Introduction
Lung cancer is characterized by uncontrolled cell
growth in lung tissues, which results in metastasis, the invasion
of tissues adjacent to the lesion and infiltration beyond the
lungs. In 2010, lung cancer accounted for >0.15 million deaths
in the USA, and >0.2 million cases are registered annually
(1). Despite surgery being the
preferred method for the removal of cancer, it cannot completely
excise the affected tissue and supplementary multi-drug
chemotherapy or radiation may be required. The current drugs of
choice for lung cancer therapy include etoposide, docetaxel,
doxorubicin (DOX), carboplatin and cisplatin (2). However, limited therapeutic action has
been demonstrated by the preferred chemotherapeutic agents for
cancer therapy.
Numerous nanoparticle (NP)-based therapies have been
approved for clinical used or have entered clinical development
over the previous two decades (3).
Liposomal drugs (4) and
polymer-drug conjugates (5) are two
leading classes of NP-based therapy and account for the majority of
the products approved for clinical use. Liposomes and polymer-NPs
each possess advantages and disadvantages. Polymeric NPs exhibit an
elevated loading capacity for hydrophobic drugs compared with
liposomes and drug release is generally dominated by polymer
degradation and drug diffusion in polymeric NPs, which can be
controlled through the use of proper polymers that exhibit a
desirable degradation rate and binding affinity with the
encapsulated drugs (6,7). Advantages of liposomal formulations
include the ability to carry hydrophilic and hydrophobic drugs
within the aqueous vesicles and lipid bilayer membranes,
respectively. The liposomal formulations also exhibit a high
biocompatibility, providing protection for the drugs from the
external environment, and easily undergo surface modification with
other molecules, including polyethylene glycol (PEG), and targeting
ligands, which achieves an improved systemic circulation lifetime
and targeted drug delivery, respectively (8,9).
However, these formulations also possess a short shelf life due to
the preparation and purification of liposomes involving relatively
complicated steps, the low loading efficiency for hydrophobic
drugs, the burst-release kinetics of encapsulated drugs and the
instability of the formulation during storage.
In the present study, lipid-coated poly
D,L-lactic-co-glycolic acid (PLGA) NPs (L-P) were prepared,
which combined the respective benefits of liposomes and polymer-NPs
and avoided their respective disadvantages. The lipid-coated NPs
comprised a biodegradable and biocompatible hydrophobic polymeric
core that was comprised of PGLA, a monolayer of phospholipids and
an outer corona layer of PEG. The biocompatibility,
biodegradability and sustained drug-release of these NPs, as well
as the easy surface modification with other molecules that include
PEG and targeting ligands, which achieves a prolonged systemic
circulation lifetime and targeted drug delivery, the excellent
stability in the blood, and, crucially, the high drug-loading
yield, makes L-Ps a promising drug delivery system (10). These properties provide the basis
for a stable, high-payload targeted drug delivery vehicle that
possesses the potential to maximize the chemotherapeutic efficacy
of anti-cancer agents on the target cancer cells.
For the targeting ligand, transferrin (TF) was
selected as a basis since the TF receptor is overexpressed in 90%
of tumors (11,12). In the present study, TF-conjugated
lipid-coated NPs (TF-LPs) were successfully prepared and
characterized. The DOX-loaded TF-LPs (TF-LP-DOX) demonstrated
elevated cytotoxicity against lung cancer cells and an improved
therapeutic effect in the lung cancer-bearing nude mice compared
with their non-targeted counterparts.
Materials and methods
Ester-terminated PLGA, with a 50:50 monomer ratio
and a viscosity of 0.50–0.85 dl/g, was purchased from Shandong Key
Laboratory of Medical Polymeric Material (Jinan, Shandong, China).
Soybean lecithin, comprising 90–95% phosphatidylcholine and
mPEG2000-DSPE and Mal-PEG2000-DSPE, was
purchased from Avanti Polar Lipids, Inc. (Alabaster, AL, USA). TF
was obtained from Sigma-Aldrich (St. Louis, MO, USA). DOX was
purchased from Zhejiang Haizheng Pharmaceutical Co., Ltd. (Taizhou,
Zheijiang, China). Other chemicals and reagents were of analytical
grade and obtained commercially.
BALB/c male athymic nude mice, ~20 g in weight, were
purchased from the Experimental Animal Center of Tianjin Medical
University (Heping, Tianjin, China). All animal experiments adhered
to the principles of care and use of laboratory animals and were
approved by the Experimental Animal Administrative Committee of
Tianjin Medical University. This study was approved by the ethics
committee of Zhengzhou University (Zhengzhou, China).
The preparation of PLGA-NPs
DOX-loaded PGLA-NPs (PGLA-NP-DOX) were prepared
using the water in oil in water double emulsion method (13,14).
Briefly, 20 mg of mPEG-PLGA was dissolved in 1 ml of methylene
chloride. Water or DOX solution (0.2 ml) was then transferred to a
centrifuge tube, and the mixture was emulsified by sonication for 3
min. The emulsion and 2 ml of 2% polyvinyl alcohol (PVA) were then
emulsified by sonication for 5 min. Subsequently, the emulsion was
slowly dropped into 10 ml of 0.6% PVA and stirred for 10 min at
room temperature. Following vacuum evaporation of the solvent, the
NPs were collected by centrifugation at 18,000 × g for 10 min at
room temperature and were washed twice using distilled water.
Preparation of the L-Ps and TF-LPs
The DOX-loaded L-Ps (LP-DOX) were prepared as
previously described (15,16). Briefly, PLGA was initially dissolved
in acetone, and lecithin and mPEG-DSPE2000 (15% of the PLGA polymer
weight; mole ratio lecithin:mPEG-DSPE2000, 7.5:2.5) were dissolved
in a 4% ethanol aqueous solution and heated to 65°C. The PLGA
acetone solution was then added into the preheated lipid aqueous
solution drop-wise (1 ml/min) under gentle stirring, which was
followed by vortexing for 3 min. The NPs were left for 2 h to
self-assemble, with continuous stirring, until the organic solvent
was evaporated. The remaining organic solvents were removed under
reduced pressure at 37°C. The final concentration of PLGA in NP
suspensions was set to 1 mg/ml with distilled water. The NPs were
used immediately, stored at 4°C, or freeze-dried in liquid nitrogen
and lyophilized for storage at −80°C for later use.
The TF-LP-DOX were prepared using the post-insertion
method (17,18). Firstly, the TF was reacted with
Traut’s reagent at a molar ratio of 1:5 to yield TF-SH. Secondly,
the TF-SH was reacted with the micelles of
DSPE-PEG2000-Mal at a molar ratio of 1:10, and then
incubated with L-P for 1 h at 37°C. The ratio of
TF-PEG2000-DSPE to lipid was 1:50. The final particles
were stored at 4°C for further experiments.
Characterization of the NPs
Size and zeta-potential
measurements
The size and zeta potential of the NPs were measured
using a dynamic light scattering detector (Zetasizer Nano-ZS90;
Malvern Instruments, Worcestershire, UK).
