Introduction
Hepatocellular carcinoma (HCC) is the most common
primary liver tumor and a major public health problem worldwide
(1), representing the third most
common cause of cancer-associated mortality (2). Although hepatic resection and
transplantation have been regarded as the optimum therapeutic
strategies, in a large number of cases, certain limitations,
including lack of donors, severity of liver disease and specific
selection criteria (Milan criteria), restrict the application of
these strategies (3,4). Consequently, these surgical methods
are appropriate in only ~30% of HCC cases (5). At present, locoregional treatments,
such as image-guided radiofrequency ablation (RFA), are promising
in the treatment of various carcinoma types, including HCC
(6). As a novel treatment strategy,
RFA possesses multiple advantages, including the capacity for
localized tumor necrosis, minimal damage to functioning liver and
the capacity for repeated treatments in case of recurrence and/or
new tumors (7). However, previous
studies have proposed that insufficient RFA may promote the
proliferation of residual HCC (8,9).
Therefore, elucidating the molecular mechanisms underlying this
undesirable effect of RFA on HCC is important.
Angiogenesis is one of the critical mechanisms
involved in the proliferation and growth of tumor cells (10). Among the numerous
angiogenesis-associated genes, vascular endothelial growth factor
(VEGF) functions as a potent angiogenic factor and is ubiquitously
expressed in various types of cancer, including HCC (9,11). A
previous study has demonstrated that VEGF overexpression in HCC is
typically associated with tumor progression, reduced median
survival and recurrence following treatment (12). In addition, a recent study has
identified that insufficient RFA induced the aggressive growth of
residual HCC mediated by VEGF overexpression (9). However, the underlying mechanisms by
which insufficient RFA enhances VEGF expression remain unknown. The
aim of the present study was to investigate the mechanisms by which
insufficient RFA promotes HCC proliferation, particularly the
intracellular signaling pathway(s) involved in VEGF
overexpression.
Materials and methods
Cell culture
The established human HCC cell line, SMMC7721, was
obtained from the American Type Culture Collection (Manassas, VA,
USA). The cells were maintained in RPMI-1640 (Corning Inc.,
Corning, NY, USA) supplemented with 10% fetal bovine serum
(Hyclone-Thermo Fisher Scientific, Waltham, MA, USA) in a
humidified atmosphere of 5% CO2 at 37°C.
Heat treatment
Insufficient RFA was simulated in vitro using
a previously described method (13). Briefly, SMMC7721 cells were seeded
into 6-well plates (5×104 cells/well). After 24 h, the
plates were sealed and submerged in a water bath at 47°C for 5 min.
Subsequently, the cultures were cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum in a humidified atmosphere
of 5% CO2 at 37°C until residual populations reached 80%
confluence. The surviving populations were propagated into the
6-well plates and exposed to the aforementioned heat treatment for
10 min. Subsequently, this process was repeated with the
stimulation time increasing with each repetition (15, 20 and then
25 min). Cells surviving the 47°C treatment regimen were designated
as the ‘47°C treatment’ group and used in subsequent experiments.
SMMC7721 cells that were not exposed to the 47°C treatment regimen
were used as the ‘control’ group. To investigate the effect of
Ca2+/calmodulin-dependent protein kinase II (CaMKII),
extracellular signal-regulated kinase (ERK) and VEGF on the growth
of residual SMMC7721 cells in the 47°C treatment group, 10 μM KN93,
a specific CaMKII inhibitor, 20 μM PD98059, a specific ERK
inhibitor, or 5 μM axitinib, a VEGF receptor antagonist (all
purchased from Sigma-Aldrich, St. Louis, MO, USA), was added to
cell cultures obtained from the ‘control’ group, termed the ‘KN93’,
‘PD98059’ and ‘axitinib’ groups, respectively, and/or the ‘47°C
treatment’ group, termed the ‘KN93 + 47°C treatment’, ‘PD98059 +
47°C treatment’ and ‘axitinib + 47°C treatment’ groups,
respectively.
Proliferation assay
Cell proliferation was analyzed using an MTT assay
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide].
