Introduction
Primary central nervous system lymphoma (PCNSL) is
clinically characterized as a rare and specific type of
non-Hodgkin’s lymphoma that is localized in the brain, spinal cord,
meninges and eyes, without the involvement of other body systems
(1–3). PCNSL exhibits an annual incidence rate
of 0.43 per 100,000 individuals in the USA (1,2). The
majority of PCNSL patients are male and the median age at diagnosis
is ~60 years, with no differences observed between ethnic groups
(1–5). It has been reported that PCNSL
accounts for 1–3% of all central nervous system (CNS) tumors and
2–3% of non-Hodgkin’s lymphoma cases, while diffuse large B-cell
lymphoma accounts for >90% of all PCNSL cases (1–4).
Diagnosis of PCNSL is most often obtained using stereotactic biopsy
(1,3–8). And
the main treatment strategies for PCNSL are chemotherapy and
radiotherapy (1–4,6–8).
Several novel therapeutic strategies have been developed, including
immunotherapy, targeted therapy or stem cell transplantation
therapy (4,6–9). PCNSL
is a highly aggressive lymphoma with a poor prognosis and a median
survival time of nine months following diagnosis (4). Due to its high malignancy and poor
prognosis, PCNSL diagnosis and treatment have been a challenge in
the neurosurgical field (1,3,4).
Therefore, the aim of the present study was to retrospectively
analyze the recent clinical data and outcomes of nine PCNSL
patients who received treatment at the First Hospital of China
Medical University (Shenyang, China) between January 2011 and
January 2014. Furthermore, combined with a literature review, the
present study reviewed and discussed the possible treatment
strategies for this malignant tumor type.
Case report
The current study included nine patients who were
diagnosed with PCNSL at the First Hospital of China Medical
University between January 2011 and January 2014. The patients
included two males and seven females patients, with ages ranging