Drug encapsulation efficiency (EE) and
drug loading coefficient
The free DOX was removed by passing through a
Sephadex G-50 column. The quantity of DOX encapsulated in the NPs
was measured by high performance liquid chromatography (HPLC;
Agilent LC1200; Agilent, Santa Clara, CA, USA). A reversed phase
Inertsil®ODS-3 column (150–4.6 mm; pore size, 5 mm; GL Sciences
Inc., Shinjuku, Japan) was used. Freeze-dried NPs (3 mg) were
dissolved in 1 ml DCM. Subsequent to the evaporation of DCM, 3 ml
mobile phase (50:50 v/v acetonitrile/water solutions) was added to
dissolve the drugs. The solution was then filtered by a 0.45 mm
polyvinylidene fluoride syringe filter for HPLC analysis. The
column effluent was detected at 227 nm using an ultraviolet/visible
detector. The EE and drug loading content were calculated as
follows: EE (%) = (amount of drug encapsulated in NPs/initial
amount of drug used in the fabrication of NPs) × 100; and drug
loading content (%) = (amount of drug encapsulated in NPs/amount of
drug encapsulated in NPs and excipients added) × 100.
Stability of NPs
To demonstrate the serum stability of lipid-coated
NPs, the particle sizes and turbidity variations of the NPs were
monitored in the presence of fetal bovine serum (FBS) (19,20).
Briefly, the NPs were mixed with an equal volume of FBS at 37°C by
gentle agitation at 36 × g. At the predetermined time-points of 1,
2, 4, 8 and 24 h, 200 μl of the sample was pipetted onto a 96-well
plate and the transmittance was measured at 750 nm using a
microplate reader (Varioskan Flash; Thermo Fisher Scientific,
Waltham, MA, USA). Another 200 μl was diluted to 1 ml using 5%
glucose solution for the particle size measurements obtained by the
Zetasizer Nano ZS90 light scattering detector (Malvern
Instruments).
In vitro drug release
The release kinetics of DOX from DOX-loaded PLGA-NP,
LP and TF-LP in phosphate-buffered saline (PBS) were evaluated
using a dialysis method for ≤4 days. The samples were individually
dispersed in 5 ml of the PBS and were placed into a cellulose
membrane dialysis tube (MW cut off, 12,000–14,000). The dialysis
tube was then placed into 195 ml of PBS and the release test was
performed at 37°C with a centrifugation rate of 320 × g. At
predetermined time points, 1 ml release medium was taken, refilled
with the same amount of the fresh medium, and concentrations of the
released drug were determined by RP-HPLC, as aforementioned.
In vitro cellular uptake
A549 cells were grown in RPMI-1640 medium (HyClone,
Logan, UT, USA) that contained 10% FBS, 100 μg/ml of streptomycin
and 100 units/ml of penicillin. The cells were maintained at 37°C
in a humidified incubator with 5% CO2.
For the quantitative study, the A549 cells were
harvested with 0.125% trypsin-EDTA solution (Invitrogen, Carlsbad,
CA, USA) and seeded into 24-well assay plates (Corning Inc.,
Corning, New York, NY, USA) at 105 viable cells/well.
Subsequent to the cells reaching confluence, the cells were
incubated with 100 μl of 10 μg/ml DOX-loaded NPs, all three types,
in the 1640-medium supplemented with 10% HyClone FBS (Thermo
Scientific) and 1% penicillin-streptomycin (Invitrogen) at 37°C for
2 or 4 h. At the designated time period, the suspension was removed
and the wells were washed three times with 1,000 μl cold PBS.
Subsequently, 50 μl of 0.5% Triton X-100 was introduced into each
well for cell lysis. The fluorescence intensity of each sample well
was measured by a microplate reader (GENios; Tecan, Männedorf,
Switzerland) with an excitation wavelength of 480 nm and an
emission wavelength of 580 nm.
For the qualitative study, A549 cells were harvested
using 0.125% trypsin-EDTA solution (Invitrogen) and seeded in
LABTEK cover glass chambers (Nalge Nunc International, Rochester,
NY, USA) having RPMI-1640 at a concentration of 5×103
viable cells/chamber. The cells were incubated overnight and were
subsequently incubated with DOX loaded NPs in the RPMI-1640
(concentration of 10 μg/ml) at 37°C. After 4 h, the cells were
washed 3 times with cold PBS and fixed by 4% paraformaldehyde for
20 min. Then, the cells were washed twice with cold PBS. The nuclei
were stained by incubating the cells with DAPI (Roche Diagnostics,
Basel, Switzerland) for an additional 10 min. The cell monolayer
was washed three times with PBS and observed by confocal laser
scanning microscopy (CLSM; Leica, Germany).
In vitro cytotoxicity and
anti-proliferation assay
Comparison between the in vitro cytotoxicity
and tumor cell proliferation of A549 cells in response to various
formulations was performed using the sulforhodamine B (SRB)
colorimetric assay. In brief, 4,000 A549 cells were seeded into
96-well plates and incubated overnight. The cells were then exposed
to serial concentrations of various DOX formulations in the culture
medium for 48 h at 37°C. Subsequently, the cells were fixed with
trichloroacetic acid, washed and stained by SRB. The absorbance was
measured at 540 nm using a 96-well plate reader (Bio-Rad
Laboratories, Hercules, CA, USA). Dose-response curves were
generated, and the concentration of drug that resulted in 50% cell
death (IC50) was calculated using Origin 7.0 software
(OriginLab, Northampton, MA, USA).
Evaluation of tumor spheroid
penetration
To prepare the three-dimensional tumor spheroids,
A549 cells were seeded at a density of 2×103 cells/200
μl per well in 96-well plates coated with 80 μl of a 2%
low-melting-temperature agarose. Seven days after the cells were
seeded, the tumor spheroids were treated with 10 μg/ml DOX-loaded
NPs. After 4 h of incubation, the spheroids were rinsed three times
with ice-cold PBS and fixed with 4% paraformaldehyde for 30 min.
The spheroids were then transferred to glass slides and covered by
glycerophosphate. The fluorescent intensity was observed by laser
scanning confocal microscopy (Leica Microsystems GmbH, Wetzlar,
Germany).
Growth inhibition of tumor spheroid
The tumor spheroids were prepared as aforementioned
for the evaluation of tumor spheroid penetration. Seven days later,
the spheroid-containing wells were treated with 0.8 mg/ml of DOX
solution, and DOX-loaded NPs. The length and width of each spheroid
was measured every day for eight days and the volume was
calculated. A volume curve was drawn to compare the effect of each
treatment with the various formulations.
In vivo imaging
The DIR-loaded NPs were utilized as previously
described to investigate the distribution of NPs in lung cancer
A549 cell-bearing nude mice. The nude mouse lung cancer xenograft
model was established by subcutaneously injecting A549 cells
(1×107 cells per animal) into the backs of 4–6 week-old
BALB/c male athymic nude mice. The DiR-loaded NPs were injected
into A549 lung cancer-bearing nude mice via intravenous
administration, and then the in vivo fluorescence imaging
was performed using the IVIS Spectrum system (Caliper Life
Sciences, Hopkinton, MA, USA).
Statistical analysis
Analysis of variance was used to assess the variance
of the whole values in each group. Statistical significance was
evaluated using Student’s t-test for the comparison between
experimental groups. P<0.05 was considered to indicate a
statistically signficant difference.
