Introduction
Cervical cancer is an major cause of mortality for
females, with the worldwide incidence of cervical cancer reaching
454,000 cases per year in 2010 (1).
Although persistent infection by human papillomavirus (HPV) is
necessary to cause cervical cancer, it is not sufficient for
disease progression (2). Other
etiological factors, including smoking, are also known to
contribute to cervical cancer (3).
Genetically, the HPV ‘early’ proteins, E6 and E7, are the primary
oncoproteins involved in cancer progression and interact with
several cellular proteins, including tumor protein 53 (TP53),
retinoblastoma 1 (RB1), Bcl-2 antagonist killer (BAK),
Fas-associated death domain (FADD) and insulin-like growth
factor-binding protein 3 (IGFBP3), involving cell adhesion,
apoptosis and the cell cycle (4–6).
DNA methylation is a well-known epigenetic marker,
as is histone modification (7). The
DNA hypomethylation and hypermethylation of several cancer-related
genes have been extensively studied (8–10). In
cervical cancer, DNA methylation also affects the expression of
several genes, including CDH1, DAPK, TIMP-3,
p16 (INK4a), FHIT and RASSF1A (11,12).
Unlike studies involving targeted candidate gene approaches, there
have been few reports regarding genome-wide methylation profiles
for investigating the methylation status of the whole genome region
by using a high-density microarray-based platform. Recently, two
differentially-methylated regions, COL25A1 and
KATNAL2, were reported as candidate biomarkers in a
genome-wide survey using a high-density microarray system (13). Furthermore, a few studies have
conducted integrated analyses of DNA methylation and RNA expression
in breast and ovarian cancer, but not in cervical cancer (14,15).
In the present study, genome-wide DNA methylation
was examined, RNA expression profiles were analyzed and an
integrated analysis of these two profiles was conducted to identify
differentially-expressed DNA methylation-regulated genes that are
involved in cervical cancer. Finally, the study identified several
novel genes associated with cervical cancer that showed
differential RNA expression by DNA methylation in tumor regions and
validated the epigenetic regulation of selected genes by treatment
with a demethylating reagent of the cervical cancer cell line
HeLa.
Materials and methods
Tissues, cell lines and reagents
Normal and tumor regions of cervical tissues were
biopsied from four patients, two pre-invasive cancer subjects (CIN
III and CIS), and two patients with cervical cancer (CxCa Ib and
CxCa IIa), and stored at the Kangdong Sacred Heart Hospital (Hallym
University, Seoul, South Korea) following international ethical
guidelines. All the patients provided written informed consent.
This study was approved by the Kangdong Sacred Heart Hospital
Institutional Review Board. Genomic DNA and RNA were purified from
frozen tissues. HeLa cells were obtained from the American Type
Culture Collection (Manassas, VA, USA) and cultured in Dulbecco’s
modified Eagle’s medium (Gibco-BRL, Grand Island, NY, USA)
supplemented with 10% fetal bovine serum, 100 U/ml penicillin and
100 μg/ml streptomycin at 37°C in a humidified CO2
incubator.
Global gene expression profiles
Total RNA was extracted using TRIzol®
(Invitrogen Life Technologies, Carlsbad, CA, USA) and further
purified using RNeasy columns (Qiagen, Hilden, Germany) according
to the manufacturer’s instructions. RNA purity and integrity were
evaluated by gel electrophoresis and the ratio of absorbance at 260
and 280 nm using the NanoDrop® ND-1000 (Thermo
Scientific, Waltham, MA, USA). Samples were further analyzed on an
Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA,
USA). Microarray analysis using human HT-12 expression v.4 bead
array (47,000 probes; Illumina, Inc., San Diego, CA, USA) was
carried out by Macrogen Co. (Seoul, South Korea) following the
manufacturer’s instructions. Raw data were extracted using the
software provided by the manufacturer [Illumina GenomeStudio
(v2011.1) Gene Expression Module (v1.9.0)] and filtered if the
P-value was <0.05, similar to signal to noise ratio, in ≥50% of
the samples analyzed. Signal values of the selected genes were
transformed using logarithms and normalized based on the quantile
method. The statistical significance of the expression data was
determined using an independent Student’s t-test and fold change,
in which the null hypothesis was such that no difference existed
between the two groups. The false discovery rate was controlled by
adjusting the P-value using the Benjamini-Hochberg algorithm. Gene
enrichment and functional annotation analysis was performed using
Panther DB (http://www.pantherdb.org/). All data
analysis and visualization of differentially expressed genes was
conducted using The R project (www.r-project.org).
Genome-wide DNA methylation profiles
Genomic DNA was purified using the DNeasy mini-kit
(Qiagen), and sample quality was evaluated using the NanoDrop
ND-1000. Intact genomic DNA was diluted to 50 ng/μl based on
Quant-iT Picogreen (Invitrogen Life Technologies) quantitation.
