Introduction
The 2008 World Health Organization (WHO)
Classification of Tumors defines refractory anemia with ring
sideroblasts associated with marked thrombocytosis (RARS-T) as a
provisional entity, with unclassifiable
myelodysplastic/myeloproliferative neoplasm (MDS/MPN) status, as
opposed to a confirmed entity (1).
Although thrombocytosis is a poor prognostic factor in MDS patients
(2), overall, RARS-T patients
exhibit a more favorable prognosis. The JAK2 V617F mutation has
been identified in ~50% of RARS-T patients and in only 2/89 cases
of typical MDS, indicating that RARS-T should be considered as a
JAK2 mutation-associated chronic MPN (3).
RARS-T is characterized by MDS characteristics and
<5% blasts in the bone marrow, and is differentiated from other
diseases by the presence of ≥15% ringed sideroblasts,
thrombocytosis and a platelet count of >450×109/l
(1). Clinical manifestations of
RARS-T include symptoms associated with anemia, leucopenia and
abnormalities of platelet function and quantity, for example,
fatigue, infection, bleeding and/or thrombosis. The International
Prognostic Scoring System (IPSS) (4), which is commonly used for MDS, is also
applicable as a prognostic tool for RARS-T. The management of
RARS-T is largely supportive, including transfusion support in
patients exhibiting symptomatic anemia and prophylaxis, and
treatment of thromboembolism. Similar to MDS, RARS-T patients
exhibiting anemia and low serum erythropoietin levels may benefit
from the administration of erythropoiesis-stimulating agents. An
ongoing clinical trial is currently studying the efficacy of
ruxolitinib, an oral JAK2 inhibitor, in JAK2 mutation-positive
RARS-T patients (ClinicalTrials.gov Identifier: NCT01895842;
http://clinicaltrials.gov/show/NCT01895842).
Furthermore, a case study has recently documented the successful
treatment of young RARS-T patients with lenalidomide (5).
MPL (6), a cellular
homologue of the oncogene v-mpl, belongs to the hematopoietic
cytokine receptor family, which is located upstream of the JAK-STAT
signaling pathway. It is reported that the MPL W515L mutation is
present in ~5% of idiopathic myelofibrosis patients and ~1% of
essential thrombocythemia patients (7). Previous studies have demonstrated that
the MPL W515L mutation is associated with an older age, a lower
hemoglobin level and higher platelet counts, however, the
association between the mutation and complications, such as
thrombosis, is not clear (8,9).
The current report presents the case of an RARS-T
patient positive for the MPL W515L mutation, but negative for the
JAK2 V617F mutation. To the best of our knowledge, this is the
first case study of an MPL W515 mutation in a patient with
RARS-T.
Case report
In August 2012, a 63-year-old female patient was
referred to Saint Mary’s Health Center (St. Louis, MO, USA) for the
hematological evaluation of macrocytic anemia and thrombocytosis. A
complete blood count (CBC) revealed the following results:
Hemoglobin, 7.6 g/dl (normal range, 12.0–15.0 g/dl); hematocrit,
24% (normal range, 36.1–44.3%); mean corpuscular volume (MCV), 109
fl (normal range, 80–100 fl); platelet count, 834×109/l
(normal range, 150–400×109/l); and a normal white blood
cell and differential count 8.2 ×109/l (normal range,
4–10 ×109/l). An iron panel demonstrated an elevated
ferritin level (214 mg/l; normal range, 12–150 ng/ml) and increased
iron saturation (>95%; normal range, 15–55%). Upon examination,
the patient was completely asymptomatic. The patient’s prior CBC,
from September 2010, demonstrated similar macrocytic anemia
(hemoglobin, 9.5 g/dl; MCV, 102 fl) and thrombocytosis (platelet
count, 701×109/l), however, the patient had not been
referred for hematological evaluation at that time.
