Introduction
Janus kinase 2 (JAK2) is a cytoplasmic tyrosine
kinase that has roles in hematopoietic growth factor signaling
pathways such as those stimulated by erythropoietin and
interleukin-3. An acquired mutation of G to T at nucleotide 1,849
of JAK2, JAK2 V617F, constitutively activates
signaling in hematopoietic stem cells and leads to the development
of myeloproliferative neoplasms (MPN) such as polycythemia vera
(PV), essential thrombocythemia (ET), and primary myelofibrosis
(PMF). Indeed, JAK2 mutation is included in the diagnostic
criteria of these three diseases (1).
Although sequence analysis is currently the standard
method for the detection of JAK2 mutation, this technique is
not suitable for clinical examinations in hospital laboratories as
it requires expensive equipment and takes time. Moreover, the
sensitivity of the detection limit of sequencing is not less than
10–20% (2). Therefore, the use of
various PCR-based methods such as allele-specific PCR (AS-PCR),
PCR-restriction fragment length polymorphism (PCR-RFLP) and
high-resolution melting analysis (HRM) has been investigated.
Recently, the quenching probe method (QP) has been attracting
attention as a novel technique capable of detecting single
nucleotide polymorphisms and mutations (3).
In QP, an oligonucleotide with a cytosine modified
by fluorescent dye at the 3′ end (Q probe) is used as the probe.
Following completion of PCR, a melting curve analysis is performed.
At low temperature, the probes hybridize with PCR products and
their fluorescence is quenched by an electron transfer to adjacent
guanine bases in the PCR products. As the temperature is raised,
the probes dissociate from the PCR products and the fluorescent
signal increases. Since the probes dissociate from unmatched
products at lower temperatures than perfectly matched products, it
is possible to detect mutations.
In this study, we performed a comparative evaluation
of the efficiency and sensitivity of four PCR-based methods to
detect JAK2 V617F. To the best of our knowledge, this is the
first study to demonstrate the efficiency of the QP method for the
detection of JAK2 V617F using a standard thermal cycler.
Materials and methods
Cells and DNA extraction
Two leukemia cell lines were used. HEL, an erythroid
leukemia cell line with a homozygous JAK2 V617F mutation,
was supplied by the Japanese Collection of Research Bioresources
(Ibaraki, Japan). KOPT-K1, a T-lymphoblastic leukemia cell line
with wild-type JAK2, was donated by Drs. Harashima and Orita
(Fujisaki Cell Center, Japan). To investigate the sensitivity of
the detection limit, samples of HEL cells and KOPT-K1 cells mixed
at various ratios were used. We obtained bone marrow samples from
16 MPN patients (six PV, eight ET, one PMF, and one unclassifiable
MPN) with informed consent. Blood samples from two normal
volunteers were used as a control. DNA was extracted from cells
following the spin column method using QIAamp DNA Blood Mini kit
(Qiagen, Germantown, MD, USA).