Microarray analysis using Infinium Human Methylation 450K BeadChip
(Illumina, Inc.) was carried out by Macrogen Co., followed by the
manufacturer’s instructions. Image processing and raw data
extraction were performed using GenomeStudio software [Illumina
GenomeStudio (v2011.1) Methylation Module (v1.9.0)] following the
manufacturer’s instructions. The β-value was calculated by
subtracting the background signals using negative controls on the
array and determining the ratio of the methylated signal intensity
against the sum of the methylated and unmethylated signals. A
β-value of 0–1.0 was reported as a significant percentage of
methylation, 0–100%, respectively, for each CpG site (16). Array CpG probes with a detection
P-value of ≥0.05, similar to the signal to noise ratio, in >50%
samples were filtered out. A filtering criterion was applied for
data analysis; a good signal value was required to obtain a
detection P-value of <0.05. Filtered data was normalized using
the quantile method. Differential methylation was determined based
on |Δ_mean| ≥0.2 (difference in methylation signal = average β of
case - average β of control).
Integrated analysis of DNA methylation
and gene expression and selection of differentially-expressed mRNA
and CpGs
Statistically significant mRNA and CpGs were
identified by |fold change| ≥2 and |Δ_mean| ≥0.2, respectively.
Next, putative mRNA-CpG target pairs with regulatory associations
were extracted. This approach assumed that DNA methylation is
negatively correlated with the mRNA expression of its targets.
Negative correlation was identified between putative pairs of mRNA
and CpGs. The novelty of selected genes with cervical cancer was
confirmed in cervical cancer gene database (CCDB; http://crdd.osdd.net/raghava/ccdb) (17). To reduce the bias of the present
data, which came from a limited number of samples, the expression
of selected genes was confirmed in a publicly accessible microarray
database, the gene expression database across normal and tumor
tissues (GENT; http://medicalgenome.kribb.re.kr/GENT) (18). All statistical data analysis was
conducted using R 2.15.1 (www.rproject.org).
Functional annotation clustering of
selected genes and clustering and functional annotation of Database
of Annotation, Visualization and Integrated Discovery (DAVID)
analysis
Hierarchical clustering of DNA methylation and RNA
expression profiles was conducted using Cluster 3.0 (19). Input data was analyzed by array mean
centering and followed by average linkage clustering using
correlation (centered) similarity metrics. Clustering results were
viewed using TreeView version 1.60 (Eisen Lab, Berkeley, CA, USA).
Functional annotation clustering and pathway analysis were
conducted using DAVID (20).
Functional clustering analysis of all selected genes was performed
using medium classification stringency (κ similarity: Similarity
term overlap, 3; threshold, 0.5; Classification: Initial and final
group membership, 3 and multiple linkage threshold, 0.5).
Demethylation by treatment with
5-aza-2′-deoxycytidine
HeLa cells (4.0×104) were plated in a
six-well plate and treated with various concentrations (0, 5, 10
and 20 μM) of 5-aza-2′-deoxycytidine as decitabine (Selleckchem;
Houston, TX, USA) for 96 h. Total DNA and RNA were isolated using
the DNeasy and RNeasy mini kits, respectively.
Reverse transcription (RT) quantitative
polymerase chain reaction (PCR) analyses
RT was performed with 2 μg total RNA using the
First-Strand cDNA Synthesis kit (Invitrogen Life Technologies)
following previously described procedures (21). To quantify the expression levels of
the selected genes, quantitative PCR analysis was performed using
the first cDNAs as templates and a specific primer set for each
gene, ALDH1A3 (P290199), PPP1R3C (P131676) and
CAMK2N1 (P229340) (Bioneer Co., Daejeon, Korea) following
the manufacturer’s instructions. Gene amplification was performed
using the ABI® PRISM 7900HT Sequence Detection System
(Applied Biosystems Life Technologies, Foster City, CA, USA) in a
20 μl reaction mixture containing 2 μl cDNA template (80 ng), 2.5
μl of each primer, 3 μl distilled water and 10 μl 2X Power SYBR
Green PCR Master mix (Applied Biosystems Life Technologies), which
included a dNTP mixture. PCR conditions were as follows: Initial
denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for
10 sec and 60°C for 30 sec. Sequence Detection System (SDS)
Software version 2.4 (Applied Biosystems Life Technologies) was
used to calculate Ct values for all genes. Relative expression was
calculated using the 2−ΔΔCt method.
Methylation-specific (MS)-PCR
analysis
Genomic DNA was modified using the EpiTect Bisulfite
kit (Qiagen), which converts cytosine residues to uracil, but does
not affect 5-methylcytosine, thus allowing identification of
methylated sequences in the DNA by subsequent MS-PCR analysis. For
MS-PCR analysis, MS primers (MSP) and unmethylation-specific
primers were designed using methylation identification software
(22). PCR was then performed in a
final volume of 50 μl using 100 μg bisulfite modified DNA and 10
pmol of each primer using the EpiTect MSP kit (Qiagen) under the
following conditions: Initial denaturation at 95°C for 5 min, 35
cycles of 95°C for 30 sec, 55°C for 30 sec and 72°C for 30 sec, and
a final extension step of 72°C for 5 min. PCR products (20 μl) were
visualized on 1.8% agarose gels using the Gel Documentation System
(AE-9000 E-graph; ATTO Corporation, Tokyo, Japan).
