Introduction
Mitogen-activated protein kinases (MAPKs), key
signal-transducing enzymes that are activated by a wide range of
extracellular stimuli, play an important role in a variety of
cellular processes, including proliferation, differentiation and
apoptosis (1,2). The MAPK superfamily consists of three
major subfamilies, extracellular signal-regulated kinases (ERKs),
c-Jun N-terminal kinases (JNKs) and p38 MAP kinases. It is well
established that ERK1/2 are typically stimulated by growth-related
stimuli, while JNK and p38 are primarily activated by
stress-related signals such as UV irradiation, osmotic shock and
inflammatory cytokines (1,2).
MAPKs are activated by phosphorylation on both the
threonine and tyrosine residues of a conserved T-X-Y motif within
the activation loop of the kinase. Negative regulation of MAPKs is
achieved by dephosphorylation of the T-X-Y motif by phosphatases
(3). There are ten MAPK
phosphatases (MKPs) that act as negative regulators of MAPKs. These
can be subdivided into three distinct groups (3). Subgroup I contains the inducible
nuclear MKPs, MKP-1, MKP-2, PAC1 and hVH-3, which target ERK, JNK
and p38. Subgroup II contains ERK-specific cytoplasmic MKPs,
encoded by MKP-3, MKP-4 and MKP-X. Subgroup III consists of three
p38- and JNK-specific MKPs, encoded by MKP-5/DUSP10, MKP-7 and
hVH-5.
The roles of MKPs in the regulation of MAPK pathways
in normal cells suggest that the possible pathological consequences
of either a loss or gain in the function of these enzymes are part
of the oncogenic process (4–7). In
addition, MAPK signaling plays a key role in determining the
response of tumor cells to conventional cancer therapies (7). There is increasing evidence that some
MKPs may be abnormally regulated in certain tumors (4–6).
Whether overexpression or loss of expression is a cause of, or
contributes to the malignant phenotype rather than simply being a
consequence of cell transformation remains ambiguous.
MKP-5 was identified as a phosphatase that
selectively inactivates JNK and p38, but not ERK1/2 (8–10).
MKP-5 mRNA is widely expressed in various tissues and
organs, and its expression in cultured cells is elevated by stress
stimuli, such as UV, anisomycin, osmotic stress and tumor necrosis
factor-α treatment, which are known to activate JNK or p38
(8,9,11).
MKP-5 was also identified as a gene that is induced by DNA
double-strand break in an ATM-dependent manner (12). Suppression of MKP-5 induces
oligodendrocyte differentiation by regulating ERK function,
suggesting an unidentified role in the ERK pathway (13).
To clarify whether or not MKP-5 is a genuine
regulator of the JNK and p38 pathways, we analyzed the interactions
between MKP-5 and the three MAPKs, JNK, p38 and ERK, and found that
it can also interact with ERK. Furthermore, a time course analysis
of ERK activation showed that MKP-5 induced, enhanced and prolonged
ERK phosphorylation, suggesting an MKP-5 function in the ERK
pathway. This data led us to investigate a novel MKP-5 function as
a scaffold protein. Accumulating evidence of altered expression in
parts of subgroup I and II MKPs in cancer tissues suggests that
these phosphatases can act as tumor regulators depending on the
cancer type (4–6). In contrast, involvement of subgroup
III MKPs in tumor formation and progression has not been thoroughly
examined. To obtain insight in the role of MKP-5 in tumor
formation, we examined its mRNA expression levels and found that
MKP-5 is frequently overexpressed in colon tumors.
Materials and methods
Expression vectors
pFLAG-MKP-2, pFLAG-MKP-3 and pFLAG-MKP-5 have been
previously described (10,14). pFLAG-MKP-5CS (C428S), phosphatase
dead mutant, was generated by site-directed mutagenesis using the
QuikChange site-directed mutagenesis system (Stratagene, Garden
Grove, CA, USA) and subcloned into pFLAG-CMV2. pCX4-bleo-RSK1 and
pSRα-HA-ERK2 were a gift from Dr T. Akagi (Kan Institute) and Dr M.
Karin (University of California, San Diego, CA, USA), respectively.
pCMV-β-galactosidase has been previously described (15). pSRE-Luc was obtained from
Stratagene.
Cell culture, DNA transfection and
stimulation
HeLa, HEK293 and COS-7 cells were maintained in
Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal
bovine serum (FBS) at 37°C under 5% CO2. Cells were
co-transfected with various pFLAG-MKP expression vectors together
with pSRα-HA-ERK2, pSRα-HA-JNK1 or pMT3-HA-p38α. For transient
assays, cells were transfected using the Fugene-6 transfection
reagent (Roche Diagnostics Inc., Manheim, Germany) according to the
manufacturer's recommendations. Eighteen hours later, cells were
starved for 18 h and then exposed to 10 ng/ml PMA (phorbol
12-myristate 13-acetate; Sigma-Aldrich Corp., Saint Louis, MO, USA)
for 15 min or 0.4 M sorbitol for 30 min.