between 47 and 76 years and an average age of 61.4 years. The
present study conformed to the principles outlined in the
Declaration of Helsinki (2013) and was approved by the Ethics
Committee of China Medical University. Furthermore, written
informed consent was obtained from all the patients.
Preoperative computed tomography (CT) and enhanced
magnetic resonance imaging (MRI) scans were performed in the nine
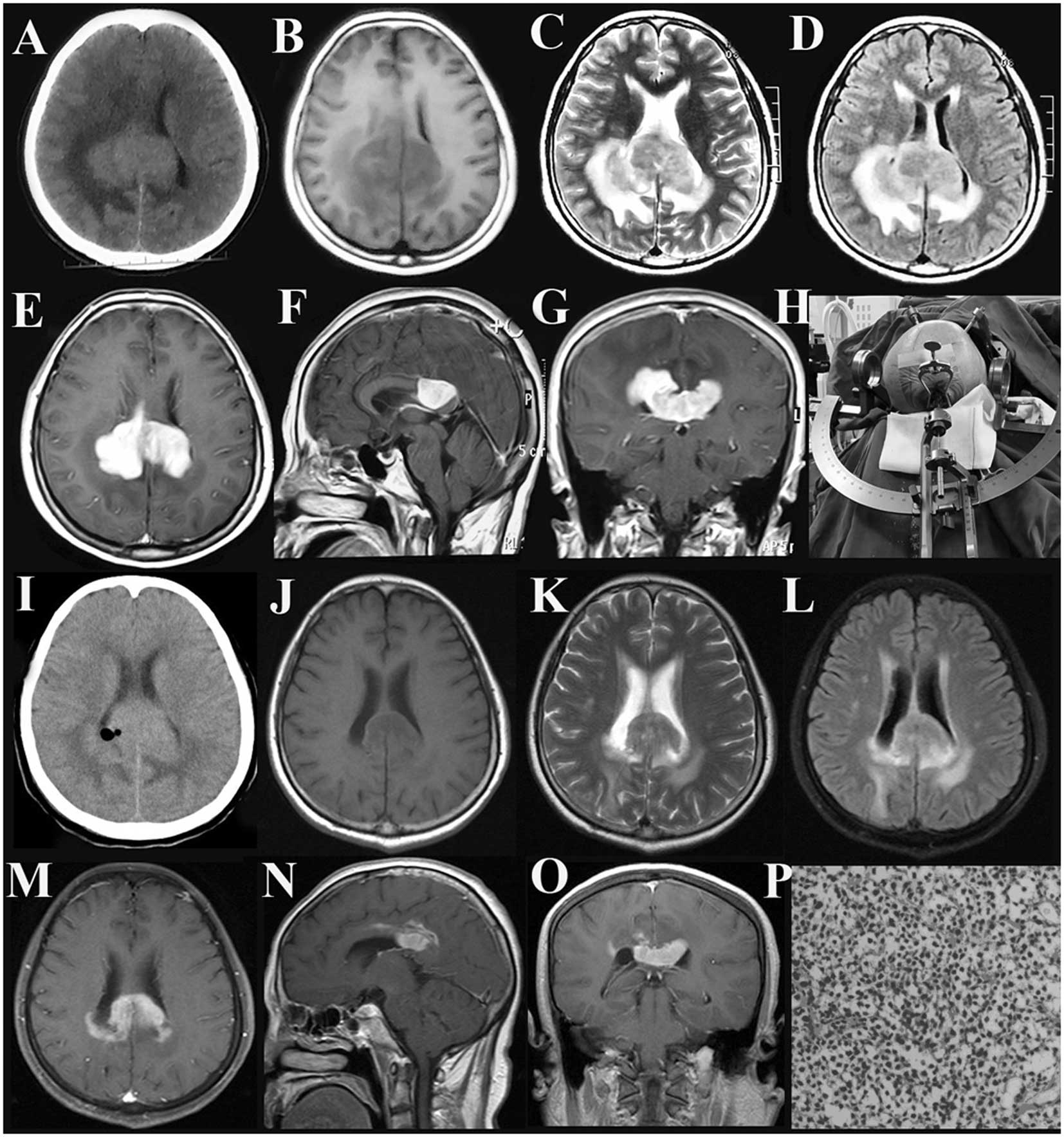
enrolled patients, which identified solitary lesions in seven
patients and multiple lesions in two patients. Three patients
exhibited a solitary lesion in the corpus callosum, one in the left
caudate nucleus, one in the left frontal lobe, one in the right
cerebellum and one in the anterior cranial fossa and canalis
opticus (Figs. 1–3). Furthermore, one patient exhibited
multiple lesions in various regions, including the left temporal
and occipital lobes, as well as in the right caudate nucleus,
whereas another patient exhibited multiple lesions only in the
ventricles. All the lesions presented a relatively high intensity
on the CT images, while hypertense lesions were observed on the
T1-weighted, T2-weighted and fluid-attenuated inversion recovery
(FLAIR) images. Following contrast enhancement, distinct, enhanced,
mass-like solid structures with typical fist-like, flame-shaped or
butterfly-shaped patterns were detected. In addition, significant
local edema was identified in the areas surrounding the lesions
(Figs. 1–3). Additionally, the disease course of all
the patients ranged between 7 days and 10 months, with an average
of 3.1 months. The detailed clinical manifestations of each patient
are listed in Table I.
 | Table IClinical manifestations, treatment
regimens, pathological characteristics and prognosis of nine
primary central nervous system lymphoma patients. |
Table I
Clinical manifestations, treatment
regimens, pathological characteristics and prognosis of nine
primary central nervous system lymphoma patients.