Briefly, a total of 3×103 trypsin-dispersed parent
SMMC7721 cells or 47°C-treated SMMC7721 cells in 0.1 ml culture
medium were seeded into 96-well plates and cultured for 24, 48 and
72 h. MTT solution was added to each well at a final concentration
of 0.5 mg/ml and incubated for 4 h. At the end of the incubation,
formazan crystals resulting from the MTT reduction assay were
dissolved through the addition of 150 μl dimethyl sulfoxide per
well. The absorbance was measured at 570 nm using an automated
ELISA plate reader (Thermo Fisher Scientific, Waltham, MA,
USA).
Western blot analysis
Cells grown in culture dishes were washed with
phosphate-buffered saline (Solarbio Science & Technology Co.,
Ltd, Beijing, China) and harvested using a cell scraper (Nest
Biotechnology Co., Ltd, Wuxi, China). Whole-cell lysates were
collected by adding freshly prepared lysis buffer [containing 150
mM NaCl, 50 mM Tris-HCl, pH 8.0, 0.1% sodium dodecyl sulfate, 1%
Triton X-100 and 1% proteinase inhibitors (1:100, Sigma-Aldrich)]
to the harvested cells, which were then incubated on ice for 30
min. Next, the cell lysates were centrifuged at 14,000 × g for 20
min at 4°C (Sorvall™ ST 16R, Thermo Fisher Scientific) and the
protein content was determined by Lowry’s method (14) using bovine serum albumin as the
standard. The samples were adjusted to contain 30 μg protein each;
subsequently, they were subjected to SDS-PAGE and
electrophoretically transferred to a polyvinylidene fluoride
membrane. Following transfer, the membrane was blocked with 5%
non-fat milk for 1 h at room temperature then incubated overnight
at 4°C with primary rabbit anti-human polyclonal anti-VEGF (1:200;
cat. no. BA0407; Wuhan Boster Biological Technology, Ltd., Wuhan,
China), anti-CaMKII (1:500; cat. no. sc-13082; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), anti-phospho-CaMKII
(1:1,000; cat. no. V1111; Promega Corporation, Madison, WI, USA),
anti-ERK (1:1,000; cat. no. sc-292838; Santa Cruz Biotechnology,
Inc.) or anti-phospho-ERK (1:1,000; cat. no. sc-101760; Santa Cruz
Biotechnology, Inc.) antibodies. Subsequently, the membrane was
incubated with polyclonal horseradish peroxidase
conjugated-secondary goat anti-rabbit antibodies (1:3,000; cat. no.
sc-2004; Santa Cruz Biotechnology, Inc.) diluted in Tris-buffered
saline and Tween-20 (containing 20 mM Tris-HCl, 150 mM NaCl, pH 7.4
and 0.1% Tween-20) for 2 h at room temperature. Finally, the
membrane was rinsed with phosphate-buffered saline and visualized
using an enhanced chemiluminescence detection kit (Thermo Fisher
Scientific). The western blot bands were quantified using
QuantityOne software (version 4.6.9; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) by measuring the band intensity for each group
and normalizing against β-actin, which was used as an internal
control.
Statistical analysis
Data are expressed as the mean ± standard error of
mean from at least three independent experiments. Statistical
analysis for multiple comparisons was performed using one-way
analysis of variance followed by Fisher’s least significant
difference test. All statistical analyses were performed using SPSS
version 17.0 software (SPSS, Inc., Chicago, IL, USA). Differences
were considered to be statistically significant when P<0.05.