Results
Identification of novel genes regulated
by DNA methylation in cervical cancer
Data from DNA methylation and RNA expression was
analyzed using an integrated approach and strong cut-off values to
overcome the limited number of samples. For
differentially-methylated genes in cervical cancer tissues,
methylation loci with an absolute value ≥0.2 of the Δ_mean in the
methylation profile were selected from the data set. To select
genes showing differential expression in cancer tissues, genes with
≥2-fold changes in cancer tissues were selected. The data from
selected methylated loci and RNA expression were then merged. To
prevent the selection of genes with individual variations, genes
showing no methylation in the DNA methylation profiles and no
change in the expression profiles in one of two individuals were
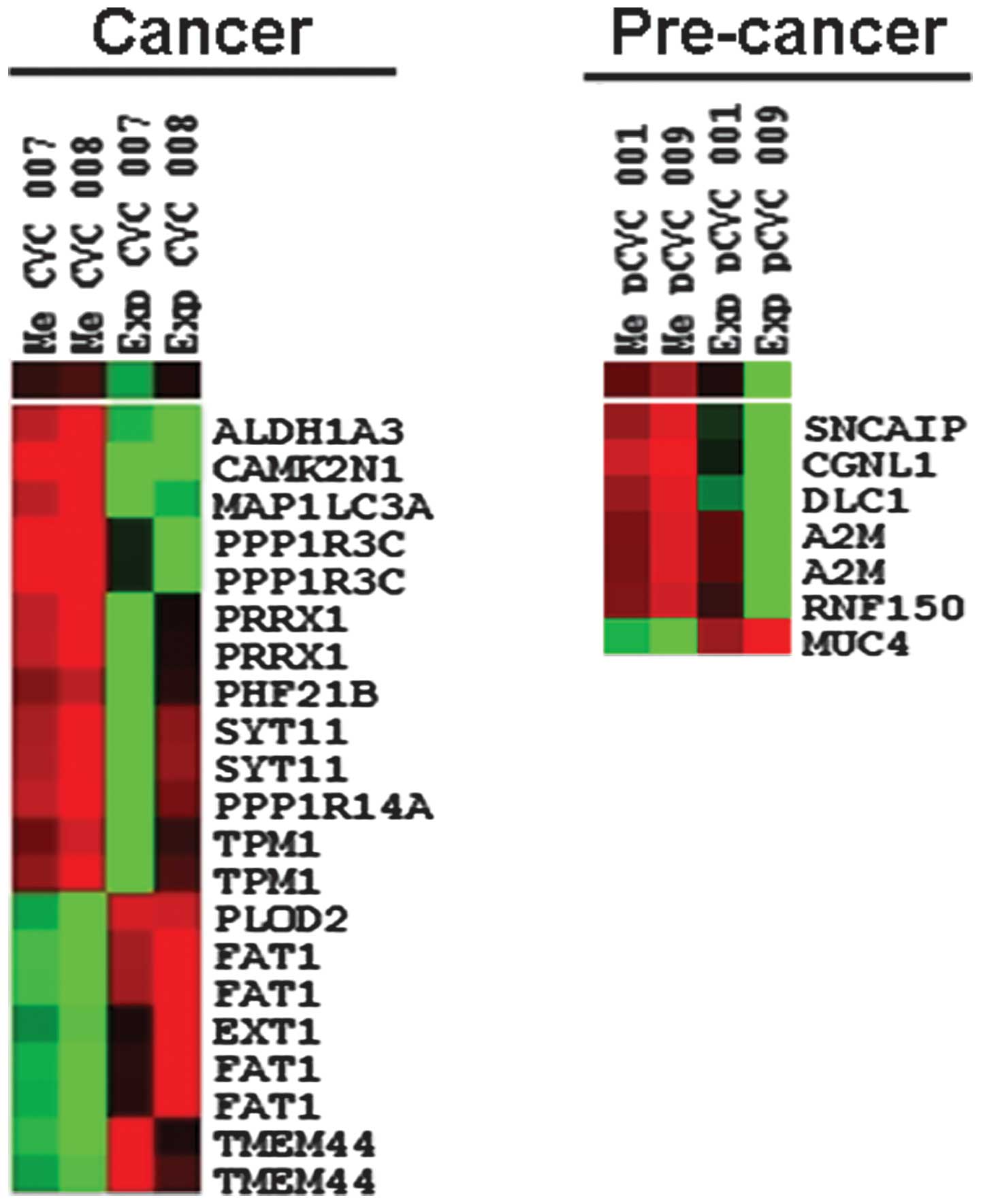
removed. Finally, nine genes that were downregulated by
hypermethylation and four genes that were upregulated by
hypomethylation in the cancer tissues were selected. Five genes
that were downregulated by hypermethylation and one gene that was
upregulated by hypomethylation in the pre-cancer tissues were also
selected (Table I).
 | Table IDifferentially-expressed and
methylated genes in tumor tissues compared with the normal tissues
of patients with cervical cancer. |
Table I
Differentially-expressed and
methylated genes in tumor tissues compared with the normal tissues
of patients with cervical cancer.