| Case | Gender/Age,
years | Clinical
manifestation | Lesion site | Treatment
regimen | Postoperative
pathological diagnosis (immunohistochemical staining) | Follow-up,
months/outcome |
|---|
| 1 | F/68 | Headache and
dizziness, for one month nausea and vomiting accompanied by
unsteady gait for one week | Right brachium
pontis | Craniotomy,
dexamethasone and radiotherapy | NHBL [CD3
(sporadic+), CD20 (+), CD34 (−), S-100 (−), vimentin (−), CK (−),
Ki67 (+>50%), GFAP (−), Pax-5 (+)] | 27/Mortality |
| 2 | F/58 | Deteriorated memory
for three months, reduced ability to speak for 10 days | Left frontal
lobe | Craniotomy and
dexamethasone | NHBL [CD34 (−),
pan-CK (−), GFAP (−), Ki-67 (+>75%), S-100 (−), vimentin (−),
CD20 (+), CD3 (−), CD38 (low+), CD68 (sporadic+), CD138 (−), IDH1
(−), Pax-5 (++)] | 12/Lost |
| 3 | F/60 | Headache and
dizziness for three months | Left caudate
nucleus | Craniotomy and
dexamethasone | NHBL/marginal-zone
B-cell lymphoma [CK (−), vimentin (−), GFAP (+), S-100 (−),
synaptophysin (−), CD3 (low+), CD20 (−), Pax-5 (+), Ki67
(+>30%), CD138 (−), κ (+), λ (−), CD5 (low+), CD23 (−), cyclin
D1 (−)] | 9.8/Mortality |
| 4 | M/47 | Headache for 10
months; headache became worse and was accompanied by difficult
movement of the left extremity for two weeks | Splenium of the
corpus callosum | Stereotactic biopsy,
MP, MTX and radiotherapy | Diffuse large B-cell
lymphoma, germinal center subtype [CD20 (low+), CD21 (+), CD3
(sporadic+), CD56 (−), pan-CK (−), GFAP (+), Ki-67 (+75%), P53 (−),
P63 (sporadic+), Pax-5 (+), S-100 (−), synaptophysin (−), TTF-1
(−), vimentin (±), Bcl-6 (sporadic+), CD10 (+), MUM1 (+)] | 10/Mortality |
| 5 | F/49 | Headache and
dizziness, deteriorated memory, slow response for six months | Genu of corpus
callosum | Craniotomy, MP and
MTX | Diffuse large B-cell
lymphoma [CK (−), vimentin (+), GFAP (+), S-100 (local+),
synaptophysin (local+), P53 (++), CD3 (sporadic+), CD20 (+), Pax-5
(+), Ki67 (+50%), Bcl-6 (+), CD10 (−), MUM1 (+)] | 13/Remission |
| 6 | F/76 | Declined olfactory
sensation for one month and visual decline for two weeks | Floor of the anterior
cranial fossa, canalis opticus | Neuroendoscopic
removal of the majority of the tumor, MP and MTX | Diffuse large B-cell
lymphoma, activated B-cell subtype [CK (+), CD3 (−), CD20 (−),
Pax-5 (+), CD30 (−), Ki-67 (+>50%) CD56 (−), CD99 (−),
synaptophysin (−), Bcl-6 (−), CD10 (−), MUM1 (+), GFAP (−)] | 8/Remission |
| 7 | M/69 | Intermittent
headaches and dizziness for three months, unclear speech for three
days | Left temporal and
occipital lobe, right caudate nucleus | Stereotactic biopsy,
MP and MTX | Diffuse large B-cell
lymphoma, activated B-cell subtype [Bcl-6 (−), CD10 (−), CD20 (++),
CD3 (+), CD30 (−), CK (−), EMA (−), GFAP (−), Ki-67(80%), MUM1 (+),
Pax-5 (+), S-100 (−), synaptophysin (−), TTF-1 (−), vimentin (+),
CD79a (±)] | 6 |
| 8 | F/65 | Headache and
dizziness for 10 days | Temporal and frontal
horn of lateral ventricle, fourth ventricle | Craniotomy, MP,
dexamethasone and MTX | NHBL, activated
B-cell subtype [CD3 (+), CD20 (+), Pax-5 (+), CD10 (−), Bcl-6 (−),
MUM1 (+), CD30 (−), Ki-67 (+>80%), CD15 (−), CD68 (+)] | 5 |
| 9 | F/61 | Difficult movement of
the left extremity for one week | Splenium of the
corpus callosum | Stereotactic biopsy,
dexamethasone and MTX | NHBL [CD20 (+), CD3
(sporadic+), pan-CK (−), GFAP (−), Ki-67 (+80%), NSE (+), S-100
(−), synaptophysin (−), vimentin (+), AFP (+), hCG (−), PLAP (−),
Pax-5 (+), Bcl-6 (+), CD10 (−), MUM1 (+)] | 5 |
The pathological patient specimens were collected in
the Department of Neurosurgery of the First Hospital of China