Detection of expressed proteins
Cell lysates were prepared as previously described
(10). Samples were separated on
10% SDS-PAGE gels and transferred to nitrocellulose membranes.
Expression levels of HA-tagged MAPKs and FLAG-tagged MKPs were
monitored using anti-HA (12CA5; Roche Diagnostics, Inc.) and
anti-FLAG M2 (Sigma-Aldrich Corp.) monoclonal antibodies,
respectively. ERK2 activation was monitored by an
anti-phospho-ERK1/2 antibody (Thr202/Tyr204; Cell Signaling
Technology, Inc., Danvers, MA, USA). The amount and activation of
RSK1 (p90 ribosomal S6 kinase1) were monitored using anti-RSK (BD
Biosciences, Franklin Lakes, NJ, USA) and anti-phospho-p90RSK
(Thr359/Ser363; Cell Signaling Technology, Inc.) antibodies,
respectively. Signals were detected by enhanced chemiluminescence
using the ECL reagent (Amersham Pharmacia Biotech, Piscataway, NJ,
USA) and Fuji LAS 4000 mini (Fuji Film Co., Tokyo, Japan).
Cell staining
HeLa cells on coverslips coated with Vitrogen 100
(Collagen Biochemical, Palo Alto, CA, USA) were co-transfected with
pFLAG-MKP-5 and pSRα-HA-ERK2. Transfected cells were fixed in PBS
(phosphate-buffered saline) containing 3.7% formaldehyde for 10 min
and then permeabilized with PBS containing 0.5% Triton X-100 for 5
min. After incubation in PBS containing 3% BSA (PBS-B) for 2 h,
cells were incubated overnight at 4°C in PBS-B containing an
anti-FLAG polyclonal antibody (provided by Dr K. Yamashita,
Kanazawa University) to detect FLAG-tagged proteins and an anti-HA
antibody to detect HA-tagged proteins. After three PBS washes,
cells were incubated for 20 min at 37°C with AlexaFluor
488-conjugated goat anti-mouse IgG (H+L) antibody (Molecular
Probes, Eugene, OR, USA) or AlexaFluor 546-conjugated goat
anti-rabbit IgG (H+L) antibody (Molecular Probes) in PBS-B. After
three PBS washes, coverslips were mounted with PBS containing 90%
glycerol. Fluorescence was visualized using a fluorescence
microscope.
Reporter analysis
HEK293 cells were transfected with pSRE-Luc together
with pCMV-β-galactosidase and MKP expression vectors. Eighteen
hours later, cells were serum-starved for 12 h, and then treated
with 10 ng/ml PMA. Six hours later, cells were harvested and washed
with PBS twice, and then lysed with LCβ lysis buffer (Toyo Ink,
Tokyo, Japan). Cell lysates were obtained using a freeze-thaw
treatment and centrifugation. Luciferase activity in the lysates
was measured with Picagene (Toyo Ink) using MicroLumat Plus LB96V
(Belthold, Bad Wildbad, Germany). Luciferase activities were
normalized to the activity of the co-expressed β-galactosidase,
which was assayed as previously described (15).
Quantitative reverse transcription
PCR
Total RNA was prepared from the specimens with
Isogen reagent (Nippon Gene, Tokyo, Japan) according to
manufacturer's instructions. cDNA was synthesized using an
oligo(dt)12–18 primer with Superscript III reverse transcriptase
(Invitrogen, Carlsbad, CA, USA) and applied to quantitative
real-time PCR (qRT-PCR) using LightCycler 480 (Roche Diagnostics,
Inc.) with the universal probe library and probe master kit (Roche
Diagnostics, Inc.). The level of MKP-5 mRNA in tumors was
expressed as a ratio relative to one surrounding a non-cancerous
region.
Statistical analysis
The expression of MKP-5 mRNA in the colon and
lung specimens was analyzed by t-test. A difference between the
non-cancerous and the cancer portion with P<0.05 was
statistically significant.