Results
VEGF overexpression is triggered by 47°C
heat treatment and blocked by ERK and CaMKII
To investigate how insufficient RFA triggers VEGF
overexpression, specific pharmacological inhibitors of ERK
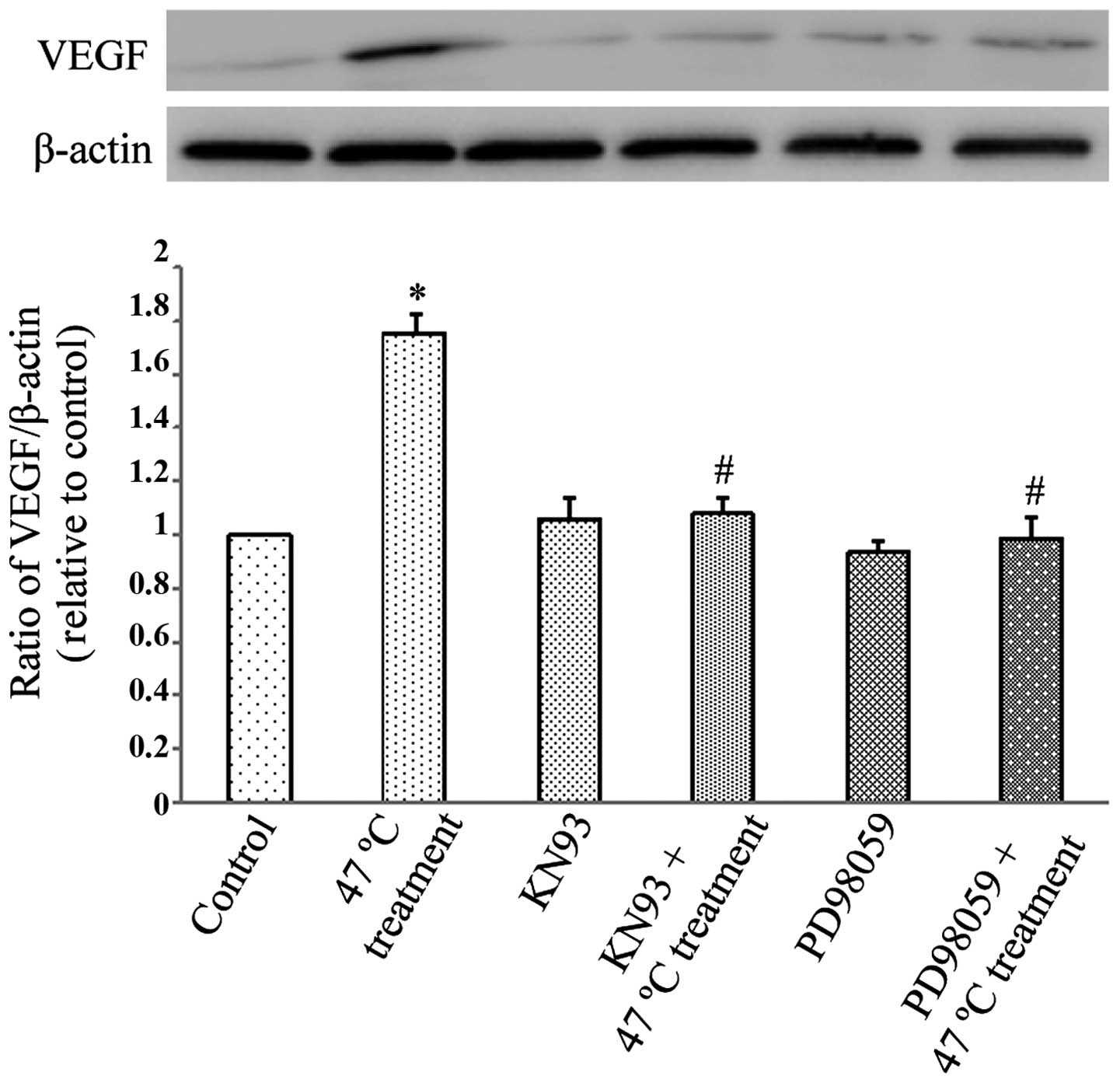
(PD98059) and CaMKII (KN93) were used. Fig. 1 demonstrates that the 47°C treatment
regimen significantly upregulated VEGF protein expression in
SMMC7721 cells (P<0.05), and this effect was significantly
reduced by pretreatment with PD98059 and KN93 (P<0.05).
Therefore, this experiment demonstrated that the 47°C treatment
regimen induced VEGF overexpression through the activation of ERK
and CaMKII signaling pathways.
