foundation of China (nos. 30772133 and 81072150) and the
Independent Innovation Foundation of Shandong University (IIFSDU)
(no. 2009JQ007) to Professor Q. Yang.
References
|
1
|
Krcova Z, Ehrmann J, Krejci V, Eliopoulos
A and Kolar Z: Tpl-2/Cot and COX-2 in breast cancer. Biomed Pap Med
Fac Univ Palacky Olomouc Czech Repub. 152:21–25. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jezierska A and Motyl T: Matrix
metalloproteinase-2 involvement in breast cancer progression: a
mini-review. Med Sci Monit. 15:RA32–RA40. 2009.PubMed/NCBI
|
|
3
|
Gasparini G: Therapy of breast cancer with
molecular targeting agents. Ann Oncol. 16:iv28–iv36. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Osborne C, Wilson P and Tripathy D:
Oncogenes and tumor suppressor genes in breast cancer: potential
diagnostic and therapeutic applications. Oncologist. 9:361–377.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Su ZZ, Kang DC, Chen YM, et al:
Identification and cloning of human astrocyte genes displaying
elevated expression after infection with HIV-1 or exposure to HIV-1
envelope glycoprotein by rapid subtraction hybridization, RaSH.
Oncogene. 21:3592–3602. 2002. View Article : Google Scholar
|
|
6
|
Hu G, Wei Y and Kang Y: The multifaceted
role of MTDH/AEG-1 in cancer progression. Clin Cancer Res.
15:5615–5620. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li J, Zhang N, Song LB, et al: Astrocyte
elevated gene-1 is a novel prognostic marker for breast cancer
progression and overall patient survival. Clin Cancer Res.
14:3319–3326. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kang D, Su Z, Sarkar D, Emdad L, Volsky D
and Fisher P: Cloning and characterization of HIV-1-inducible
astrocyte elevated gene-1, AEG-1. Gene. 353:8–15. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brown DM and Ruoslahti E: Metadherin, a
cell surface protein in breast tumors that mediates lung
metastasis. Cancer Cell. 5:365–374. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hu G, Chong R, Yang Q, et al: MTDH
activation by 8q22 genomic gain promotes chemoresistance and
metastasis of poor-prognosis breast cancer. Cancer Cell. 15:9–20.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Su Z: Customized rapid subtraction
hybridization (RaSH) gene microarrays identify overlapping
expression changes in human fetal astrocytes resulting from human
immunodeficiency virus-1 infection or tumor necrosis factor-α
treatment. Gene. 306:67–78. 2003.
|
|
12
|
Emdad L: Activation of the nuclear factor
κB pathway by astrocyte elevated gene-1: implications for tumor
progression and metastasis. Cancer Res. 66:1509–1516. 2006.
|
|
13
|
Thirkettle HJ, Girling J, Warren AY, et
al: LYRIC/AEG-1 is targeted to different subcellular compartments
by ubiquitinylation and intrinsic nuclear localization signals.
Clin Cancer Res. 15:3003–3013. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu RC, Qin J, Hashimoto Y, et al:
Regulation of SRC-3 (pCIP/ACTR/AIB-1/RAC-3/TRAM-1) coactivator
activity by IκB kinase. Mol Cell Biol. 22:3549–3561.
2002.PubMed/NCBI
|
|
15
|
Lee SG, Su ZZ, Emdad L, Sarkar D and
Fisher PB: Astrocyte elevated gene-1 (AEG-1) is a target gene of
oncogenic Ha-ras requiring phosphatidylinositol 3-kinase and c-Myc.
Proc Natl Acad Sci USA. 103:17390–17395. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee SG, Jeon HY, Su ZZ, et al: Astrocyte
elevated gene-1 contributes to the pathogenesis of neuroblastoma.