Results and Discussion
Characterization of the NPs
Particle size, size distribution, drug
encapsulation efficiency and drug-loading efficiency
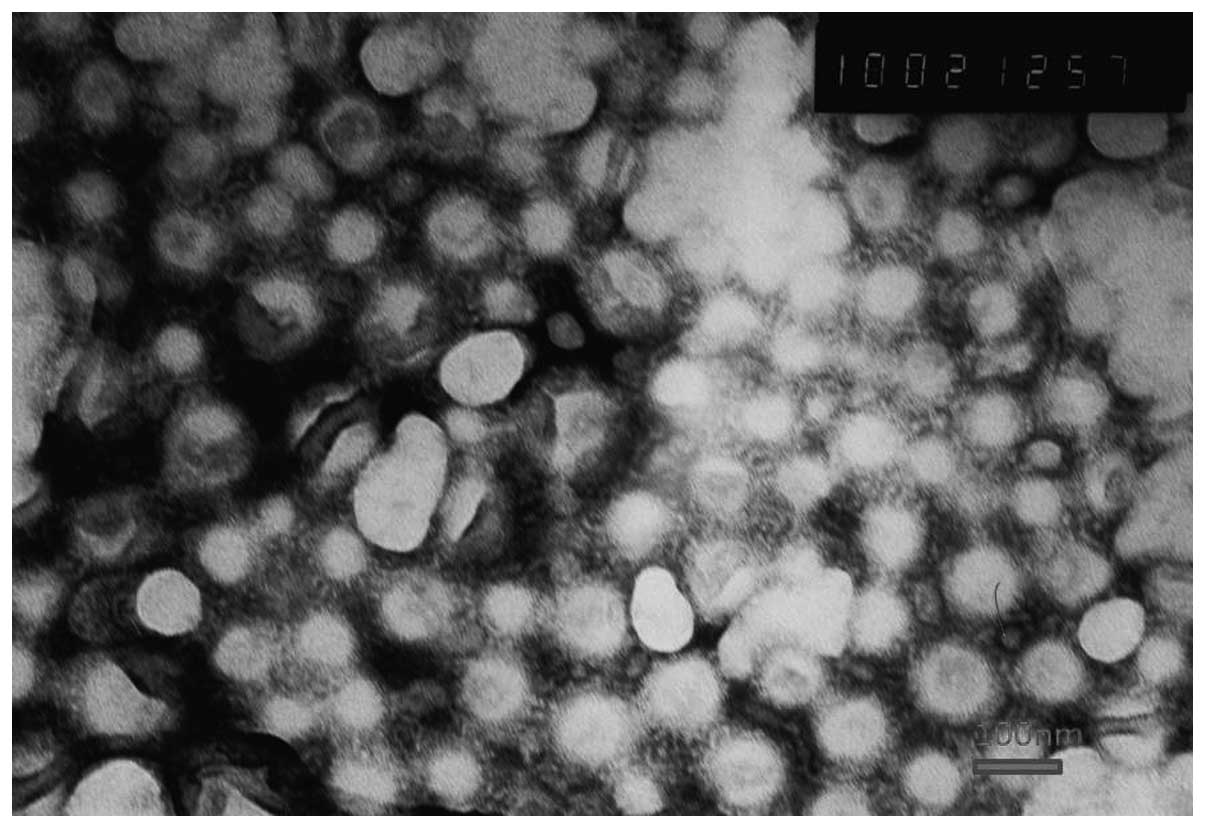
Transmission electron microscopy was used to observe
the shape and surface morphology of the investigated NPs (Fig. 1). The NPs were all revealed by
microscopy to exhibit a uniform spherical appearance that indicated
the successful formation of the lipid-coated NPs. The conventional
DOX-loaded NPs were, on average, ~110 nm in diameter, with a PDI of
0.200 (Table I). In order to
justify the clinical application of NPs, the drug encapsulation
efficiency (EE) is crucial. The EE of the three types of NPs
formulations are reported in Table
I. These EE values are reasonable and confirm the effectiveness
of lipid-coated NPs for loading anticancer drugs. Evidently, the
present formulation system reveals the potential for a useful and
practical drug delivery carrier with an appropriate size, stability
and drug loading capacity.
 | Table ICharacteristics of DOX-loaded PLGA-NP,
L-P and TF-LP (n=3). |
Table I
Characteristics of DOX-loaded PLGA-NP,
L-P and TF-LP (n=3).
| Group | Particle size,
nm | Polydispersity | Zeta-potential,
mV | Encapsulation
efficiency, % |
|---|
| PLGA-NP | 93±8.8 | 0.197 | −21.37±1.51 | 85.75±2.55 |
| L-P | 111±11.4 | 0.180 | −22.16±1.88 | 94.29±1.94 |
| TF-LP | 108±12.5 | 0.212 | −21.32±1.91 | 92.48±2.57 |
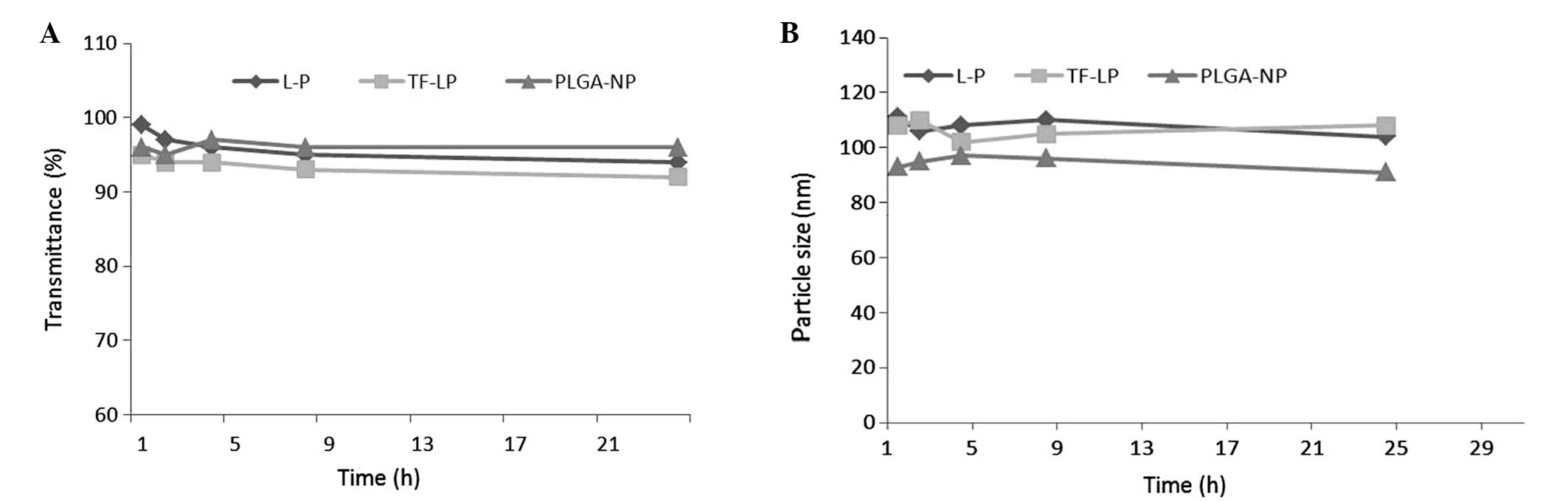
Stability of DOX-loaded NPs
As particle stability in physiological conditions is
a prerequisite for the further application of NPs in vivo,
50% FBS was employed to mimic the in vivo conditions.