Medical University. Following pathological diagnosis, the patients
adopted the following chemotherapy and radiotherapy treatment
regimens: Two patients underwent craniotomy for tumor removal and
were administered with 10 mg/day dexamethasone for six days
(craniotomy + dexamethasone); one patient received craniotomy for
tumor removal in combination with 10 mg/day dexamethasone for six
days and 40 Gy whole-brain radiotherapy (WBRT; craniotomy +
dexamethasone + WBRT); two patients received craniotomy in
combination with 80 mg methylprednisolone (MP) twice a day (b.i.d)
for three days, followed by 40 mg MP b.i.d. for three days, and 3
g/m2 methotrexate (MTX; craniotomy + MP + MTX); two
patients received stereotactic biopsy in combination with MP and
MTX (stereotactic biopsy + MP + MTX); one patient received
stereotactic biopsy in combination with MP, MTX and radiotherapy
(stereotactic biopsy + MP + MTX + radiotherapy); and one patient
received neuroendoscopic surgery in combination with MP and MTX
(neuroendoscopic surgery + MP + MTX). Following surgery, all the
patients underwent a systemic examination, including whole-body
lymph node scanning and chest and abdominal CT scans. In addition,
four patients received bone marrow aspiration biopsies and one
patient underwent positron emission tomography (PET). The
possibility of secondary CNS lymphoma was ruled out as no primary
lesions were detected in other body systems of the patients.
Postoperative pathological examination confirmed the
diagnosis of non-Hodgkin’s B-cell lymphoma in all the patients,
including diffuse large cell lymphoma in four patients,
marginal-zone B-cell lymphoma in one patient, activated B-cell
subtype in three patients and small B-cell lymphoma in one patient.
The detailed pathological and immunochemical results are listed in
Table I.
The follow-up duration range was 5–27 months with an
average duration of 10.1 months. After the initial three months of
follow-up, the clinical symptoms of all the patients were
significantly improved. The tumor disappeared in seven patients,
while a marked reduction in the tumor size was observed in two
patients. However, six patients presented tumor recurrence within
the follow-up period; among them, three patients succumbed to the
disease, two survived and one was lost to follow-up after 12 months
(Table I).
Discussion
PCNSL is highly aggressive, and the most common
histological subtype of PCNSL is diffuse large B-cell lymphoma,
with a male to female ratio of 1.2–1.7:1 among PCNSL patients
(1,3,5). In
the present study, all the PCNSL patients exhibited B-cell
lymphoma, accounting for ~0.2% of the total number of patients who
were diagnosed with cranial tumors (9/4059 patients) during the
time period of the present study in the Department of Neurosurgery
of the First Hospital of China Medical University. The incidence
rate has exhibited an overall increasing trend in the past 30 years
(1,10). This increase is associated with
various factors, including the increased number of hospitalized
patients, improved diagnostic technologies, increased incidence of
HIV infection and administration of immunosuppressive agents
following organ transplantation. Of these influencing factors,
immunodeficiency is a high risk factor for PCNSL (1,6). For
instance, previous studies have identified that the incidence rate
of lymphomas is significantly higher in acquired immune deficiency
syndrome (AIDS) patients compared with patients not suffering from
AIDS (1,10). In particular, the incidence rate of
CNS lymphomas reached 5% in AIDS patients prior to the
administration of effective antiretroviral therapy, but
significantly declined following therapy (1,4).