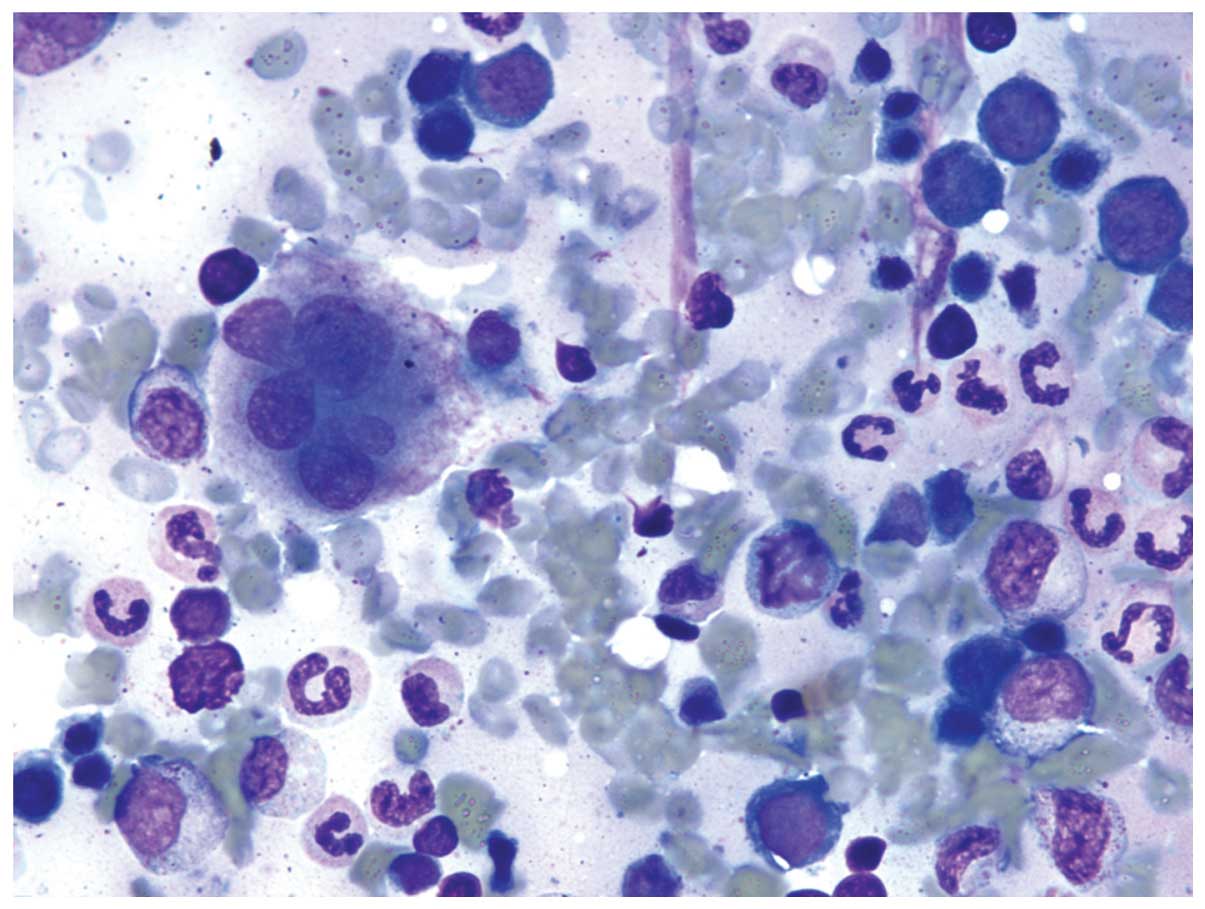
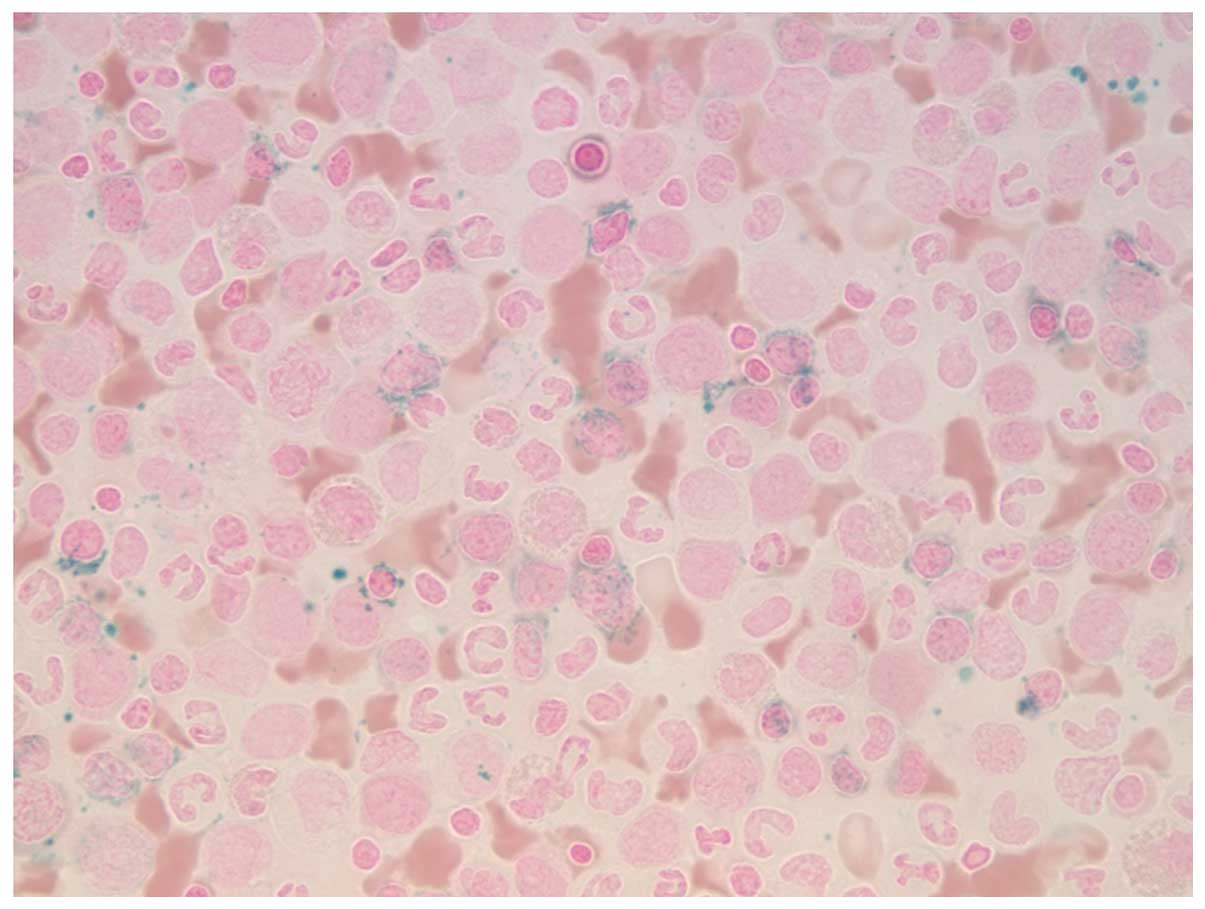
A bone marrow aspiration and biopsy demonstrated
marked erythroid hyperplasia, trilineage dyspoiesis
(Fig. 1) and increased ring
sideroblasts (Fig. 2) compared with
the erythroid precursors; 44% of the precursors were ring
sideroblasts. Cytogenetic and fluorescence in situ
hybridization analysis of the patient was positive for an MPL W515L
mutation and an isolated chromosome 13q deletion, however, there
was no evidence of a chromosome 5q deletion, JAK2 mutation or
BCR-ABL fusion gene. According to the WHO Classification of Tumors
(1), the patient was diagnosed with
RARS-T, with an MPL W515L mutation, a chromosome 13q deletion and
an IPSS Score of 0.5 (intermediate-1 risk).
The patient commenced subcutaneous epoetin α therapy
(60,000 units/week) from November 2012, however, following a
suboptimal response, epoetin α therapy was terminated and
subcutaneous darbepoetin α therapy (300 μg/week) commenced.
Furthermore, the patient was administered with 81 mg aspirin per
day for the treatment of thrombus prophylaxis. From November 2012
until the writing of this study, the patient’s hemoglobin
concentration (range, 8.0–10.0 g/dl) and platelet count (range,
600–770×109/l) have remained stable. At present, the
patient is asymptomatic, transfusion-independent, continues to work
and maintains a good performance status. A repeat bone marrow
biopsy in June 2013 revealed stable hematological results and no
evidence of disease progression.
Discussion
An MPL W515L mutation and isolated chromosome 13q
deletion is rare in an RARS-T patient negative for a JAK2 mutation
and 5q deletion. A search of the literature reveals a number of
studies regarding MPL W515 mutations in MDS with sideroblastic
change, however, it does not reveal any studies on MPL W515
mutations in typical RARS-T. Schnittger et al (10) reported a case of an MPL W515
mutation with features of ringed sideroblasts and thrombocytosis;
however, the patient was not anemic and exhibited an asymptomatic
benign course. Another study reported the case of a JAK-2
mutation-negative and MPL W515 mutation-positive patient exhibiting
grade 2 myelofibrosis; however, no details regarding clinical
course, laboratory results or bone marrow biopys results were
provided (3).