Results
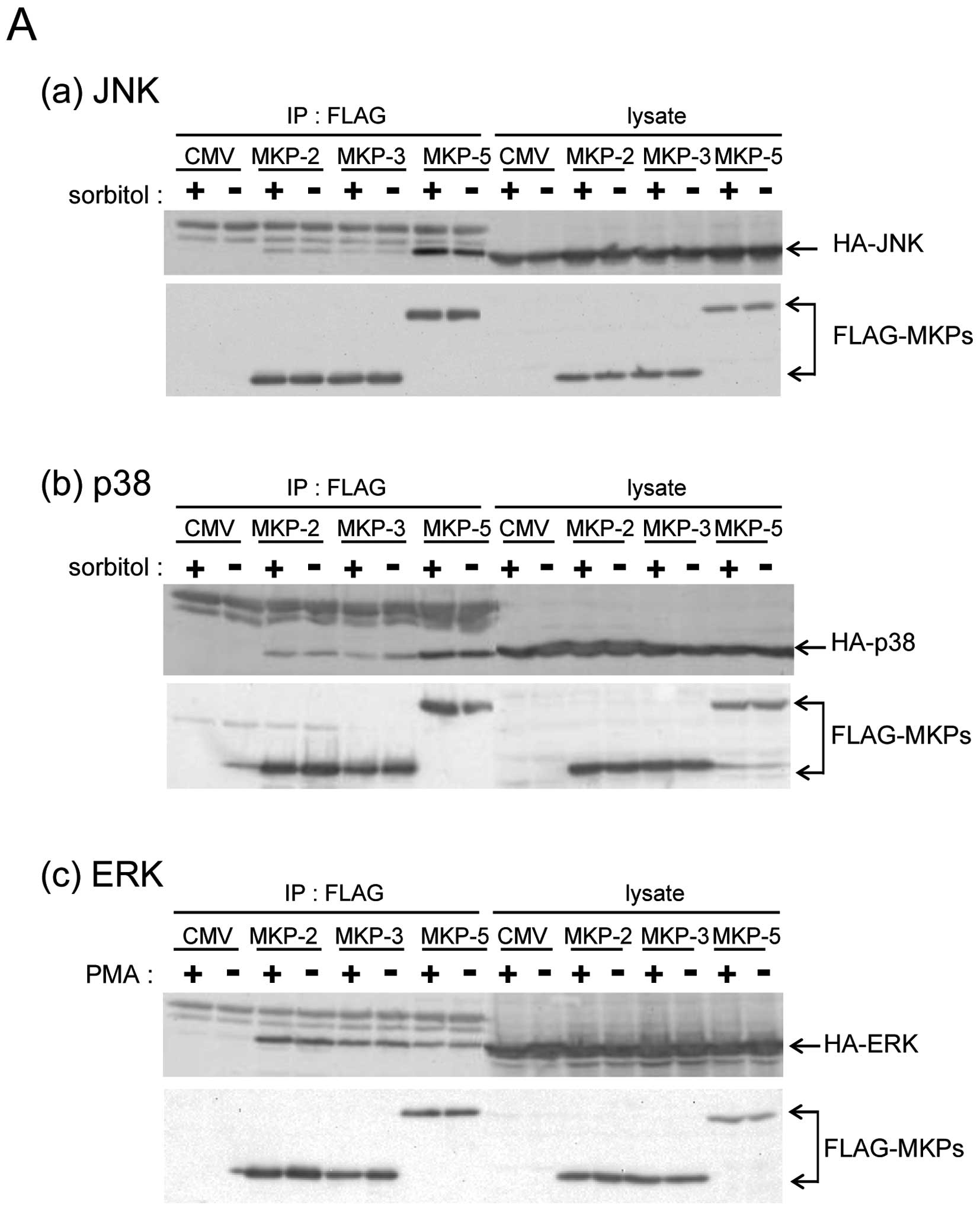
MKP-5 interacts with ERK
To clarify whether MKP-5 only regulates the JNK and
p38 pathways, we compared the interaction of MKP-5 with JNK, p38
and ERK, using the immunoprecipitation method. Compared with
FLAG-MKP-2 and FLAG-MKP-3 (belonging to the MKP-subgroup I and II,
respectively) FLAG-MKP-5 had a much higher binding specificity to
HA-JNK1 and HA-p38α (Fig. 1A-a and
b), in accordance with a previous report (3). Interestingly, HA-ERK was
co-immunoprecipitated with FLAG-MKP-5, although MKP-5 did not
possess ERK phosphatase activity (8,9). The
amount of HA-ERK2 that co-immunoprecipitated with FLAG-MKP-5 was
similar to the amounts that co-immunoprecipitated with FLAG-MKP-2
and FLAG-MKP-3, both of which preferentially dephosphorylate ERK
(Fig. 1A-c). These results indicate
that MKP-5 binds ERK similarly to ERK phosphatases such as MKP-2
and MKP-3. Next, we examined the co-localization of FLAG-MKP-5 and
HA-ERK in cells (Fig. 1B).
Immunostaining with anti-HA antibody showed that after PMA
treatment HA-ERK2 was localized primarily in the nucleus (small
arrow heads). However, in cells where FLAG-MKP-5 was co-expressed,
HA-ERK2 was detected in the cytoplasm (wide arrow heads). This
suggests that MKP-5 retains ERK in the cytoplasm even after PMA
treatment.