The clinical manifestations of PCNSL are closely
associated with various factors, including the location and number
(solitary or multiple) of lesions. Previous studies have revealed
that multiple lesions occur in ~34% of PCNSL patients (1,5) and
are commonly detected in the frontal lobe. Furthermore, patients
with lesions in deep brain tissue, including the corpus callosum,
basal ganglia, the area surrounding the ventricles, the brainstem
or the cerebellum, accounted for ~40% of PCNSL patients (1,3,6–9).
In the present study, seven patients exhibited a solitary lesion
and two presented multiple lesions, with the lesion sites being
consistent with the aforementioned high-occurrence sites. A recent
literature review identified that ~50% of patients exhibited
clinical manifestations involving motor and sensory dysfunctions,
30% developed personality changes, 55% experienced headaches, ~35%
suffered from nausea, ~10% suffered from vomiting and ~20% of cases
were accompanied by uveitis (1). In
the present study, five patients experienced headaches, three
developed impaired speaking capability, three had limb movement
disorders, one suffered from nausea and vomiting, two demonstrated
declined memory and responsiveness and one exhibited declined
olfactory and visual sensation. These symptoms are consistent with
the aforementioned manifestation distribution pattern.
The imaging manifestations of CNS lymphomas
demonstrate a relative specificity. The tumor mass predominantly
displays a relatively high density (occasionally exhibiting a
density similar to the surrounding area) on CT images, with even
enhancement observed upon enhanced scanning. In addition, MRI
typically demonstrates an equal or low signal on the T1-weighted
image and a high signal on the T2-weighted and FLAIR images.
Enhanced scanning demonstrates a distinct, even enhancement, with a
fist-like, flame-shaped or a unique butterfly-shaped structure in
patients with lesions in the corpus callosum (5,6–9). In
the present study, the enhanced MRI scans of the patients with
lesions in the cerebellum or left frontal lobe and the patients
with multiple lesions demonstrated a flame-shaped and fist-like
structure, respectively. Furthermore, the enhanced MRI of the three
patients with a single lesion in the corpus callosum presented a
butterfly-shaped structure. These typical imaging manifestations
are conducive to a preliminary diagnosis of a CNS lymphoma.
Regarding the differential diagnosis of CNS lymphomas, the common
clinical focus is the differential diagnosis from multifocal or
multicentric glioma, multiple sclerosis and inflammation. Although
specific scan sequences of magnetic resonance spectroscopy,
diffusion-weighted images or PET may be used, the authors of the
present study consider pathological examination to be the gold
standard for the differential diagnosis of CNS lymphomas.
Pathological examination has revealed that the
majority of PCNSLs are highly malignant (high grade) and the most
common PCNSL type is diffuse large B-cell lymphoma (1,4,8,9).
Additionally, light microscope observations have indicated that
tumor cells present a diffuse distribution, relatively consistent
cell size and small cytoplasmic area. The tumor cell nuclei are
medium in size and round or oval in shape, with one or more
nucleoli and the presence of mitotic nuclei. The specific
immunohistochemical markers of tumor cells are cluster of
differentiation (CD)20-positive and CD138-negative (1). In addition, other markers, including
multiple myeloma oncogene-1, B-cell lymphoma-6, pan-B-cell
antibody, CD19, CD20, CD79α and paired box 5, are typically
positive (1). In patients with a
clinically normal immune function, the Epstein-Barr virus is
predominantly negative, and the mutation frequency of
proto-oncogenes, including Pim-1, RhoH/TTF and c-Myc, is two to
five-fold higher than in patients with extracranial diffuse large
B-cell lymphomas (1). Perivascular
large B-cell lymphoma is a pathologically high-malignancy type of
PCNSL; however, its occurrence is rare. In addition, low-malignancy
(low-grade) PCNSL is rare and predominantly occurs in the spinal
cord, with immunocytoma and marginal-zone B-cell lymphoma
(mucosa-associated lymphoid tissue type) as two common types.