Particle sizes and transmittance variations as important parameters
were monitored in the present study to explore the serum stability
of NPs. As reported in Fig. 2, the
particle sizes and transmittance have hardly changed for L-P and
TF-LP over 24 h, indicating that there was no aggregation in the
presence of serum.
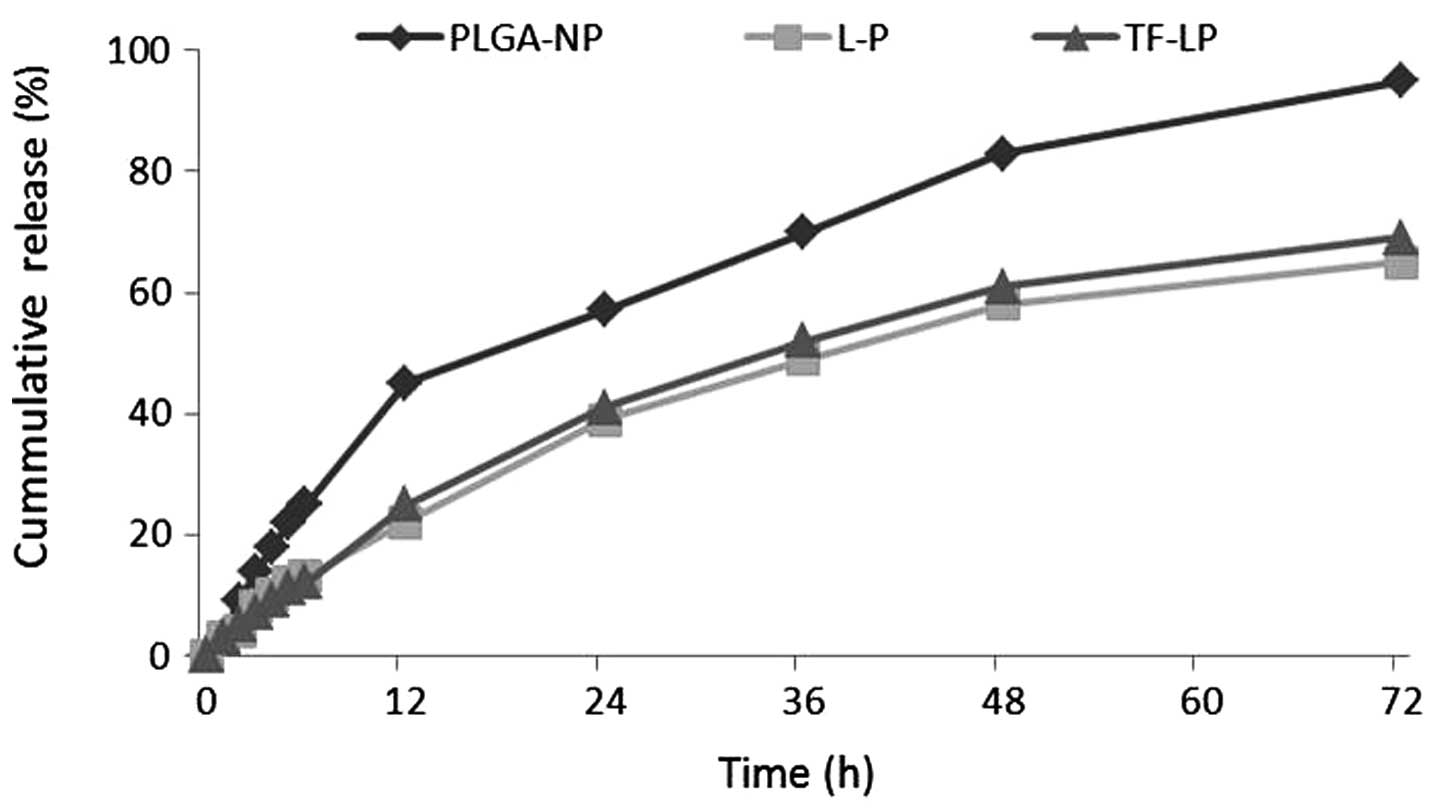
In vitro drug release
The present study investigated the release of DOX
in vitro from PLGA-NP, L-P and TF-LP. The release profile of
these three groups is shown in Fig.
3. DOX was released at a higher rate from PLGA-NPs compared
with the other groups. As shown in Fig.
3, PLGA-NPs demonstrated almost 95% drug release within three
days. Conversely, L-Ps and TF-LPs produced only ~65% leakage within
three days.
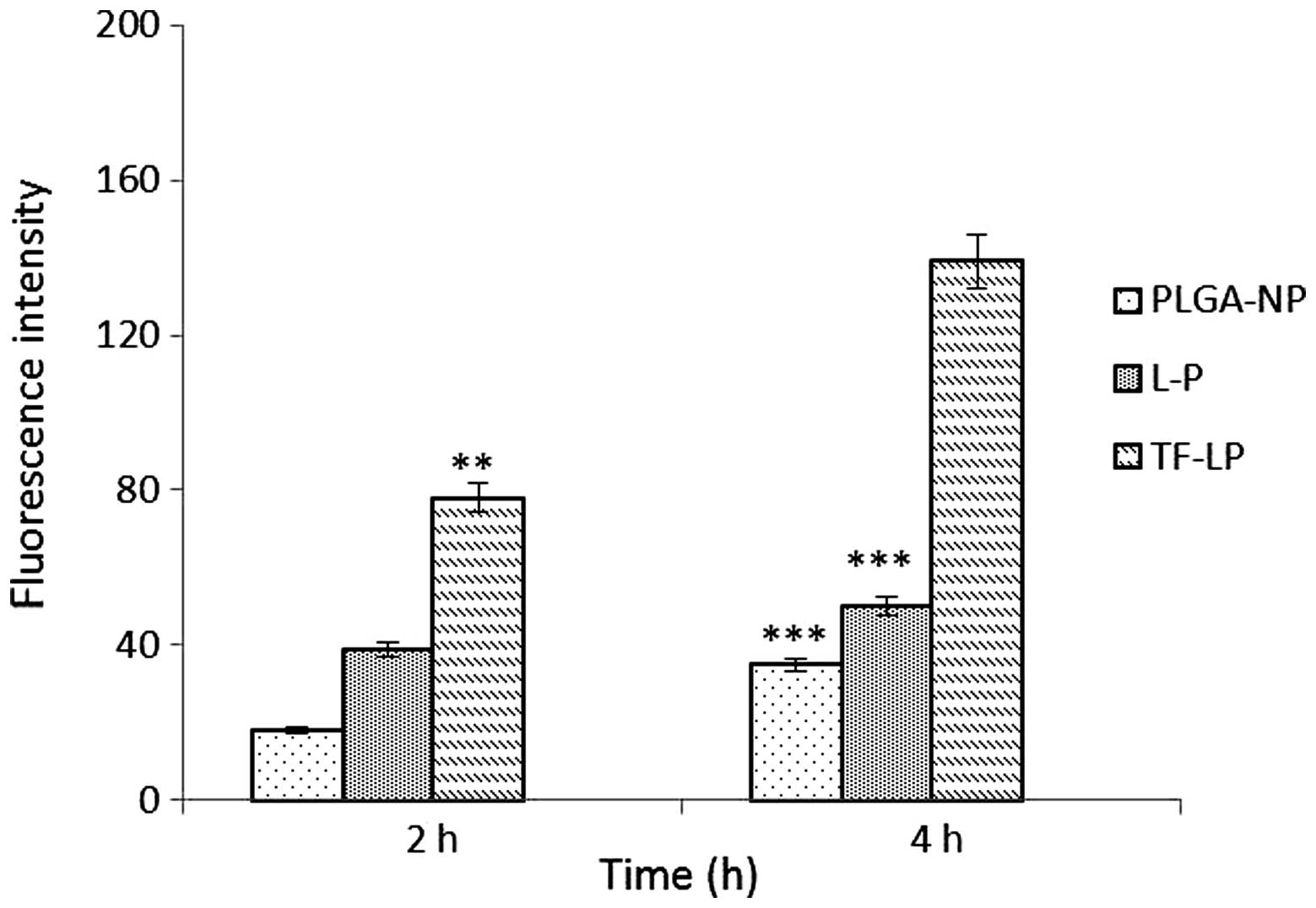
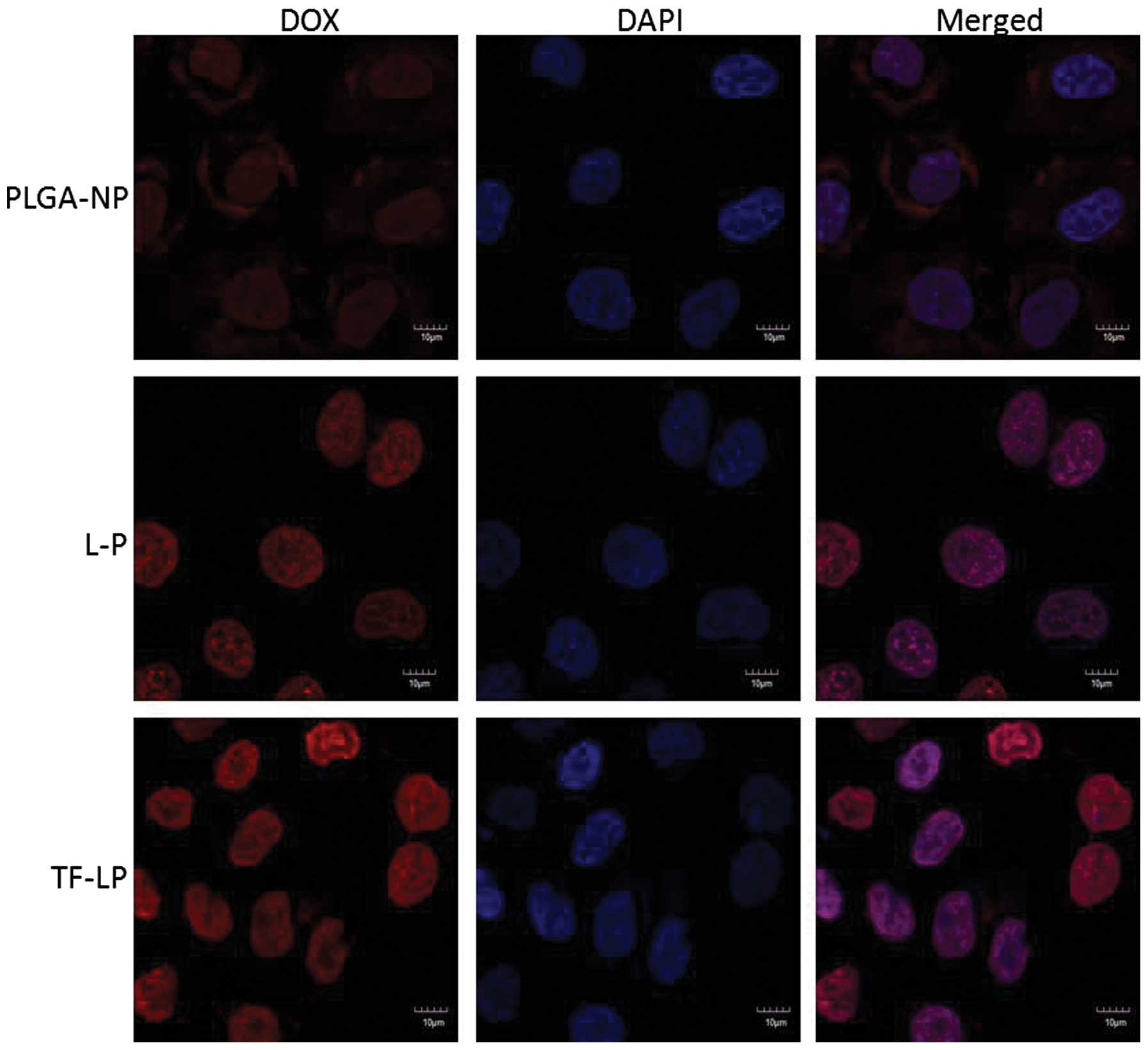
Cellular uptake
The A549 cells were able to take up the DOX-loaded
PLGA-NP, L-P and TF-LP at various capacities (Fig. 4). The TF-LP uptake was ~2.8 and 4.1
times higher compared with L-P and PLGA-NP, respectively. A similar
result was obtained for the targeting capacity of TF receptors. The
fluorescence intensity of TF-LP in the A549 cells was significantly
higher when compared with PLGA-NP and L-P (P<0.001). The
quantitative results indicated analogous results to the
fluorescence imaging shown in Fig.
5. Due to the existence of lipids analogous to cell membrane
components on the surface of L-P, the uptake of the L-P in A549
cells is facilitated by the mutual interaction between L-P and the
cell membrane, resulting in an elevated uptake efficiency compared
with PLGA-NP. For TF-LP, the receptor-mediated endocytosis (RME)
may facilitate the cellular uptake, resulting in an increased
uptake efficiency compared with L-P.