AS-PCR
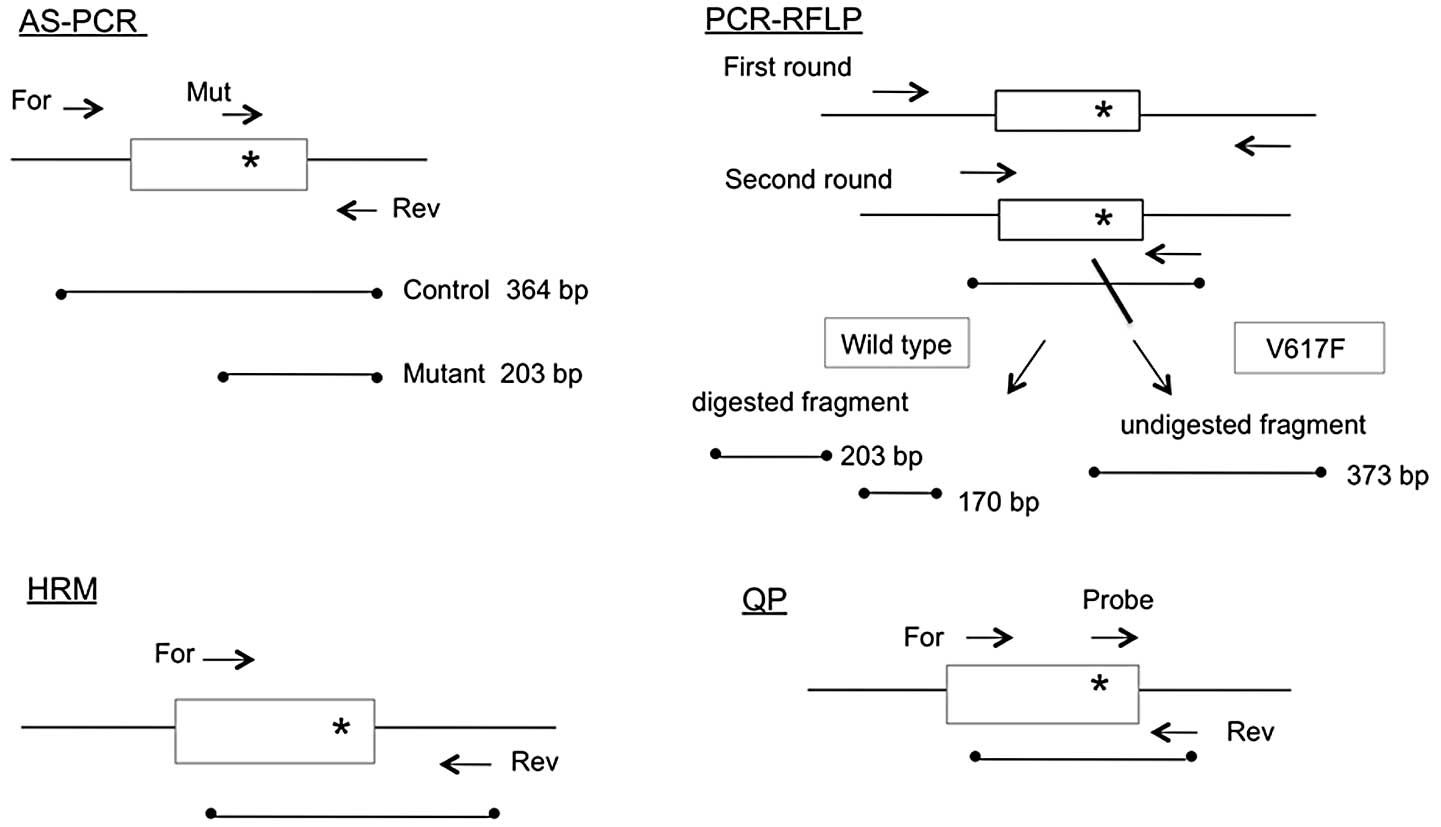
The schematic outlines of all four PCR methods are
shown in Fig. 1, while the primer
sequences used are presented in Table
I. The AS-PCR method used in this study was modified from a
previously reported protocol (4)
and the mutant primer contained an intentional mismatch at the
third nucleotide from the 3′ end to improve specificity.
Amplifications were performed in a 50 μl reaction volume with 0.2
μg DNA, 1.5 mM MgCl2, 0.2 mM dNTP, 0.5 μM primers, and 1
unit Taq polymerase (Takara, Japan), and the resulting PCR
products were electrophoresed in a 2% agarose gel and stained with
ethidium bromide. The assay was repeated at least three times to
ascertain the reproducibility of this method.
 | Table IPrimers, probes and PCR protocols. |
Table I
Primers, probes and PCR protocols.