Oncogene. 28:2476–2484. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thirkettle HJ, Mills IG, Whitaker HC and
Neal DE: Nuclear LYRIC/AEG-1 interacts with PLZF and relieves
PLZF-mediated repression. Oncogene. 28:3663–3670. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sarkar D, Park ES, Emdad L, Lee SG, Su ZZ
and Fisher PB: Molecular basis of nuclear factor-κB activation by
astrocyte elevated gene-1. Cancer Res. 68:1478–1484. 2008.
|
|
19
|
Yoo BK, Emdad L, Su ZZ, et al: Astrocyte
elevated gene-1 regulates hepatocellular carcinoma development and
progression. J Clin Invest. 119:465–477. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kikuno N, Shiina H, Urakami S, et al:
Knockdown of astrocyte-elevated gene-1 inhibits prostate cancer
progression through upregulation of FOXO3a activity. Oncogene.
26:7647–7655. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Khuda II, Koide N, Noman AS, et al:
Astrocyte elevated gene-1 (AEG-1) is induced by lipopolysaccharide
as toll-like receptor 4 (TLR4) ligand and regulates TLR4
signalling. Immunology. 128:e700–e706. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee SG, Su ZZ, Emdad L, Sarkar D, Franke
TF and Fisher PB: Astrocyte elevated gene-1 activates cell survival
pathways through PI3K-Akt signaling. Oncogene. 27:1114–1121.
2007.PubMed/NCBI
|
|
23
|
Yu C, Chen K, Zheng H, et al:
Overexpression of astrocyte elevated gene-1 (AEG-1) is associated
with esophageal squamous cell carcinoma (ESCC) progression and
pathogenesis. Carcinogenesis. 30:894–901. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Emdad L, Lee SG, Su ZZ, et al: Astrocyte
elevated gene-1 (AEG-1) functions as an oncogene and regulates
angiogenesis. Proc Natl Acad Sci USA. 106:21300–21305. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Doerfler W, Hohlwey U, Müller K, Remus R,
Heller H and Hertz J: Foreign DNA integration perturbations of the
genome oncogenesis. Ann NY Acad Sci. 945:276–288. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li J, Yang L, Song L, et al: Astrocyte
elevated gene-1 is a proliferation promoter in breast cancer via
suppressing transcriptional factor FOXO1. Oncogene. 28:3188–3196.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu JM, Yuan Y, Huan J and Shen ZY:
Inhibition of breast and brain cancer cell growth by BCCIPalpha, an
evolutionarily conserved nuclear protein that interacts with BRCA2.
Oncogene. 20:336–345. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Meng XB, Lu HM and Shen ZY: BCCIP
functions through p53 to regulate the expression of p21Waf1/Cip1.
Cell Cycle. 3:1457–1462. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ash S, Yang D and Britt D: LYRIC/AEG-1
overexpression modulates BCCIPα protein levels in prostate tumor
cells. Biochem Biophys Res Commun. 371:333–338. 2008.PubMed/NCBI
|
|
30
|
Meng X, Fan J and Shen Z: Roles of BCCIP
in chromosome stability and cytokinesis. Oncogene. 26:6253–6260.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Emdad L, Sarkar D, Su Z, et al: Astrocyte
elevated gene-1: recent insights into a novel gene involved in
tumor progression, metastasis and neurodegeneration. Pharmacol
Ther. 114:155–170. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kunnumakkara A, Anand P and Aggarwal B:
Curcumin inhibits proliferation, invasion, angiogenesis and
metastasis of different cancers through interaction with multiple
cell signaling proteins. Cancer Lett. 269:199–225. 2008. View Article : Google Scholar
|
|
33
|
Qiao M, Sheng SJ and Pardee AB: Metastasis
and Akt activation. Cell Cycle. 7:2991–2996. 2008. View Article : Google Scholar
|
|
34
|
Cao Y: Opinion: emerging mechanisms of
tumour lymphangiogenesis and lymphatic metastasis. Nat Rev Cancer.
5:735–743. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu L, Wu J, Ying Z, et al: Astrocyte
elevated gene-1 upregulates matrix metalloproteinase-9 and induces
human glioma invasion. Cancer Res. 70:3750–3759. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar
|
|
37
|
Matsuo Y, Ochi N, Sawai H, et al:
CXCL8/IL-8 and CXCL12/SDF-1α co-operatively promote invasiveness
and angiogenesis in pancreatic cancer. Int J Cancer. 124:853–861.