The JAK-2 V617F mutation occurs in ~50% of RARS-T
patients and appears to be predictive of a lower mortality rate
compared with the mutation-negative group (3). A clinical trial of ruxolitinib, an
oral JAK-2 inhibitor, is currently ongoing with the aim of
investigating its efficacy in the treatment of MDS patients who
carry the JAK2 V617F mutation (NCT01895842). MPL, a cellular
homologue of the v-MPL oncogene, is located upstream of the
JAK-STAT signaling pathway (11).
The MPL W515L mutation induces constitutive, cytokine-independent
activation of the JAK-STAT signaling pathway, and may be
significant in the pathogenesis of RARS-T (12). However, it remains unclear whether
the MPL W515L mutation is associated with an improved prognosis in
RARS-T patients and whether it acts as a target for JAK2 pathway
inhibitors, including ruxolitinib. In the case presented in the
current study, the patient remained asymptomatic and
transfusion-independent with no disease progression at the one year
follow-up, indicating that the MPL W515L mutation is associated
with a favorable prognosis. However, further prospective and
long-term investigation of patients exhibiting similar cytogenetic
profiles is required prior to reaching a definitive conclusion.
References
|
1
|
Vardiman JW, Bennett JM, Bain BJ, et al:
Myelodysplastic/myeloproliferative neoplasm, unclassifiable. WHO
Classification of Tumours of Haematopoietic and Lymphoid Tissues.
Swerdlow SH, Campo E, Harris NL, et al: 4th edition. IARC Press;
Lyon: pp. 85–86. 2008
|
|
2
|
Zikria J, Galili N, Tsai WY, et al:
Thrombocytosis in myelodysplastic syndromes: not an innocent
bystander. J Blood Disord Transfus. S3:0022012.
|
|
3
|
Schmitt-Graeff AH, Teo SS, Olschewski M,
et al: JAK2V617F mutation status identifies subtypes of refractory
anemia with ringed sideroblasts associated with marked
thrombocytosis. Haematologica. 93:34–40. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Greenberg P, Cox C, LeBeau MM, et al:
International scoring system for evaluating prognosis in
myelodysplastic syndromes. Blood. 89:2079–2088. 1997.PubMed/NCBI
|
|
5
|
Taylor G, Culligan D and Vickers MA:
Refractory anemia with ring sideroblasts associated with marked
thrombocytosis complicated by massive splenomegaly treated with
lenalidomide resulting in resolution of splenomegaly but severe and
prolonged pancytopenia. Case Rep Hemato. 2013:7184802013.
|
|
6
|
Vigon I, Mornon JP, Cocault L, et al:
Molecular cloning and characterization of MPL, the human homolog of
the v-mpl oncogene: identification of a member of the hematopoietic
growth factor receptor superfamily. Proc Natl Acad Sci USA.
89:5640–5644. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pardanani AD, Levine RL, Lasho T, et al:
MPL515 mutations in myeloproliferative and other myeloid disorders:
a study of 1182 patients. Blood. 108:3472–3476. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Beer PA, Campbell PJ, Scott LM, et al: MPL
mutations in myeloproliferative disorders: analysis of the PT-1
cohort. Blood. 112:141–149. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vannucchi AM, Antonioli E, Guglielmelli P,
et al: Characteristics and clinical correlates of MP 515W>L/K
mutation in essential thrombocythemia. Blood. 112:844–847. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schnittger S, Bacher U, Haferlach C, et
al: Detection of an MPLW515 mutation in a case with features of
both essential thrombocythemia and refractory anemia with ringed
sideroblasts and thrombocytosis. Leukemia. 22:453–455. 2008.
View Article : Google Scholar
|
|
11
|
Tefferi A: Novel mutations and their
functional and clinical relevance in myeloproliferative neoplasms:
JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia. 24:1128–1138.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pikman Y, Lee BH, Mercher T, et al:
MPLW515L is a novel somatic activating mutation in myelofibrosis
with myeloid metaplasia. PLoS Med. 3:e2702006. View Article : Google Scholar : PubMed/NCBI
|