| Method | Primer sequence | No. of cycles | Melting point
temperature | Ref. |
|---|
| AS-PCR | For:
5′-atctatagtcatgctgaaagtaggagaaag-3′
Mut: 5′-agcatttggttttaaattatggagtatatt-3′
Rev: 5′-ctgaatagtcctacagtgttttcagtttca-3′ | 40 | 94°C 1 min, 57°C 1
min, 72°C 1 min | 4 |
| PCR-RFLP |
| First round | For:
5′-ggtttcctcagaacgttgatgg-3′
Rev: 5′-ttgtttgggcattgtaaccttc-3′ | 35 | 94°C 1 min, 55°C 1
min, 72°C 1 min | 5 |
| Second round | For:
5′-tgctgaaagtaggagaaagtgcat-3′
Rev: 5′-tcctacagtgttttcagtttcaaaaa-3′ | 25 | 94°C 1 min, 60°C 1
min, 72°C 1 min | |
| HRM | For:
5′-agcaagctttctcacaagca-3′
Rev: 5′-ctgacacctagctgtgatcctg-3′ | 45 | 95°C 15 sec, 58°C 15
sec, 72°C 15 sec | 6 |
| QP | For:
5′-gcagcaagtatgatgagcaagctttctc-3′
Rev: 5′-gctctgagaaaggcattagaaagcctg-3′
Probe: 5′-agtatgtttctgtggagac-(BODIPY
FL)-3′ | 45 | 95°C 15 sec, 58°C 15
sec, 72°C 15 sec | 7 |
PCR-RFLP
The PCR-RFLP method used in this study was modified
from a previously reported protocol (5). The first round of nested PCR was
performed in a 50 μl reaction volume with 0.2 μg DNA sample, 1.5 mM
MgCl2, 0.2 mM dNTP, 0.5 μM primers, and 1 unit
Taq polymerase. The second round of PCR was performed in a
50 μl reaction volume containing 2 μl of the first round products.
The resulting products were digested with BsaXI (New England
Biolabs, Hitchin, UK), and the PCR products and
BsaXI-treated products were electrophoresed as described
above.
HRM
HRM was performed using a LightCycler Nano (Roche
Diagnostics, Mannheim, Germany) according to a previously reported
protocol, with modifications (6).
Amplifications were performed in a 20 μl reaction volume with 30 ng
DNA sample, 3 mM MgCl2, 0.2 μM primers and 10 μl Master
Mix [LC 480 High Resolution Melting Master (Roche)] following
incubation for 10 min at 95°C. Melting conditions were as follows:
denaturation at 95°C for 1 min, renaturation at 40°C for 1 min, and
melting at a gradient from 60–95°C with 50 fluorescent acquisitions
per 1°C. The normalized and temperature-shifted difference curves
were determined using LightCycler Nano HRM software (Roche).
QP
The QP method was performed with modifications to a
previously reported protocol (7)
using a LightCycler Nano. Following incubation for 10 min at 95°C,
amplifications were performed in a 20 μl reaction volume with 30 ng
DNA sample, 1.5 mM MgCl2, 0.2 μM primers, 0.1 μM Q
probe, and 10 μl reaction mix [LC 480 Genotyping Master (Roche)].
The Q probe was labeled with BODIPY FL at its 3′ end, and had a
sequence complementary to the mutant allele. Following completion
of PCR, the temperature was maintained at 95°C for 1 min, 40°C for
1 min, and then gradually increased to 90°C at the rate of
0.05°C/sec, during which the fluorescence signal was continually
acquired. The curves for degree of fluorescence increase (dF/dT)
were obtained from the data using LightCycler Nano software
(Roche).
Direct sequencing
To confirm the G to T mutation at nucleotide 1,849,
the PCR products from selected samples were sequenced using a
3130xl Genetic Analyzer and BigDye v3.1 (Applied Biosystems, Foster
City, CA, USA).