2009.PubMed/NCBI
|
|
38
|
Kuwano T: Cyclooxygenase 2 is a key enzyme
for inflammatory cytokine-induced angiogenesis. FASEB J.
18:300–310. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Marrogi A, Travis W, Welsh J, et al:
Nitric oxide synthase, cyclooxygenase 2, and vascular endothelial
growth factor in the angiogenesis of non-small cell lung carcinoma.
Clin Cancer Res. 6:4739–4744. 2000.PubMed/NCBI
|
|
40
|
Liu H, Song X, Liu C, Xie L, Wei L and Sun
R: Knockdown of astrocyte elevated gene-1 inhibits proliferation
and enhancing chemo-sensitivity to cisplatin or doxorubicin in
neuroblastoma cells. J Exp Clin Cancer Res. 28:192009. View Article : Google Scholar
|
|
41
|
Lerebours F, Vacher S, Andrieu C, et al:
NF-kappa B genes have a major role in inflammatory breast cancer.
BMC Cancer. 8:412008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yoo BK, Gredler R, Vozhilla N, et al:
Identification of genes conferring resistance to 5-fluorouracil.
Proc Natl Acad Sci USA. 106:12938–12943. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yoo BK, Chen D, Su ZZ, et al: Molecular
mechanism of chemoresistance by astrocyte elevated gene-1. Cancer
Res. 70:3249–3258. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kim DH, Mohapatra G, Bollen A, Waldman FM
and Feuerstein BG: Chromosomal abnormalities in glioblastoma
multiforme tumors and glioma cell lines detected by comparative
genomic hybridization. Int J Cancer. 60:812–819. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ried TJK, Holtgreve-Grez H, et al:
Comparative genomic hybridization of formalin-fixed,
paraffin-embedded breast tumors reveals different patterns of
chromosomal gains and losses in fibroadenomas and diploid and
aneuploid carcinomas. Cancer Res. 55:5415–5423. 1995.
|
|
46
|
Bergamaschi A, Kim YH, Wang P, et al:
Distinct patterns of DNA copy number alteration are associated with
different clinicopathological features and gene-expression subtypes
of breast cancer. Genes Chromosomes Cancer. 45:1033–1040. 2006.
View Article : Google Scholar
|
|
47
|
Warr T, Ward S, Burrows J, et al:
Identification of extensive genomic loss and gain by comparative
genomic hybridisation in malignant astrocytoma in children and
young adults. Genes Chromosomes Cancer. 31:15–22. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sarkar D, Emdad L, Lee SG, Yoo BK, Su ZZ
and Fisher PB: Astrocyte elevated gene-1: far more than just a gene
regulated in astrocytes. Cancer Res. 69:8529–8535. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Micalizzi DS, Farabaugh SM and Ford HL:
Epithelial-mesenchymal transition in cancer: parallels between
normal development and tumor progression. J Mammary Gland Biol
Neoplasia. 15:117–134. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Trimboli AJ, Fukino K, de Bruin A, et al:
Direct evidence for epithelial-mesenchymal transitions in breast
cancer. Cancer Res. 68:937–945. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Radisky ES and Radisky DC: Matrix
metalloproteinase-induced epithelial-mesenchymal transition in
breast cancer. J Mammary Gland Biol Neoplasia. 15:201–212. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Britt D, Yang D, Flanagan D, et al:
Identification of a novel protein, LYRIC, localized to tight
junctions of polarized epithelial cells. Exp Cell Res. 300:134–148.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Itoh M and Bissell MJ: The organization of
tight junctions in epithelia: implications for mammary gland
biology and breast tumorigenesis. J Mammary Gland Biol Neoplasia.
8:449–462. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ikenouchi J: Regulation of tight junctions
during the epithelium-mesenchyme transition: direct repression of
the gene expression of claudins/occludin by Snail. J Cell Sci.
116:1959–1967. 2003. View Article : Google Scholar : PubMed/NCBI
|