| Gene | Accession
number | Name | Δ value | Fold change | CCDB | GENT |
|---|
| Downregulated genes
by hypermethylation in cancer | | | | | | |
| SYT11 | NM_152280.2 | Synaptotagmin
XI | 0.32 | −2.70 | NA | Down |
|
CAMK2N1 | NM_018584.4 |
Calcium/calmodulin-dependent protein
kinase II inhibitor 1 | 0.28 | −4.95 | NA | Up |
|
ALDH1A3 | NM_000693.1 | Aldehyde
dehydrogenase 1 family, member A3 | 0.27 | −3.08 | NA | Not changed |
| PRRX1 | NM_006902.3 | Paired related
homeobox 1, transcript variant pmx-1a | 0.25 | −3.18 | NA | Down |
|
PPP1R3C | NM_005398.3 | Protein phosphatase
1, regulatory (inhibitor) subunit 3C | 0.22 | −6.18 | Down | Down, highly |
|
MAP1LC3A | NM_181509.1 |
Microtubule-associated protein 1 light
chain 3α, transcript variant 2 | 0.22 | −3.14 | NA | Down |
| PHF21B | NM_138415.1 | PHD finger protein
21B | 0.20 | −2.06 | NA | Down, highly |
| TPM1 | NM_001018004.1 | Tropomyosin 1 (α),
transcript variant 7 | 0.20 | −2.07 | NA | Down, highly |
|
PPP1R14A | NM_033256.1 | Protein phosphatase
1, regulatory (inhibitor) subunit 14A | 0.20 | −2.91 | NA | Down, highly |
| Upregulated genes
by hypomethylation in cancer | | | | | | |
| PLOD2 | NM_000935.2 | Procollagen-lysine,
2-oxoglutarate 5-dioxygenase 2, transcript variant 2 | −0.25 | 2.66 | Up | Up, highly |
| FAT1 | NM_005245.3 | FAT tumor
suppressor homolog 1 (Drosophila) | −0.23 | 3.44 | NA | NA |
| EXT1 | NM_000127.2 | Exostoses
(multiple) 1 | −0.22 | 2.40 | NA | Up |
| TMEM44 | NM_138399.3 | Transmembrane
protein 44, transcript variant 1 | −0.21 | 3.02 | NA | Up |
| Downregulated genes
by hypermethylation in pre-cancer | | | | | | |
| SNCAIP | NM_005460.2 | Synuclein, alpha
interacting protein | 0.23 | −2.39 | NA | Down |
| A2M | NM_000014.4 |
α-2-macroglobulin | 0.21 | −2.22 | Up | Down |
| CGNL1 | NM_032866.3 | Cingulin-like
1 | 0.21 | −3.14 | NA | Down, highly |
| DLC1 | NM_024767.2 | Deleted in liver
cancer 1, transcript variant 3 | 0.20 | −2.45 | Down | Down, highly |
| RNF150 | NM_020724.1 | Ring finger protein
150 | 0.20 | −2.18 | NA | Down, highly |
| Upregulated genes
by hypomethylation in pre-cancer | | | | | | |
| MUC4 | NM_018406.3 | Mucin 4, cell
surface-associated, transcript variant 1 | −0.21 | 2.94 | NA | Up |
In silico analysis of the association of
selected genes with cervical cancer
Next, the selected genes were analyzed using a
publicly available database. Firstly, it was determined whether the
genes had been reported as cervical cancer-related genes in the
CCDB. As shown in Table I, only one
of the downregulated genes in the tumor tissues, PPP1R3C,
had previously been reported to be downregulated in cervical
cancer. Almost all the selected genes were identified as novel
genes associated with cervical cancer. Secondly, to elucidate
whether the selected genes are expressed differentially in cervical
cancer, the RNA expression patterns of genes in the tissue samples
from patients with cervical cancer were analyzed using GENT. Four
genes, PPP1R3C, PHF21B, TPM1 and
PPP1R14A, were highly downregulated in the tumor tissues
compared with the normal tissues. In addition, three genes,
MAP1LC3A, PRRX1 and SYT11, were moderately
downregulated in the tumor tissues. Other genes, CAMK2N1 and
ALDH1A3, showed no change or only minimal upregulation in
the tumor tissues. These expression patterns were similar to the
RNA expression data. The same analysis was conducted using genes
upregulated by hypomethylation in cancer and using genes up- or
downregulated in pre-cancer. Seven genes were determined as novel
genes associated with cervical cancer, since they had not been
reported in the CCDB. The majority of those genes, excluding
FAT1, which appeared as not applicable in GENT, were up- or
downregulated in the tumor tissue samples of patients with cervical
cancer. GENT and CCDB data supported the observed association of
the selected genes with cervical cancer.
Functional annotation clustering of
selected genes and clustering and functional annotation of DAVID
analysis
To confirm the negative correlation between DNA
methylation and RNA expression profiles associated with the
progression of cervical cancer, 19 genes that were selected on the
basis of integrated analysis were clustered. Common methylation and
expression patterns were visualized by unsupervised hierarchical
clustering using an average linkage clustering method. As shown in
Fig. 1, a separate cluster was
formed by methylation and expression data. A negative correlation
of methylation and expression was observed using TreeView.