Results
Discrimination between wild-type and
mutant alleles
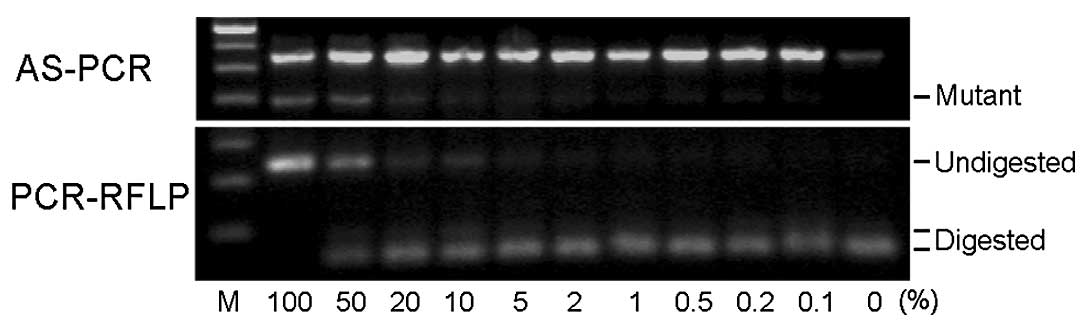
The AS-PCR method produced a 364-bp band from
wild-type and mutant alleles, as well as a 203-bp band from the
mutant allele (upper panel of Fig.
2). The nested PCR products obtained by PCR-RFLP were 373 bp in
size. Following BsaXI digestion, the products from the
wild-type allele were cut into 203-bp and 170-bp fragments while
the products from the mutant allele remained unchanged (lower panel
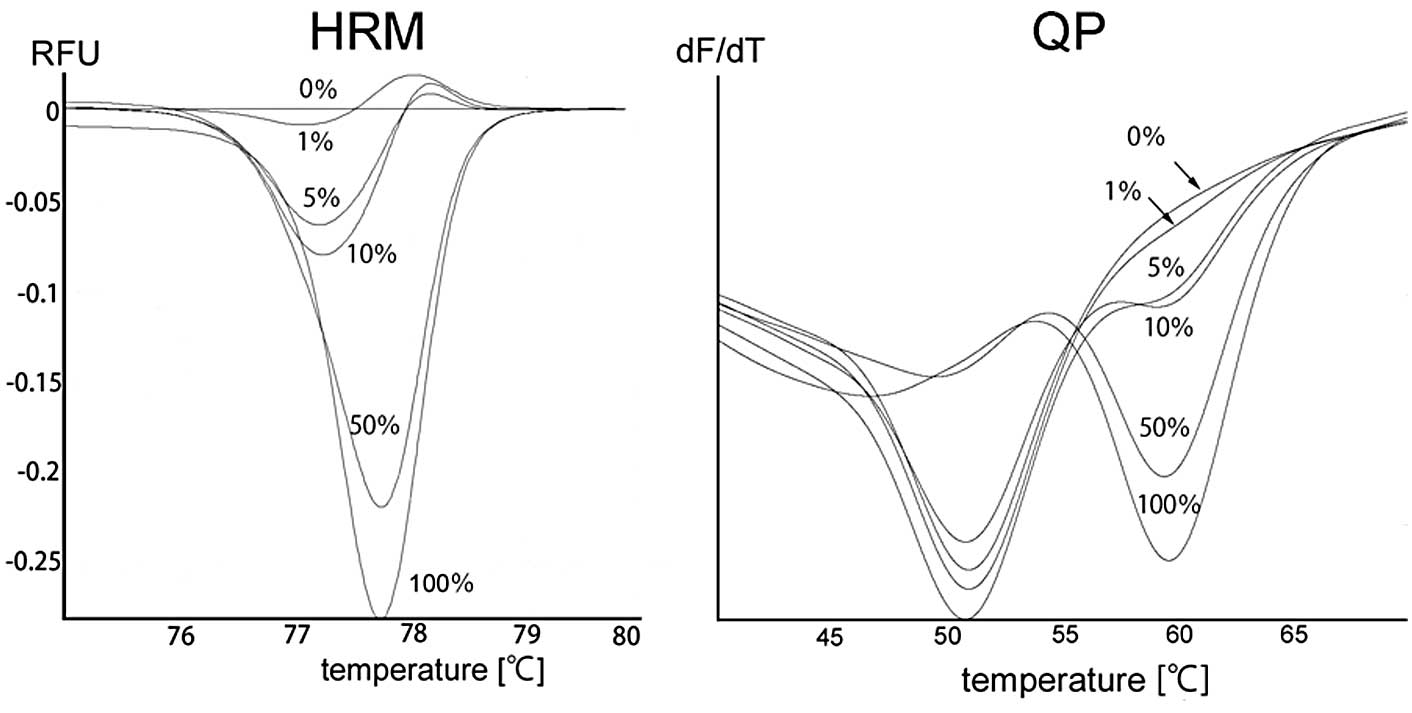
of Fig. 2). With the HRM method,
the melting profile of the wild-type control was designated as the
horizontal line to which the normalized and temperature-shifted
difference curves were drawn. The samples containing mutant
products are distinguishable by their concave-up curves (left panel
of Fig. 3). Finally, with the QP
method, the wild-type allele produced concave-up curves with the
lowest point at 51°C, while the mutant allele produced concave-up
curves with the lowest point at 60°C (right panel of Fig. 3).
Detection of the JAK2 V617F mutation in
patient samples
Of the 16 patient samples, 7 (3 PV, 3 ET, zero PMF,
and one unclassifiable MPN) were determined to be V617F-positive.
Although the results obtained by AS-PCR from a few samples were
initially discrepant with those obtained via the other methods,
this was settled by adjusting the MgCl2 concentration
and annealing temperature. The results from the four PCR-based
methods and direct sequencing were ultimately found to be
concordant.
Sensitivity of detection
We examined the sensitivity of the detection limit
using DNA extracted from samples containing mixtures of HEL and
KOPT-K1 cells in various ratios. Fig.
2 shows representative results from AS-PCR and PCR-RFLP. The
numbers indicate the percentage of HEL cells in the given sample.
Although the mutant bands obtained by AS-PCR from low percentage
samples are not clearly visible in this photograph, the bands from