Marginal-zone B-cell lymphomas predominantly occur in female
patients (male to female ratio, 1:4) and have an average age of
onset of ~49 years and a relatively satisfactory clinical prognosis
(1). The present study included one
female patient with marginal-zone B-cell lymphoma who did not
receive postoperative radiotherapy and succumbed to the disease
10.5 months after surgery.
Primary CNS T-cell lymphoma is rare; thus, the
present study did not include any T-cell lymphoma patients. The
majority of reported cases are clinically sporadic, accounting for
~2% of PCNSL patients (1,3,4,8,9).
The age of onset is ~60 years, and its clinical manifestations and
prognosis are similar to B-cell lymphoma (1). CD4-positivity is the major
immunohistochemical marker of primary CNS T-cell lymphoma, although
other markers may include positive staining for pan T antigens
(including CD3 and CD5) (1,4). In addition, a rarer pathological type
of PCNSL exists, termed histiocytic sarcoma, which is highly
malignant and has poor clinical outcomes (1).
Currently, the diagnostic criteria of PCNSL are
relatively definitive, consisting of three major elements: i) The
lesions are localized in the brain, spinal cord, meninges and eyes;
ii) the lesions in the aforementioned locations are pathologically
diagnosed as lymphoma; and iii) the lesions do not involve other
parts of the body (1,3–5,8). In
the present study, pathological specimens were predominantly
collected in the Department of Neurosurgery using stereotactic
lesion biopsy, craniotomy for lesion biopsy and cellular
pathological examination of cerebrospinal fluid. Stereotactic
biopsy is the most common approach and should be the first choice
for achieving a definitive pathological diagnosis prior to surgery
in patients who are highly suspected to suffer from PCNSL (8).
The effectiveness of PCNSL treatment remains
unsatisfactory (1,3,4).
Existing studies regarding PCNSL treatment strategies are
predominantly retrospective treatment analyses, whereas prospective
randomized controlled studies with a large sample size are lacking.
Therefore, the optimization of treatment selection requires
additional systemic studies to be performed. Currently, the
generally accepted treatment strategy for PCNSL is a
chemotherapy-based comprehensive treatment (1–4,7–9,11–22).
The majority of studies have indicated that the main purpose of the
neurosurgical procedure is to obtain a pathological specimen and
that no clear association exists between the complete removal of
the lesion and patient prognosis (1,7–9,13).
However, a previous study identified that the overall survival rate
of patients who underwent complete resection or incomplete
resection of the tumors was significantly higher compared with
patients who received only stereotactic biopsy and particularly in
patients with a solitary lesion (14). The majority of researchers consider
that chemotherapy (single- or multiple-agent chemotherapy) with or
without radiotherapy should be performed at an early stage once the
tumor pathological properties are identified (1–4,6–10,12,13,18,19).
The most common chemotherapeutic agent used in single-agent
chemotherapy is MTX, which is recommended to be administration at a
high dose of MTX (2.5–3.5 g/m2, up to a maximal dose of
8 g/m2) (1,3,10,12,13,16,18).