Interaction between CaMKII and ERK
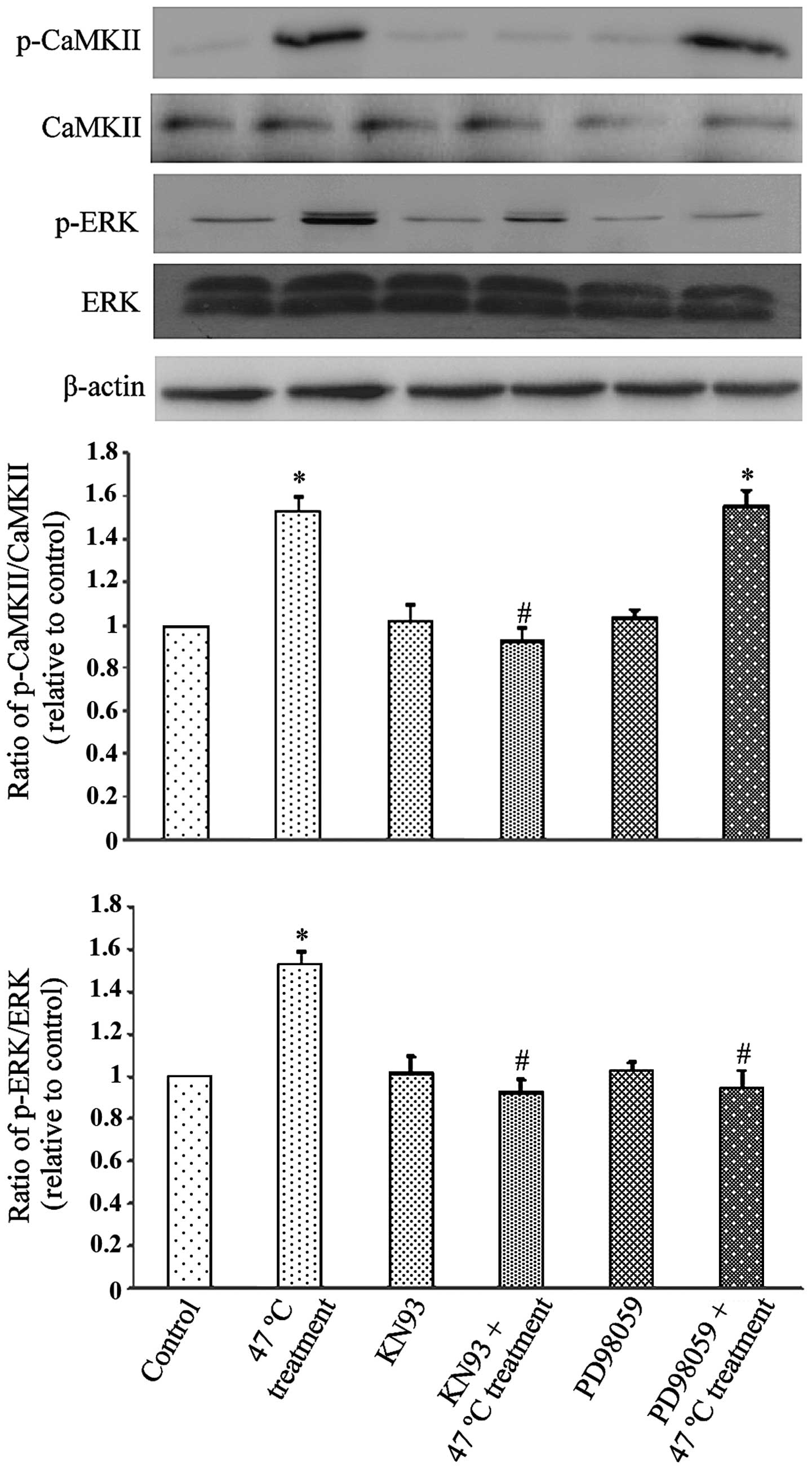
The possible interaction between CaMKII and ERK
following the 47°C treatment was investigated. As Fig. 2 demonstrates, the 47°C treatment
triggered a significant increase in ERK activation
(phosphorylation; P<0.05), which was inhibited by PD98059 and
KN93 (P<0.05). The 47°C treatment also triggered a significant
increase in CaMKII phosphorylation (P<0.05), which was only
blocked by KN93 (P<0.05) and not by the inhibitor of ERK,
PD98059. These observations indicate that CaMKII activation is
required for ERK activation following the 47°C heat treatment, but
not vice versa.