In vitro cytotoxicity and
anti-proliferation assay
The cytotoxic effects of the various DOX
formulations on A549 cells are summarized in Table II. The efficacy of DOX-loaded NPs
was improved by modification with TF. In particular, TF-LP resulted
in decreases of 33.8 and 64.8% in the IC50 values
compared with L-P and PLGA-NPs after 48-h incubation with A549
cells, respectively.
| Table IICytotoxicity against A549 of various
DOX formulations in vitro after 48 h incubation. |
Table II
Cytotoxicity against A549 of various
DOX formulations in vitro after 48 h incubation.
| Formulations | IC50 in
A549 cells, μg/ml |
|---|
| DOX | 0.06200 |
| PLGA-NP-DOX | 0.00938 |
| LP-DOX | 0.00650 |
| TF-LP-DOX | 0.00330 |
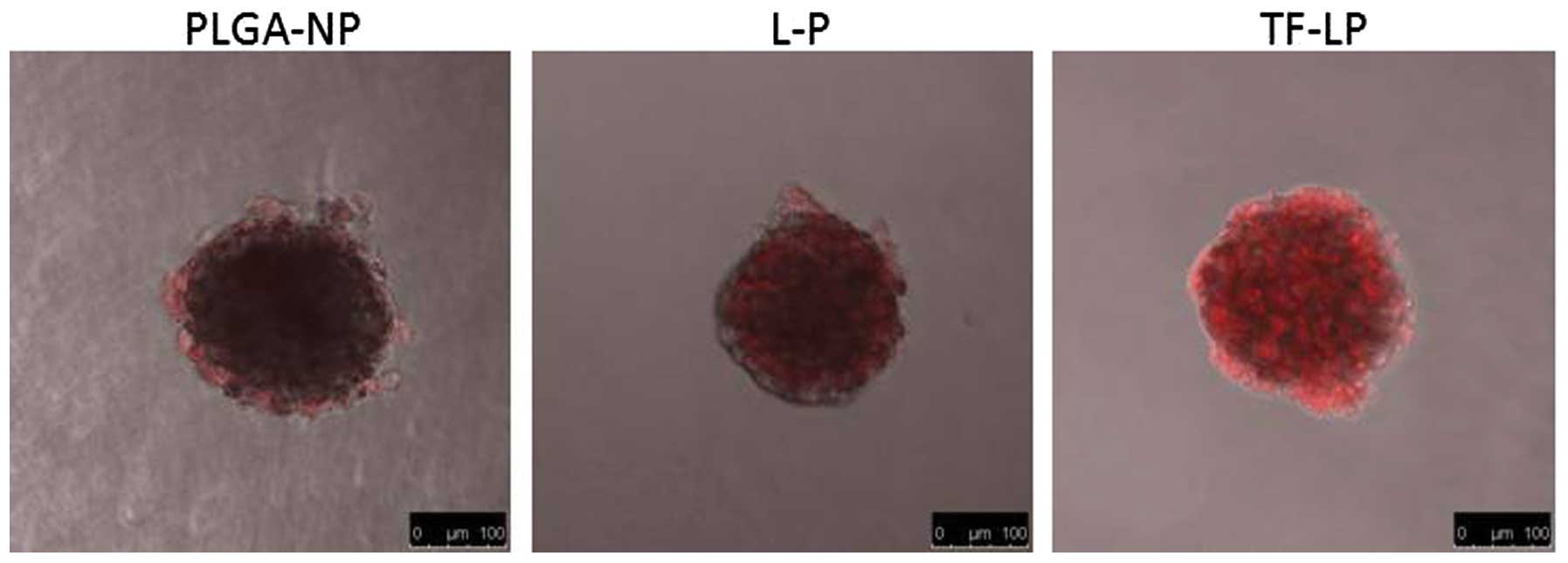
Evaluation of tumor spheroid
penetration
There are hypoxic and avascular regions in numerous
solid tumors. As delivery systems exhibit poor permeation, a low
quantity of the drug accesses the interior of solid tumors. Tumor
spheroids were prepared as they lack blood vessels, which mimics
the in vivo status of tumors (21–23).
The tumor spheroid is an invaluable tool for the evaluation of the
solid tumor penetration effect of NPs. Confocal laser scanning
microscopy images of 3D tumor spheroids 4 h subsequent to the
application of DOX-loaded PLGA-NP, L-P and TF-LP are shown in
Fig. 6. The present results
indicated that the presence of TF-targeting ligand enhanced solid
tumor penetration.
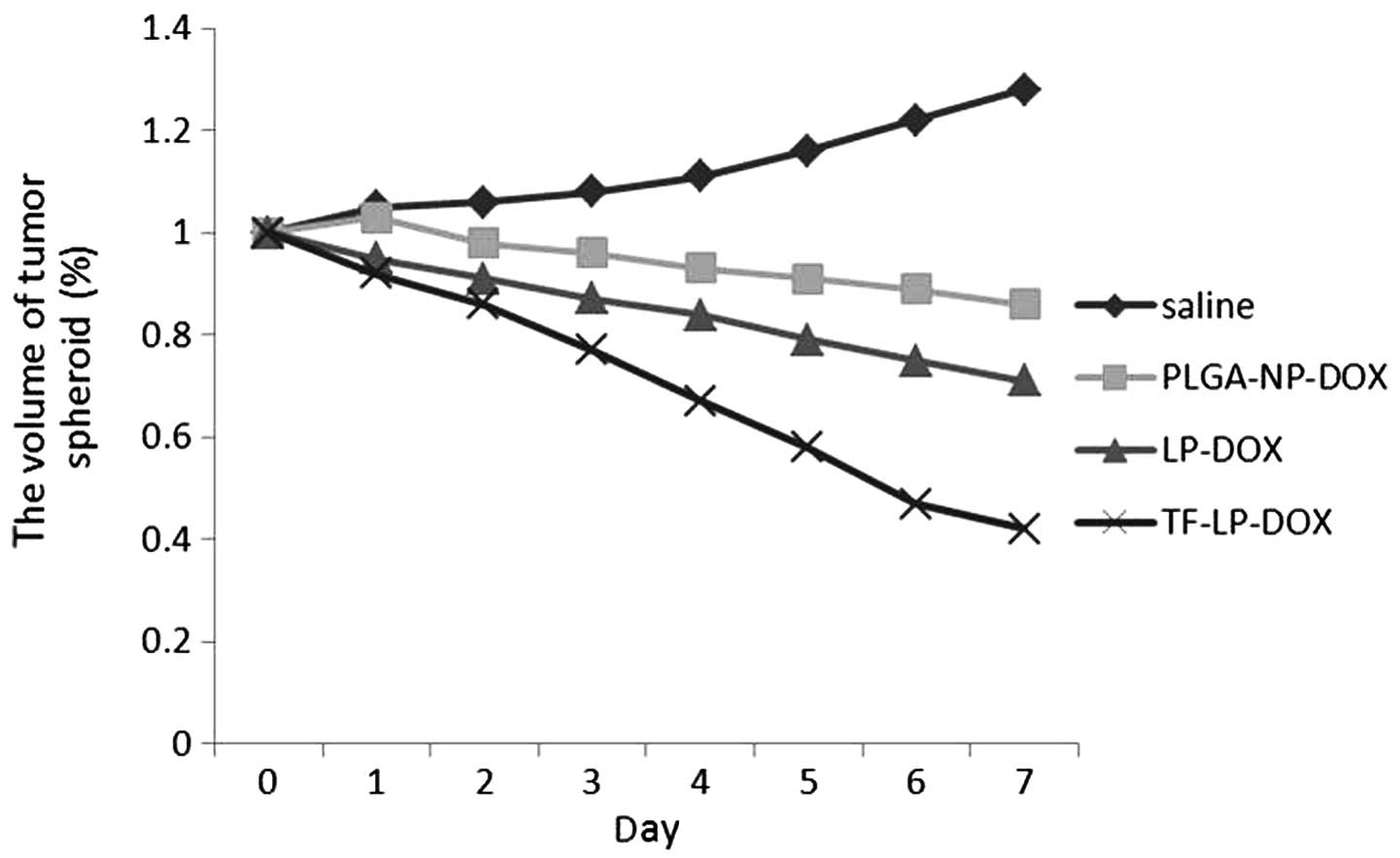
Growth inhibition of tumor spheroids
The present study also investigated the effect of
various treatments on the growth of tumor spheroids. The volume
ratios of the in vitro tumor spheroids subsequent to
treatment with saline, PLGA-NPs, L-Ps and TF-LPs at the final DOX
concentration of 0.25 mg/ml are shown in Fig. 7. In the absence of any drug, the
tumor spheroids were observed to continue to increase in size and
volume, reaching 128% of the primary volume after seven days. A
marked reduction in the volume of tumor spheroids was observed in
all DOX formulations after seven days of treatment, indicating that
the tumor spheroids were sensitive to DOX. The percentage change in
the ratios of tumor spheroid volumes on day seven was almost 86, 71
and 42% for the PLGA-NP, L-P and TF-LP groups, respectively. The
present results indicated that the inhibitory effects of DOX on the
3D tumor spheroids was significantly improved by TF-LP. Solid
tumors contain high-pressure regions with few vessels. This in
vivo status was successfully imitated as the tumor spheroids
lacked blood vessels, and the elevated inhibitory effect indicates
that TF-LP may improve the in vivo therapeutic effect of
chemotherapeutic agents.