the 0.2–0.5% mutant samples were visible on a transilluminator. The
PCR-RFLP method detected mutants in 1–2% samples. As shown in the
left panel of Fig. 3, the samples
containing 1–5% mutant allele were distinguishable by their
concave-up curves on HRM. Finally, QP detected the mutant allele in
samples containing 5% mutant, as evidenced by the concave-up curves
with the lowest point at 60°C of the samples (right panel of
Fig. 3).
Discussion
In this study, we compared the efficiency of four
PCR-based methods to detect JAK2 V617F (Table II). AS-PCR was found to be the most
sensitive method of the four, although this method did have
weaknesses. Firstly, this method was initially found to produce
false-positive results in a small number of samples due to the
annealing of the mutant primer to the wild-type allele. Secondly,
it is not possible to estimate the mutant allele burden using this
method. To cope with this weakness, another AS-PCR was carried out
according to a previously reported protocol using two primers for
the mutant allele and wild-type allele individually, in addition to
the outer forward and reverse primers (8). However, the sensitivity and
reproducibility were not as good with this protocol, likely due to
competition of the primers (data not shown).
| Table IIComparison of PCR methods. |
Table II
Comparison of PCR methods.
| Method | Sensitivity (%) | Time (h) | Strengths | Weaknesses |
|---|
| AS-PCR | 0.2–0.5 | 4.5 | High sensitivity | Possible false
positive |
| PCR-RFLP | 1–2 | 9.5 | High specificity | Time consuming |
| HRM | 1–5 | 2.0 | Fast results | Difficult to identify
the mutant curve at low percentages |
| QP | 5 | 2.0 | Easy to recognize the
mutant curve | Requires high quality
DNA |
The PCR-RFLP method did not result in false-positive
and false-negative results in this study. Furthermore, this method
was able to approximately estimate the mutant allele burden by
comparing the density of the digested bands and undigested band.