Effect of PD98059, KN93 and axitinib on
SMMC7721 cell proliferation
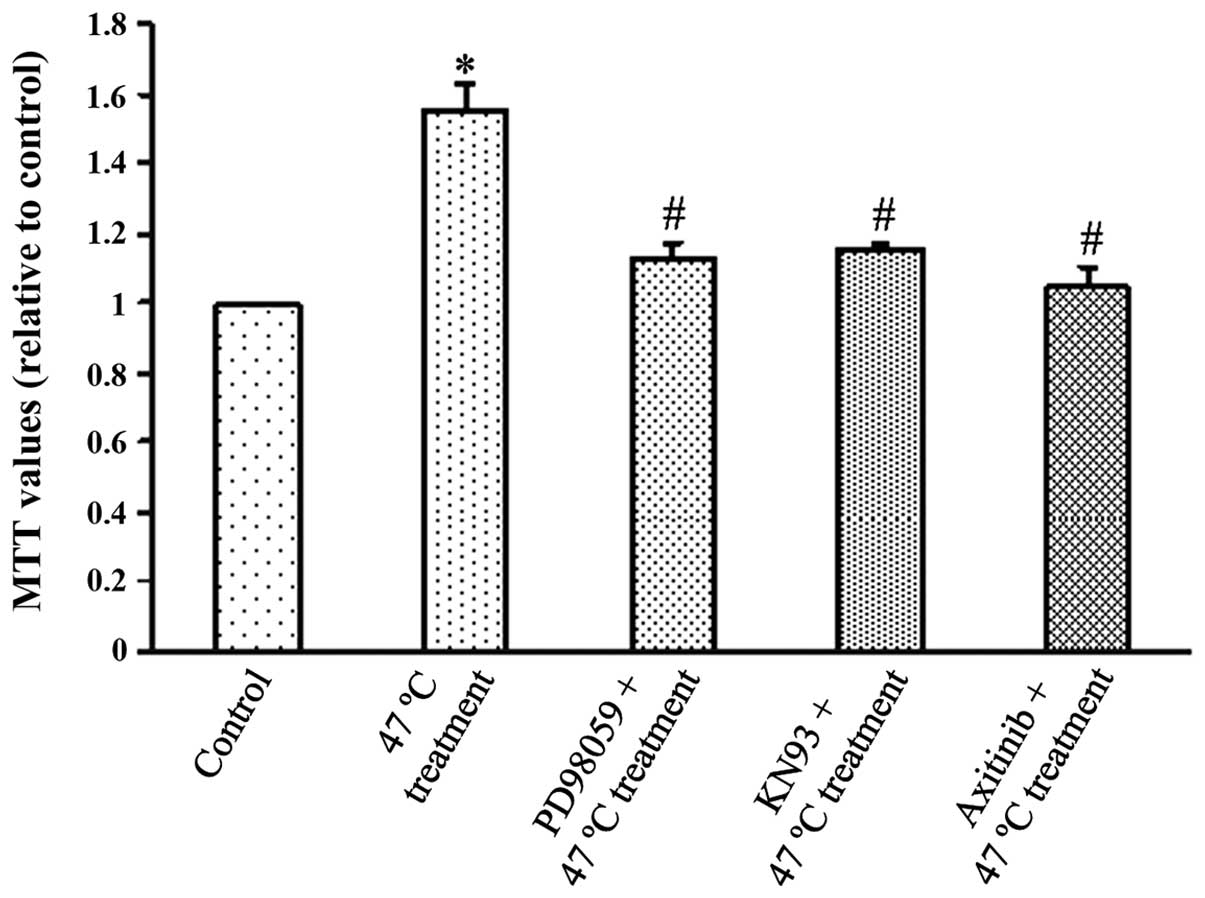
Fig. 3 demonstrates
that SMMC7721 cells in the 47°C treatment group exhibited
significantly higher viability compared with the control group at
48 h after the 47°C treatment (P<0.05), indicating that the 47°C
heat stimulation increased cell proliferation. Notably, PD98059 or
KN93 pretreatment inhibited this effect (P<0.05), indicating
that ERK and CaMKII may act to increase tumour cell proliferation.