Functional annotation clustering was conducted using DAVID with
default conditions to analyze the biological significance of the 19
selected genes. This showed that the selected genes were classified
into five clusters to be enriched in the cell junction,
membrane/plasma membrane, cytoskeletal part, glycoprotein and
ion/metal binding (Table II).
| Table IIFunctional annotation clustering
(gene ontology) of 19 selected genes showing five clusters. |
Table II
Functional annotation clustering
(gene ontology) of 19 selected genes showing five clusters.
| Annotation
cluster | Enrichment
category | Count | P-value | False discovery
ratea |
|---|
| 1 | | | | |
| GOTERM_CC_FAT | Cell junction | 4 |
1.70×10−2 |
8.20×10−1 |
| GOTERM_CC_FAT | Plasma membrane
part | 7 |
2.20×10−2 |
6.80×10−1 |
| GOTERM_CC_FAT | Plasma
membrane | 7 |
2.10×10−1 |
9.30×10−1 |
| 2 | | | | |
| GOTERM_CC_FAT | Cytoskeletal
part | 4 |
8.10×10−2 |
8.10×10−1 |
| GOTERM_CC_FAT | Cytoskeleton | 4 |
1.90×10−1 |
9.20×10−1 |
| GOTERM_CC_FAT | Intracellular
non-membrane-bound organelle | 4 |
5.60×10−1 | 1.00 |
| GOTERM_CC_FAT | Non-membrane-bound
organelle | 4 |
5.60×10−1 | 1.00 |
| 3 | | | | |
|
SP_PIR_KEYWORDS | Membrane | 9 |
2.00×10−1 |
9.60×10−1 |
| GOTERM_CC_FAT | Integral to plasma
membrane | 3 |
3.80×10−1 |
9.90×10−1 |
| GOTERM_CC_FAT | Intrinsic to plasma
membrane | 3 |
3.90×10−1 |
9.90×10−1 |
|
UP_SEQ_FEATURE | Topological domain:
Cytoplasmic | 5 |
4.00×10−1 | 1.00 |
|
UP_SEQ_FEATURE | Transmembrane
region | 6 |
5.10×10−1 | 1.00 |
|
SP_PIR_KEYWORDS | Transmembrane | 6 |
5.20×10−1 |
9.90×10−1 |
| GOTERM_CC_FAT | Intrinsic to
membrane | 7 |
6.00×10−1 | 1.00 |
|
UP_SEQ_FEATURE | Topological domain:
Extracellular | 3 |
7.50×10−1 | 1.00 |
| GOTERM_CC_FAT | Integral to
membrane | 6 |
7.60×10−1 | 1.00 |
| 4 | | | | |
|
UP_SEQ_FEATURE | Glycosylation site:
N-linked (GlcNAc) | 6 |
3.50×10−1 | 1.00 |
|
SP_PIR_KEYWORDS | Signal | 5 |
3.60×10−1 |
9.90×10−1 |
|
UP_SEQ_FEATURE | Signal peptide | 5 |
3.70×10−1 | 1.00 |
|
SP_PIR_KEYWORDS | Glycoprotein | 6 |
3.80×10−1 |
9.90×10−1 |
|
UP_SEQ_FEATURE | Disulfide bond | 3 |
7.70×10−1 | 1.00 |
|
SP_PIR_KEYWORDS | Disulfide bond | 3 |
7.80×10−1 | 1.00 |
| 5 | | | | |
|
SP_PIR_KEYWORDS | Metal-binding | 4 |
5.40×10−1 |
9.90×10−1 |
| GOTERM_MF_FAT | Metal ion
binding | 5 |
8.00×10−1 | 1.00 |
| GOTERM_MF_FAT | Cation binding | 5 |
8.10×10−1 | 1.00 |
| GOTERM_MF_FAT | Ion binding | 5 |
8.20×10−1 | 1.00 |
| GOTERM_MF_FAT | Transition metal
ion binding | 3 |
8.90×10−1 | 1.00 |
RT-PCR analysis for the validation of DNA
methylation by MSP and RNA expression using the cervical cancer
cell line HeLa, following treatment with 5-aza-2′deoxycytidine
To further confirm whether the selected genes were
regulated by DNA methylation, the HeLa cells were treated with a
demethylating reagent, and the methylation status and RNA
expression level was measured by MS-PCR using each primer set
(Table III) and RT-quantitative
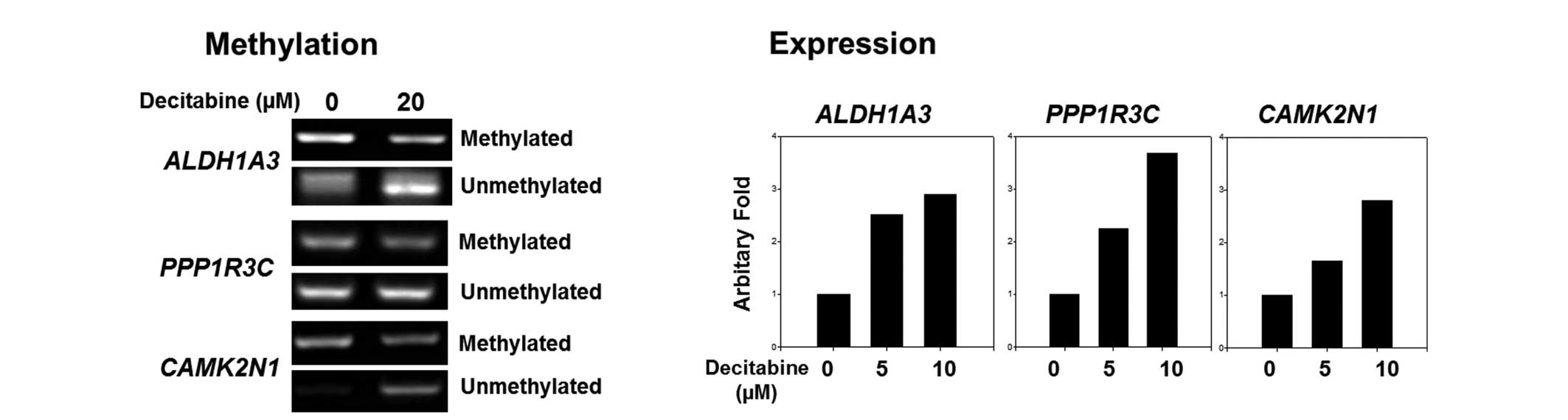
PCR analysis, respectively. Methylation of selected genes,
CAMK2N1, ALDH1A3 and PPP1R3C, was found to be
decreased, while demethylation was increased following treatment
with the demethylating reagent, as expected. Additionally, RNA
expression was increased dose-dependently as a result of the
decreased methylation status (Fig.