MTX may be administered concurrently with other agents, including
vincristine, procarbazine, temozolomide (150 mg/m2/day),
rituximab (375 mg/m2) and dexamethasone (12 mg)
(15–17,22),
or in combination with whole-brain radiotherapy (WBRT) (1). The response rate to single-agent
chemotherapy using MTX is 52–88%, to multiple-agent chemotherapy
combining MTX and other chemotherapeutic agents is 70–94%, and to
WBRT is ~90% (15). The two-year
survival rate of the patients who received single- and
multiple-agent MTX chemotherapy combined with WBRT was 58–72% and
43–73%, respectively (15). A
previous study has demonstrated that an increase in radiation dose
and radiotherapy area did not appear to affect the overall patient
survival rate (19). Due to the
high radiation dose of up to 45 Gy used in the majority of studies,
the neurotoxicity incidence was relatively high (83% within one
year among patients aged >60 years), resulting in a marked
deterioration in patient cognitive function and quality of life
(20). Recent studies identified
that patients who underwent high-dose chemotherapy combined with
autologous stem cell transplantation exhibited a three-year
survival rate of 60% and an event-free survival rate of 53%
(16,17). In newly-diagnosed young patients,
the outcomes of this treatment strategy were superior (21). In the present study, all the
patients were administered hormones following craniotomy or
stereotactic biopsy due to a previous finding that lesions in the
majority of patients are sensitive to hormones. The underlying
mechanism is that hormones induce tumor cell apoptosis and reduce
the surrounding edema of the tumor body through binding to
glucocorticoid receptors on the surface of tumor cells. A previous
study reported that this hormone-induced effect may last for 6–60
months (11); however, the majority
of studies have indicated that hormones only temporarily relieve
the symptoms (3,4,9,21).
Therefore, it was proposed that all the patients in the present
study should receive postoperative chemotherapy and radiotherapy.
Six patients were selected to undergo subsequent high-dose MTX
chemotherapy, while two patients underwent subsequent radiotherapy
(regional + whole-brain). The follow-up data indicated a short-term
improvement of the clinical manifestations in all the patients, as
evidenced completely undetectable lesions in seven patients and
significant lesion reduction in two patients. However, six out of
seven patients subsequently exhibited tumor recurrence and the
recurred tumors progressed rapidly, with three patients succumbing
to the disease during the follow-up period. These data indicate a
poor prognosis for the PCNSL patients. Currently, the treatment
strategies for PCNSL, in particular recurrent PCNSL, remains
unsatisfactory.
In conclusion, PCNSL is a rare tumor type in the
CNS. Compared with the extracranially-localized large B-cell
lymphoma, the effectiveness of the PCNSL treatment is
unsatisfactory, which may be due to the lack of sufficient
knowledge regarding the biological properties of PCNSL. In
addition, the poor permeability of the existing chemotherapeutic
agents through the blood-brain barrier may be responsible for the
poor prognosis of PCNSL patients. Currently, comprehensive
treatment strategies based on a combination of stereotactic biopsy,
chemotherapy and radiotherapy are recommended. Developments in
targeted biological and gene therapy strategies are expected to
improve the treatment efficacy for PCNSL in the future.
Acknowledgements
The present study was supported by the Liaoning
Provincial Natural Science Foundation of China (grant no.
2013021075 awarded to Bo Qiu), the Fund for Scientific Research of
the First Hospital of China Medical University (grant no. fsfh1304,
awarded to Bo Qiu) and the National Natural Science Foundation of
China (grant no. 31100770, awarded to Jun Wang). The authors would
like to thank the staff of the Department of Neurosurgery of the
First Hospital of China Medical University (Shenyang, China) for
their technical assistance.
References
|
1
|
Rubenstein J, Ferreri AJ and Pittaluga S:
Primary lymphoma of the central nervous system: epidemiology,
pathology and current approaches to diagnosis, prognosis and
treatment. Leuk Lymphoma. 49(Suppl 1): 43–51. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yoon JH, Kang HJ, Kim H, et al: Successful
treatment of primary central nervous system lymphoma without
irradiation in children: single center experience. J Korean Med
Sci. 27:1378–1384. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bhagavathi S and Wilson JD: Primary
central nervous system lymphoma. Arch Pathol Lab Med.
132:1830–1834. 2008.PubMed/NCBI
|
|
4
|
Phillips EH, Fox CP and Cwynarski K:
Primary CNS lymphoma. Curr Hematol Malig Rep. 9:243–253. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Haque S, Law M, Abrey LE and Young RJ:
Imaging of lymphoma of the central nervous system, spine, and
orbit. Radiol Clin North Am. 46:339–361. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nayak L and Batchelor TT: Recent advances
in treatment of primary central nervous system lymphoma. Curr Treat
Options Oncol. 14:539–552. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Perini GF, Campregher PV, Santos FP and
Hamerschlak N: Primary central nervous system lymphoma: what a
neurologist/neurosurgeon should know? Arq Neuropsiquiatr.