Axitinib, a VEGF receptor (VEGFR) antagonist, also inhibited
SMMC7721 cell viability (P<0.05), demonstrating that elevated
VEGF may exert its growth promoting effects through VEGFR.
Discussion
As a result of its multiple advantages, RFA is
currently widely used for HCC treatment as an alternative to
traditional surgery. However, mounting clinical and experimental
evidence has revealed that RFA treatment may cause rapid growth of
any residual HCC (8,15). However, the underlying mechanisms
and factors that mediate this rapid growth of residual HCC
following RFA remain unclear. Several mediators have been proposed
to be involved in this undesirable process, including increased
VEGFA expression (9). VEGFA is the
most important angiogenic molecule of the VEGF family, which
includes VEGFA, VEGFB, VEGFC, VEGFD and placental growth factor
(PLGF) (9). The present study did
not differentiate between the VEGF subtypes, but identified a
similar enhanced expression of total VEGF following a 47°C
treatment regimen, which was used to simulate RFA. In addition, the
specific VEGFR inhibitor, axitinib (16), was found to greatly suppress 47°C
heat stimulated tumor cell proliferation. Therefore, VEGF
upregulation that was triggered by the heat stimulation exerted its
proliferative effect by binding to and activating its receptor,
VEGFR.
Subsequently, the underlying mechanism of this
enhanced VEGF expression was investigated. The Raf/MEK/ERK
signaling pathway is downstream of Ras activation, and tyrosine
phosphorylation of these signaling molecules is essential to cancer
cell proliferation (17). A recent
study identified that HCC exhibited increased expression of
phospho-ERK and enhanced proliferation following insufficient RFA
(13). In addition, the specific
ERK inhibitor, PD98059, significantly suppressed the malignancy of
HCC in a previous study (13), as
well as in the present study, demonstrating a critical role of ERK
in insufficient RFA-induced HCC malignancy. The present study,
demonstrated that VEGF overexpression following the 47°C treatment
regimen was significantly downregulated by PD98059, demonstrating
that upregulated VEGF expression by heat stimulation was
ERK-dependent.
CaMKII is a ubiquitous mediator of
Ca2+-associated signaling that phosporylates various
substrates to coordinate and regulate Ca2+-mediated
modifications in cellular function (18). ERK is one of the targets of CaMKII
and the CaMKII-ERK cascade has been identified in a recent study
(19). Furthermore, a previous
study demonstrated that the CaMKII-mediated activation of ERK
contributed to cell proliferation of papillary thyroid carcinoma
(20). Based on these observations,
the present study hypothesized that the ERK activation triggered by
the 47°C heat treatment may be CaMKII-dependent. ERK
phosphorylation was inhibited by PD98059 (an ERK inhibitor) and
KN93 (a specific inhibitor of CaMKII); however, PD98059 did not
affect CaMKII phosphorylation. CaMKII may operate upstream of ERK,
as this study initially hypothesized. Furthermore, the present
study confirmed that upregulated VEGF expression and tumor cell
proliferation were significantly decreased by KN93.
In conclusion, the present study described a
possible mechanism of the growth promoting effects of RFA on
residual HCC. RFA promoted residual HCC growth through increasing
VEGF expression via CaMKII-induced ERK activation. These results
may improve the understanding of the mechanisms of residual HCC
progression and relapse, and thus provide novel, effective targets
for the prevention and treatment of this undesirable effect during
RFA therapy.
References
|
1
|
Venook AP, Papandreou C, Furuse J and de
Guevara LL: The incidence and epidemiology of hepatocellular
carcinoma: a global and regional perspective. Oncologist. 15(Suppl
4): 5–13. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lau WY, Lai EC and Lau SH: The current
role of neoadjuvant/adjuvant/chemoprevention therapy in partial
hepatectomy for hepatocellular carcinoma: a systematic review.