2). These results indicated that the genes were originally
methylated in cervical cancer cells and showed increased
demethylation followed by increased RNA expression following
treatment with a demethylating reagent.
| Table IIIPrimers sequences for
methylation-specific polymerase chain reaction. |
Table III
Primers sequences for
methylation-specific polymerase chain reaction.
| Gene | Nature of sequence
primer | Sense | Antisense | Product size,
bp |
|---|
| CAMK2N1 | Methylated |
AATTAGGAGGGGACGTTAAAATC |
ACATATATCCCTAACAAACAACGAA | 185 |
| Unmethylated |
GAATTAGGAGGGGATGTTAAAATT |
ACATATATCCCTAACAAACAACAAA | 186 |
| ALDH1A3 | Methylated |
GTTCGTTTATTGACGAAATTTTTTC |
AAAAACCGTACGCTTCTACGAC | 134 |
| Unmethylated |
TTGTTTATTGATGAAATTTTTTTGG |
ACAAAAAACCATACACTTCTACAAC | 135 |
| PPP1R3C | Methylated |
ATTTGTTTTTAAGTACGTGATTCGA |
GATACCCAAATAACTCTCTACACGTC | 153 |
| Unmethylated |
ATTTGTTTTTAAGTATGTGATTTGA |
AATACCCAAATAACTCTCTACACATC | 153 |
Discussion
In the present study, genome-wide DNA methylation
and RNA expression profiling was conducted to identify novel genes
associated with cervical cancer. An integrated analysis of the
methylation and expression profiles was conducted and
differentially-expressed genes regulated by DNA methylation were
selected. To determine whether these genes were associated with
cervical cancer, CCDB and GENT were used. The selected genes were
also confirmed as novel epigenetic markers by treating HeLa cells
with a demethylating reagent and measuring DNA methylation status
and RNA expression.
To identify novel genes associated with cervical
cancer, the majority of studies have focused on genome-wide RNA
expression analysis (23,24). DNA methylation is another natural
mechanism of regulating specific gene expression (7). Using high-throughput analysis,
including microarray-based systems, global DNA methylation analyses
have been used to investigate the DNA methylation status in the
whole genome (13). However, not
all DNA methylation regulates RNA expression associated with cancer
initiation and progression. DNA methylation and RNA expression
should be analyzed on a genome-wide scale simultaneously using the
same clinical samples. Thus, an integrated analysis of DNA
methylation and RNA expression profiles was conducted in the
present study using a genome-wide microarray-based system. As shown
in Fig. 1, the selected genes
showed a negative correlation for methylation and expression
profiles. This result supports the fact that the selected genes
showing differential expression between normal and tumor tissues
are regulated by the DNA methylation status. Two publicly available
databases were used to analyze the candidate genes selected from
the present study. Firstly, to identify the novelty of selected
genes with cervical cancer, a search was applied to observe whether
the candidate genes were already reported in the CCDB or not. A
total of 15 of the selected 19 genes were identified as novel genes
associated with cervical cancer (Table
I). Secondly, to overcome the shortage of sample numbers, the
expression of the selected genes in cervical cancer was further
analyzed using the microarray database (GENT). A total of 16 of the
selected 19 genes showed positive expression patterns with the
present data (Table I). These
results from the two databases indicate that the selected genes may
be novel genes associated with cervical cancer and involved in the
progression of cervical cancer.