In vivo near-infrared (NIR) imaging
A NIR reflection fluorescence probe
1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide
(DIR) was encapsulated in each NP to trace the NP delivery behavior
in mice. As shown in Fig. 8, the
signal intensity in the tumor of TF-LPs at 24 h was stronger
compared with the other groups, which indicated an elevated lung
cancer-targeting property of TF-LPs. There were NIR reflection
fluorescent signals in the excised tumor of each group, and the
intensity of the fluorescence in the TF-LP group was the strongest
compared with all the other groups, indicating an increase in the
delivery of drug to the tumor. Control animals injected with saline
solution produced no fluorescent signals, which confirmed that the
observed fluorescent signal in the experimental groups was derived
from the NPs. It was also observed that there was a slight
difference between the fluorescence intensity in the groups treated
with L-P and PLGA-NP. The biodistribution experiments (Fig. 8) indicated that TF-LP, as a drug
delivery carrier in vivo, was also able to specifically
target therapeutic agents to tumors that overexpress the TF
receptor via TF. It was hypothesized that the high accumulation
effect and the strongest fluorescence intensity of the TF-LP group
were achieved by the following mechanisms, involving two steps.
First, three formulations accumulated in the tumor site and reached
high concentrations in the tumor, due to the enhanced permeability
and retention effect (24).
Secondly, it was hypothesized that TF-LP, which bound to and was
internalized in tumor cells via ligand-receptor interactions, may
lead to a promising accumulation in tumors compared with the other
non-targeting formulations. The non-targeting formulations remained
in the interstitial space and were easily identified, decomposed
and phagocytosed, thereby resulting in drug release outside the
cancer cells (25). In the present
study, the effect of the treatment with TF-LP in vivo and
the biodistribution of DIR-loading TF-LP in the A549-bearing nude
mice indicated that TF-LP may be a novel and potent drug delivery
system for targeting lung cancer and reducing the side-effects of
chemotherapeutic agents to a considerable extent.
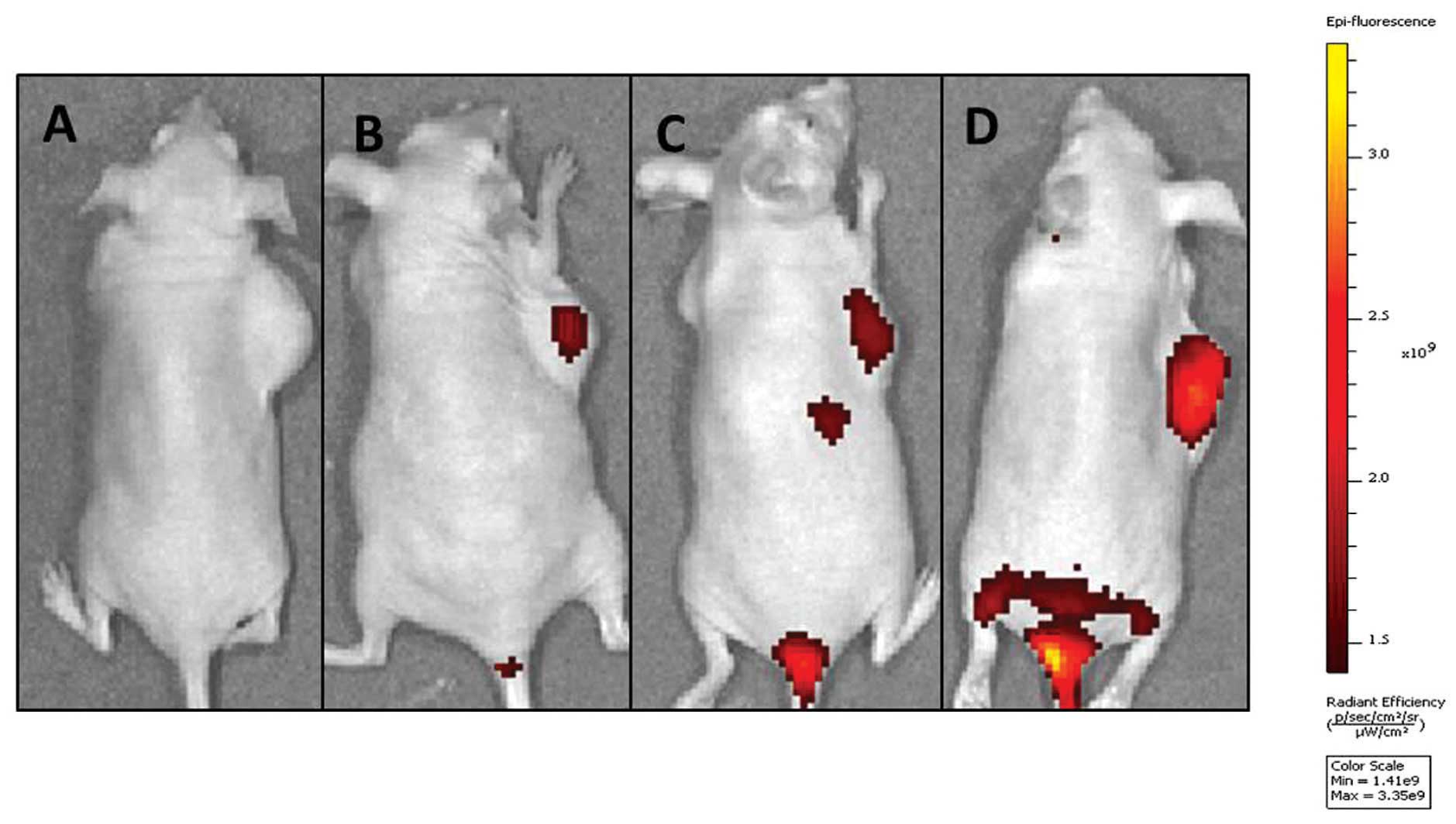 | Figure 8In vivo investigation. Image of
the mice that were anesthetized 24 h after intravenous injection of
various types of
1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine
iodide-loaded nanoparticles, respectively. Mice were injected with
(A) saline, (B) PLGA-NP, (C) L-P or (D) TF-LP. Imaging revealed
that the accumulation of nanoparticles in the tumor was highest for
TF-LP, when compared with the other nanoparticles. PLGA-NP,
lipid-coated poly D,L-lactic-co-glycolic acid nanoparticles;
L-P, lipid-coated PGLA-NPs; TF-LP, transferrin-modified LPs. |
Conclusion
The present study successfully synthesized a
targeted-NP drug-delivery platform that was specific to lung cancer
cells using TF and biomaterials approved by the Food and Drug
Administration. The particle size, surface charge, and drug loading
yield drug release rate, which are factors that may be controlled
for specific therapeutic applications, were characterized. The data
from the present in vitro DOX release experiments revealed
that lipid-coated NPs undergo a sustainable, controlled release of
DOX. The targeting specificity of the synthesized NPs was
demonstrated, along with the enhanced cytotoxicity of the NPs
against target cells and tumor spheroids compared with the
non-targeted cells. In addition, the DOX-loaded TF-LP exhibited
evident antitumor effects in lung cancer-bearing mice. The present
platform exhibits considerable therapeutic potential due to the
effective delivery of a variety of chemotherapeutic agents to lung
cancer tumors in a targeted manner.