Hepatobiliary Pancreat Dis Int. 8:124–133. 2009.PubMed/NCBI
|
|
4
|
Menon KV, Hakeem AR and Heaton ND: Review
article: liver transplantation for hepatocellular carcinoma - a
critical appraisal of the current worldwide listing criteria.
Aliment Pharmacol Ther. 40:893–902. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bryant R, Laurent A, Tayar C, van Nhieu
JT, Luciani A and Cherqui D: Liver resection for hepatocellular
carcinoma. Surg Oncol Clin N Am. 17:607–633. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lencioni R: Loco-regional treatment of
hepatocellular carcinoma. Hepatology. 52:762–773. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lau WY and Lai EC: The current role of
radiofrequency ablation in the management of hepatocellular
carcinoma: a systematic review. Ann Surg. 249:20–25. 2009.
View Article : Google Scholar
|
|
8
|
Obara K, Matsumoto N, Okamoto M, et al:
Insufficient radiofrequency ablation therapy may induce further
malignant transformation of hepatocellular carcinoma. Hepatol Int.
2:116–123. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kong J, Kong J, Pan B, et al: Insufficient
radiofrequency ablation promotes angiogenesis of residual
hepatocellular carcinoma via HIF-1α/VEGFA. PloS One. 7:e372662012.
View Article : Google Scholar
|
|
10
|
Zhao K, Song X, Huang Y, et al: Wogonin
inhibits LPS-induced tumor angiogenesis via suppressing
PI3K/Akt/NF-κB signaling. Eur J Pharmacol. 737:57–69. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kerbel RS: Tumor angiogenesis. N Engl J
Med. 358:2039–2049. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kaseb AO, Morris JS, Hassan MM, et al:
Clinical and prognostic implications of plasma insulin-like growth
factor-1 and vascular endothelial growth factor in patients with
hepatocellular carcinoma. J Clin Oncol. 29:3892–3899. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dong S, Kong J, Kong F, et al:
Insufficient radiofrequency ablation promotes
epithelial-mesenchymal transition of hepatocellular carcinoma cells
through Akt and ERK signaling pathways. J Transl Med. 11:2732013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lowry OH, Rosebrough NJ, Farr AL and
Randall RJ: Protein measurement with the Folin phenol reagent. J
Biol Chem. 193:265–275. 1951.PubMed/NCBI
|
|
15
|
Yamada S, Utsunomiya T, Morine Y, et al:
Expressions of hypoxia-inducible factor-1 and epithelial cell
adhesion molecule are linked with aggressive local recurrence of
hepatocellular carcinoma after radiofrequency ablation therapy. Ann
Surg Oncol. 21(Suppl 3): S436–S442. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Santoni M, Berardi R, Amantini C, et al:
Role of natural and adaptive immunity in renal cell carcinoma
response to VEGFR-TKIs and mTOR inhibitor. Int J Cancer.
134:2772–2777. 2014. View Article : Google Scholar
|
|
17
|
Xue L, Li M, Chen T, et al: PE-induced
apoptosis in SMMC-7721 cells: Involvement of Erk and Stat
signalling pathways. Int J Mol Med. 34:119–129. 2014.PubMed/NCBI
|
|
18
|
Maier LS: Role of CaMKII for signaling and
regulation in the heart. Front Biosci (Landmark Ed). 14:486–496.
2009. View Article : Google Scholar
|
|
19
|
Russo E, Salzano M, De Falco V, et al:
Calcium/Calmodulin-dependent protein kinase II and its endogenous
inhibitor alpha in medullary thyroid cancer. Clin Cancer Res.
20:1513–1520. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rusciano MR, Salzano M, Monaco S, et al:
The Ca2+-calmodulin-dependent kinase II is activated in papillary
thyroid carcinoma (PTC) and mediates cell proliferation stimulated
by RET/PTC. Endocr Relat Cancer. 17:113–123. 2010. View Article : Google Scholar
|