Gene ontology analysis showed that the first cluster
covered two categories, the cell junction and the plasma membrane
(Table II). Four genes,
CAMK2N1, CGNL1, DLC1 and SYT11, the
expression of which was lower in the tumor tissues than in the
normal tissues, were enriched in the cell junction. Generally,
proteins associated with the cell junction suppress cell
proliferation and stimulate cell differentiation. Downregulation of
these genes has been linked to epithelial-mesenchymal transition
and cancer (25). Seven genes,
including three additional genes, FAT1, MUC4 and
TPM1, were categorized in the plasma membrane. It has been
reported that the MUC family genes, MUC1,
MUC16 and MUC4, are upregulated in cervical squamous
cell carcinoma (26,27). Similarly to this previous study, the
MUC4 gene was shown to be upregulated by hypomethylation in
the pre-cancer tissues compared with that in the normal tissues in
the present study (Table I). Based
on these analyses, the selected genes were clustered into
enrichment categories associated with cancer progression.
Treatment with demethylating reagent decreased the
methylation of selected genes and increased the demethylation
(Fig. 2). Furthermore, gene
expression was increased. These results support the fact that the
selected genes are regulated by DNA methylation. Methylation of
ALDH1A3 was originally reported in lung cancer and was
identified as a methylation marker for other types of cancer,
including breast, prostate, colon and brain tumors (28,29).
However, to the best of our knowledge, there have been no studies
regarding the methylation of ALDH1A3 in cervical cancer. Two
other genes, PPP1R3C and CAMK2N1, were downregulated
by hypermethylation in cervical cancer. The epigenetic regulation
of these genes was validated in the present study (Fig. 2). Based on the findings of a
previous study, the PPP1R3C gene is downregulated in
cervical cancer (30); its
methylation status has been reported in melanoma, colon and
prostate cancers, but not in cervical cancer (31,32).
Similar to the present results, the PPP1R3C gene is highly
downregulated in cervical cancer, as per the GENT database. These
results are consistent with the present data, including the
downregulation and hypermethylation of the PPP1R3C gene. The
CAMK2N1 gene is a calcium/calmodulin-dependent protein
kinase II inhibitor 1, and its expression is negatively correlated
with the progression of human colon cancer. The silencing of
CAMK2N1 expression increases tumor growth and cell cycle
progression (33). There have been
no studies examining CAMK2N1 and methylation in cancer. It
can be postulated that the CAMK2N1 gene is regulated by DNA
methylation, since CpG islands are located in the first exon and
intron in the CAMK2N1 gene, according to the University of
California Santa Cruz genome browser (http://genome.ucsc.edu). Based on these data,
CAMK2N1 may play important roles in cervical cancer
progression through epigenetic regulation.
In conclusion, in the present study, 19 genes
associated with cervical cancer were identified showing
differential RNA expression by DNA methylation using the integrated
analysis of data from RNA expression and DNA methylation arrays.
The present study also determined that a number of the 19 genes
have not previously been reported in association with cervical
cancer and validated the epigenetic regulation of three genes,
ALDH1A3, PPP1R3C and CAMK2N1, by MS-PCR and
RT-quantitative PCR. Taken together, these results suggest that the
selected genes may be involved in the progression or initiation of
cervical cancer through the differential expression by DNA
methylation. The functional role of these genes or their potential
as novel DNA methylation markers remains to be examined.
Acknowledgements
This study was supported by a grant from the Hallym
University Medical Center Research Fund (no. 01-2009-15), an
intramural grant from the Korea National Institute of Health (no.
2010-N73004-00) and the Korea Center for Disease Control and
Prevention (no. 4845-301).
References
|
1
|
Forouzanfar MH, Foreman KJ, Delossantos
AM, et al: Breast and cervical cancer in 187 countries between 1980
and 2010: a systematic analysis. Lancet. 378:1461–1484. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Woodman CB, Collins SI and Young LS: The
natural history of cervical HPV infection: unresolved issues. Nat
Rev Cancer. 7:11–22. 2007. View
Article : Google Scholar
|
|
3
|
Jensen KE, Schmiedel S, Frederiksen K, et
al: Risk for cervical intraepithelial neoplasia grade 3 or worse in
relation to smoking among women with persistent human
papillomavirus infection. Cancer Epidemiol Biomarkers Prev.
21:1949–1955. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Werness BA, Levine AJ and Howley PM:
Association of human papillomavirus types 16 and 18 E6 proteins
with p53. Science. 248:76–79. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dyson N, Howley PM, Münger K and Harlow E:
The human papilloma virus-16 E7 oncoprotein is able to bind to the
retinoblastoma gene product. Science. 243:934–937. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Whiteside MA, Siegel EM and Unger ER:
Human papillomavirus and molecular considerations for cancer risk.
Cancer. 113(10 Suppl): 2981–2994. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Esteller M: Epigenetics in cancer. N Engl
J Med. 358:1148–1159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Feinberg AP and Vogelstein B:
Hypomethylation distinguishes genes of some human cancers from
their normal counterparts. Nature. 301:89–92. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Herman JG, Merlo A, Mao L, et al:
Inactivation of the CDKN2/p16/MTS1 gene is frequently associated
with aberrant DNA methylation in all common human cancers. Cancer
Res. 55:4525–4530. 1995.PubMed/NCBI
|
|
10
|
Silva TD, Vidigal VM, Felipe AV, et al:
DNA methylation as an epigenetic biomarker in colorectal cancer.