However, the weakness of this method is that it is time consuming,
taking 9.5 h to obtain results. While it was possible to obtain
results after the first round of PCR and subsequent enzyme
digestion from cell line samples, detection in patient samples
required nested PCR.
By contrast, HRM and QP are able to produce results
in 2 h as there is no need for gel electrophoresis with these
methods. However, high purity DNA samples are required; crude DNA
samples did not produce clear results due to non-specific PCR
products that caused the vacillation of the curves. Therefore, a
spin-column method should be used for DNA extraction rather than a
phenol/chloroform or agglutination partition method. When HRM and
QP were compared, it was easier to recognize the curves derived
from the mutant alleles using the QP method as the curves created
by the wild-type allele and mutant allele have different positions
on the temperature axis. Moreover, it is possible to roughly
estimate allele burden quantification following QP by comparing the
sizes of the curves at 51°C to those at 60°C, as well as the
proportions of two curves from a given patient sample to that shown
in Fig. 3.
Taken together, AS-PCR was found to be the best
method for the detection of JAK2 V617F in terms of
sensitivity, while QP was the best method in terms of promptness
and ease of interpretation. In clinical settings, JAK2 V617F
assays are mainly used for diagnosis at presentation rather than
for detecting minimal residual diseases. As cells expressing the
mutant allele usually account for more than 10% of the samples in
these cases, high sensitivity is not necessarily required. We
therefore suggest that QP would be the preferable method for
JAK2 V617F detection in clinical examination in
hospitals.
Acknowledgements
We thank Dr N. Murakami and Dr T. Fukuda (Tokyo
Medical and Dental University) for their assistance in obtaining
samples from the patients. This study was supported in part by a
Grant-in-Aid for Scientific Research (C) from the Japan Society for
the Promotion of Science (No. 18690522).
References
|
1
|
Thiele J, Kvasnycka HM, Orazi A, Tefferi A
and Birgegard G: Polycythemia vera. WHO Classification of Tumours
of Haematopoietic and Lymphoid Tissues. Swerdlow SH, Campo E,
Harris NL, Jaffe ES, Pileri ES, Stein H, Thiele J and Vardiman JW:
IARC Press; Geneva: pp. 40–43. 2008
|
|
2
|
James C, Ugo V, Le Couédic JP, et al: A
unique clonal JAK2 mutation leading to constitutive signalling
causes polycythaemia vera. Nature. 434:1144–1148. 2005. View Article : Google Scholar
|
|
3
|
Crockett AO and Wittwer CT:
Fluorescein-labeled oligonucleotides for real-time PCR: using the
inherent quenching of deoxyguanosine nucleotides. Anal Biochem.
290:89–97. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Baxter EJ, Scott LM, Campbell PJ, et al:
Acquired mutation of the tyrosine kinase JAK2 in human
myeloproliferative disorders. Lancet. 365:1054–1061. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Horn T, Kremer M, Dechow T, et al:
Detection of the activating JAK2 V617F mutation in
paraffin-embedded trephine bone marrow biopsies of patients with
chronic myeloproliferative diseases. J Mol Diagn. 8:299–304. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Er TK, Lin SF, Chang JG, et al: Detection
of the JAK2 V617F missense mutation by high resolution melting
analysis and its validation. Clin Chim Acta. 408:39–44. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tanaka R, Kuroda J, Stevenson W, et al:
Fully automated and super-rapid system for the detection of
JAK2V617F mutation. Leuk Res. 32:1462–1467. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jones AV, Kreil S, Zoi K, et al:
Widespread occurrence of the JAK2 V617F mutation in chronic
myeloproliferative disorders. Blood. 106:2162–2168. 2005.
View Article : Google Scholar : PubMed/NCBI
|