References
|
1
|
Jinturkar KA, Anish C, Kumar MK, et al:
Liposomal formulations of Etoposide and Docetaxel for p53 mediated
enhanced cytotoxicity in lung cancer cell lines. Biomaterials.
33:2492–2507. 2012. View Article : Google Scholar
|
|
2
|
Sengupta S, Tyagi P, Velpandian T, et al:
Etoposide encapsulated in positively charged liposomes:
pharmacokinetic studies in mice and formulation stability studies.
Pharmacol Res. 42:459–464. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li R, Zhang Q, Wang XY, et al: A targeting
drug delivery system for ovarian carcinoma: transferring modified
lipid coated paclitaxel-loaded nanoparticles. Drug Res (Stuttg).
64:541–547. 2014. View Article : Google Scholar
|
|
4
|
Oh S, Kim BJ, Singh NP, et al: Synthesis
and anti-cancer activity of covalent conjugates of artemisinin and
a transferrin-receptor targeting peptide. Cancer Lett. 274:33–39.
2009. View Article : Google Scholar
|
|
5
|
Hu CM and Zhang L: Therapeutic
nanoparticles to combat cancer drug resistance. Curr Drug Metab.
10:836–841. 2009. View Article : Google Scholar
|
|
6
|
Torchilin VP: Recent advances with
liposomes as pharmaceutical carriers. Nat Rev Drug Discov.
4:145–160. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang AZ, Bagalkot V, Vasilliou CC, et al:
Superparamagnetic iron oxide nanoparticle-aptamer bioconjugates for
combined prostate cancer imaging and therapy. ChemMedChem.
3:1311–1315. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nakatsuji T, Kao MC, Zhang L, et al: Sebum
free fatty acids enhance the innate immune defense of human
sebocytes by upregulating beta-defensin-2 expression. J Invest
Dermatol. 130:985–994. 2010. View Article : Google Scholar
|
|
9
|
Hu CM, Kaushal S, Tran Cao HS, et al:
Half-antibody functionalized lipid-polymer hybrid nanoparticles for
targeted drug delivery to carcinoembryonic antigen presenting
pancreatic cancer cells. Mol Pharm. 7:914–920. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kibria G, Hatakeyama H, Ohga N, et al:
Dual-ligand modification of PEGylated liposomes shows better cell
selectivity and efficient gene delivery. J Control Release.
153:141–148. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sharma G, Modgil A, Sun C and Singh J:
Grafting of cell-penetrating peptide to receptor-targeted liposomes
improves their transfection efficiency and transport across
blood-brain barrier model. J Pharm Sci. 101:2468–2478. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wadia JS and Dowdy SF: Protein
transduction technology. Curr Opin Biotechnol. 13:52–56. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang H, Zhao Y, Wu Y, et al: Enhanced
anti-tumor efficacy by co-delivery of doxorubicin and paclitaxel
with amphiphilic methoxy PEG-PLGA copolymer nanoparticles.
Biomaterials. 32:8281–8290. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cui Y, Xu Q, Chow PK, et al:
Transferrin-conjugated magnetic silica PLGA nanoparticles loaded
with doxorubicin and paclitaxel for brain glioma treatment.
Biomaterials. 34:8511–8520. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chan JM, Zhang L, Yuet KP, et al:
PLGA-lecithin-PEG core-shell nanoparticles for controlled drug
delivery. Biomaterials. 30:1627–1634. 2009. View Article : Google Scholar
|
|
16
|
Zhang L, Chan JM, Gu FX, et al:
Self-assembled lipid-polymer hybrid nanoparticles: a robust drug
delivery platform. ACS Nano. 2:1696–1702. 2008. View Article : Google Scholar
|
|
17
|
Yang X, Koh CG, Liu S, et al: Transferrin
receptor-targeted lipid nanoparticles for delivery of an antisense
oligodeoxyribonucleotide against Bcl-2. Mol Pharm. 6:221–230. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chiu SJ, Liu S, Perrotti D, et al:
Efficient delivery of a Bcl-2-specific antisense
oligodeoxyribonucleotide (G3139) via transferrin receptor-targeted
liposomes. J Controlled Release. 112:199–207. 2006. View Article : Google Scholar
|
|
19
|
Jiang T, Zhang Z, Zhang Y, et al:
Dual-functional liposomes based on pH-responsive cell-penetrating
peptide and hyaluronic acid for tumor-targeted anticancer drug
delivery. Biomaterials. 33:9246–9258. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maeda N, Takeuchi Y, Takada M, et al:
Anti-neovascular therapy by use of tumor neovasculature-targeted
long-circulating liposome. J Control Release. 100:41–52. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lewis CE and Pollard JW: Distinct role of
macrophages in different tumor microenvironments. Cancer Res.
66:605–612. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fukumura D, Xu L, Chen Y, Gohongi T, et
al: Hypoxia and acidosis independently up-regulate vascular
endothelial growth factor transcription in brain tumors in vivo.
Cancer Res. 61:6020–6024. 2001.PubMed/NCBI
|
|
23
|
Jain RK: Delivery of molecular and
cellular medicine to solid tumors. Adv Drug Deliv Rev. 64(Suppl):
353–365. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang X, Yang L, Chen ZG and Shin DM:
Application of nanotechnology in cancer therapy and imaging. CA
Cancer J Clin. 58:97–110. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen X, Wang X, Wang Y, et al: Improved
tumor-targeting drug delivery and therapeutic efficacy by cationic
liposome modified with truncated bFGF peptide. J Control Release.
145:17–25. 2010. View Article : Google Scholar : PubMed/NCBI
|