71:254–257. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brastianos PK and Batchelor TT: Primary
central nervous system lymphoma: overview of current treatment
strategies. Hematol Oncol Clin North Am. 26:897–916. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gallop-Evans E: Primary central nervous
system lymphoma. Clin Oncol (R Coll Radiol). 24:329–338. 2012.
View Article : Google Scholar
|
|
10
|
Olson JE, Janney CA, Rao RD, et al: The
continuing increase in the incidence of primary central nervous
system non-Hodgkin lymphoma: a surveillance, epidemiology, and end
results analysis. Cancer. 95:1504–1510. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kürtüncü M, Tüzün E, Durmuş H, Mutlu M,
Akman-Demir G and Eraksoy M: Primary cerebral lymphoma with a
5-year remission to single-agent corticosteroids. Leuk Lymphoma.
50:1552–1553. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jezersek Novakovic B: Treatment outcomes
and survival in patients with primary central nervous system
lymphomas treated between 1995 and 2010 - a single centre report.
Radiol Oncol. 46:346–353. 2012. View Article : Google Scholar
|
|
13
|
Bellinzona M, Roser F, Ostertag H, Gaab RM
and Saini M: Surgical and removal of primary central nervous system
lymphomas (PCNSL) presenting as space occupying lesions: a series
of 33 cases. Eur J Surg Oncol. 31:100–105. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Weller M, Martus P, Roth P, Thiel E and
Korfel A; German PCNSL Study Group. Surgery for primary CNS
lymphoma? Challenging a paradigm. Neuro Oncol. 14:1481–1484. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ferreri AJ, Abrey LE, Blay JY, et al:
Summary statement on primary central nervous system lymphomas from
the Eighth International Conference on Malignant Lymphoma, Lugano,
Switzerland, June 12 to 15, 2002. J Clin Oncol. 21:2407–2414. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schorb E, Kasenda B, Atta J, et al:
Prognosis of patients with primary central nervous system lymphoma
after high-dose chemotherapy followed by autologous stem cell
transplantation. Haematologica. 98:765–770. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pennese E, Vergine C, Matera R, Dargenio
M, Forese P and Di Renzo N: Complete response induced by
fotemustine given as single agent in a patient with primary central
nervous system non-Hodgkin aggressive lymphoma relapsed after
high-dose chemotherapy and autologous stem cell support. Leuk
Lymphoma. 52:2188–2189. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Thiel E, Korfel A, Martus P, et al:
High-dose methotrexate with or without whole brain radiotherapy for
primary CNS lymphoma (G-PCNSL-SG-1): a phase 3, randomised,
non-inferiority trial. Lancet Oncol. 11:1036–1047. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ferreri AJ, Verona C, Politi LS, Chiara A,
Perna L, Villa E and Reni M: Consolidation radiotherapy in primary
central nervous system lymphomas: impact on outcome of different
fields and doses in patients in complete remission after upfront
chemotherapy. Int J Radiat Oncol Biol Phys. 80:169–175. 2011.
View Article : Google Scholar
|
|
20
|
Correa DD, Shi W, Abrey LE, et al:
Cognitive functions in primary CNS lymphoma after single or
combined modality regimens. Neuro Oncol. 14:101–108. 2012.
View Article : Google Scholar :
|
|
21
|
Illerhaus G: Primary CNS lymphoma. Dtsch
Med Wochenschr. 138:2515–2518. 2013.(In German). PubMed/NCBI
|
|
22
|
Omuro A, Taillandier L, Chinot O, et al;
ANOCEF Group (French Neuro-Oncology Association). Primary CNS
lymphoma in patients younger than 60: can whole-brain radiotherapy
be deferred? J Neurooncol. 104:323–330. 2011. View Article : Google Scholar
|