Oncol Lett. 6:1687–1692. 2013.PubMed/NCBI
|
|
11
|
Jeong DH, Youm MY, Kim YN, et al: Promoter
methylation of p16, DAPK, CDH1, and TIMP-3 genes in cervical
cancer: correlation with clinicopathologic characteristics. Int J
Gynecol Cancer. 16:1234–1240. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Neyaz MK, Kumar RS, Hussain S, et al:
Effect of aberrant promoter methylation of FHIT and RASSF1A genes
on susceptibility to cervical cancer in a North Indian population.
Biomarkers. 13:597–606. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lendvai A, Johannes F, Grimm C, et al:
Genome-wide methylation profiling identifies hypermethylated
biomarkers in high-grade cervical intraepithelial neoplasia.
Epigenetics. 7:1268–1278. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun Z, Asmann YW, Kalari KR, et al:
Integrated analysis of gene expression, CpG island methylation, and
gene copy number in breast cancer cells by deep sequencing. PLoS
One. 6:e174902011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li M, Balch C, Montgomery JS, et al:
Integrated analysis of DNA methylation and gene expression reveals
specific signaling pathways associated with platinum resistance in
ovarian cancer. BMC Med Genomics. 2:342009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Irizarry RA, Ladd-Acosta C, Carvalho B, et
al: Comprehensive high-throughput arrays for relative methylation
(CHARM). Genome Res. 18:780–790. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Agarwal SM, Raghav D, Singh H and Raghava
GP: CCDB: a curated database of genes involved in cervix cancer.
Nucleic Acids Res. 39:D975–979. 2011. View Article : Google Scholar :
|
|
18
|
Shin G, Kang TW, Yang S, et al: GENT: gene
expression database of normal and tumor tissues. Cancer Inform.
10:149–157. 2011.PubMed/NCBI
|
|
19
|
Eisen MB, Spellman PT, Brown PO and
Botstein D: Cluster analysis and display of genome-wide expression
patterns. Proc Natl Acad Sci USA. 95:14863–14868. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang da W, Sherman BT, Tan Q, et al: The
DAVID Gene functional classification tool: a novel biological
module-centric algorithm to functionally analyze large gene lists.
Genome Biol. 8:R1832007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim JM, Sohn HY, Yoon SY, et al:
Identification of gastric cancer-related genes using a cDNA
microarray containing novel expressed sequence tags expressed in
gastric cancer cells. Clin Cancer Res. 11:473–482. 2005.PubMed/NCBI
|
|
22
|
Li LC and Dahiya R: MethPrimer: designing
primers for methylation PCRs. Bioinformatics. 18:1427–1431. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chao A, Wang TH, Lee YS, et al: Molecular
characterization of adenocarcinoma and squamous carcinoma of the
uterine cervix using microarray analysis of gene expression. Int J
Cancer. 119:91–98. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Song JY, Lee JK, Lee NW, et al: Microarray
analysis of normal cervix, carcinoma in situ, and invasive cervical
cancer: identification of candidate genes in pathogenesis of
invasion in cervical cancer. Int J Gynecol Cancer. 18:1051–1059.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kavanagh E, Tsapara A, Matter K and Balda
MS: Tight junctions and the regulation of epithelial cell
proliferation and gene expression. Tight Junctions. Springer; pp.
101–115. 2006, View Article : Google Scholar
|
|
26
|
Togami S, Nomoto M, Higashi M, et al:
Expression of mucin antigens (MUC1 and MUC16) as a prognostic
factor for mucinous adenocarcinoma of the uterine cervix. J Obstet
Gynaecol Res. 36:588–597. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Munro EG, Jain M, Oliva E, et al:
Upregulation of MUC4 in cervical squamous cell carcinoma:
pathologic significance. Int J Gynecol Pathol. 28:127–133. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shames DS, Girard L, Gao B, et al: A
genome-wide screen for promoter methylation in lung cancer
identifies novel methylation markers for multiple malignancies.
PLoS Med. 3:e4862006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang W, Yan W, You G, et al: Genome-wide
DNA methylation profiling identifies ALDH1A3 promoter methylation
as a prognostic predictor in G-CIMP-primary glioblastoma. Cancer
Lett. 328:120–125. 2013. View Article : Google Scholar
|
|
30
|
Wong YF, Cheung TH, Tsao GS, et al:
Genome-wide gene expression profiling of cervical cancer in Hong
Kong women by oligonucleotide microarray. Int J Cancer.
118:2461–2469. 2006. View Article : Google Scholar
|
|
31
|
Bonazzi VF, Irwin D and Hayward NK:
Identification of candidate tumor suppressor genes inactivated by
promoter methylation in melanoma. Genes Chromosomes Cancer.
48:10–21. 2009. View Article : Google Scholar
|
|
32
|
Kim JH, Dhanasekaran SM, Prensner JR, et
al: Deep sequencing reveals distinct patterns of DNA methylation in
prostate cancer. Genome Res. 21:1028–1041. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang C, Li N, Liu X, Zheng Y and Cao X: A
novel endogenous human CaMKII inhibitory protein suppresses tumor
growth by inducing cell cycle arrest via p27 stabilization. J Biol
Chem. 283:11565–11574. 2008. View Article : Google Scholar : PubMed/NCBI
|