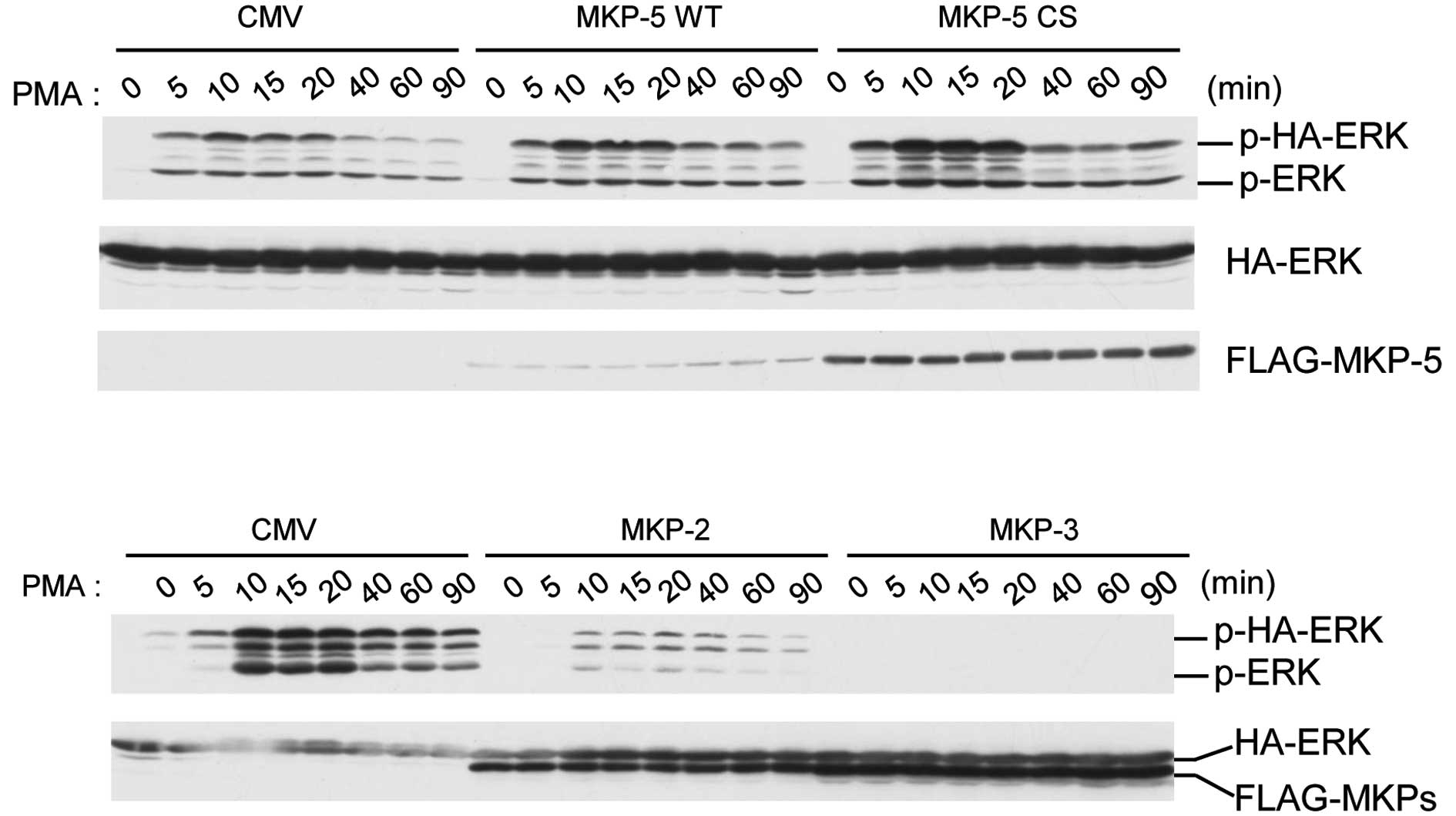
MKP-5 induces enhanced and prolonged
PMA-stimulated ERK phosphorylation, without requiring its enzyme
activity
To clarify the physiological meaning of the
interaction between MKP-5 and ERK, we analyzed the time-course of
PMA-stimulated ERK in the presence of MKP-5 in COS-7 cells
(Fig. 2). The phosphorylation
levels of HA-ERK2 reached maximal levels 15 min after PMA
stimulation, and decreased to basal levels by 90 min (Fig. 2, upper panel). However, when
FLAG-MKP-5 or FLAG-MKP-5CS, a phosphatase dead mutant, were
co-expressed, we observed enhanced and prolonged phosphorylation of
HA-ERK2 (Fig. 2, upper panel). In
contrast, co-expression of MKP-2 and MKP-3 suppressed ERK
activation for at least 90 min after PMA treatment (Fig. 2, lower panel). These results
indicate that FLAG-MKP-5 upregulates ERK phosphorylation and that
this effect does not require MKP-5 enzyme activity.
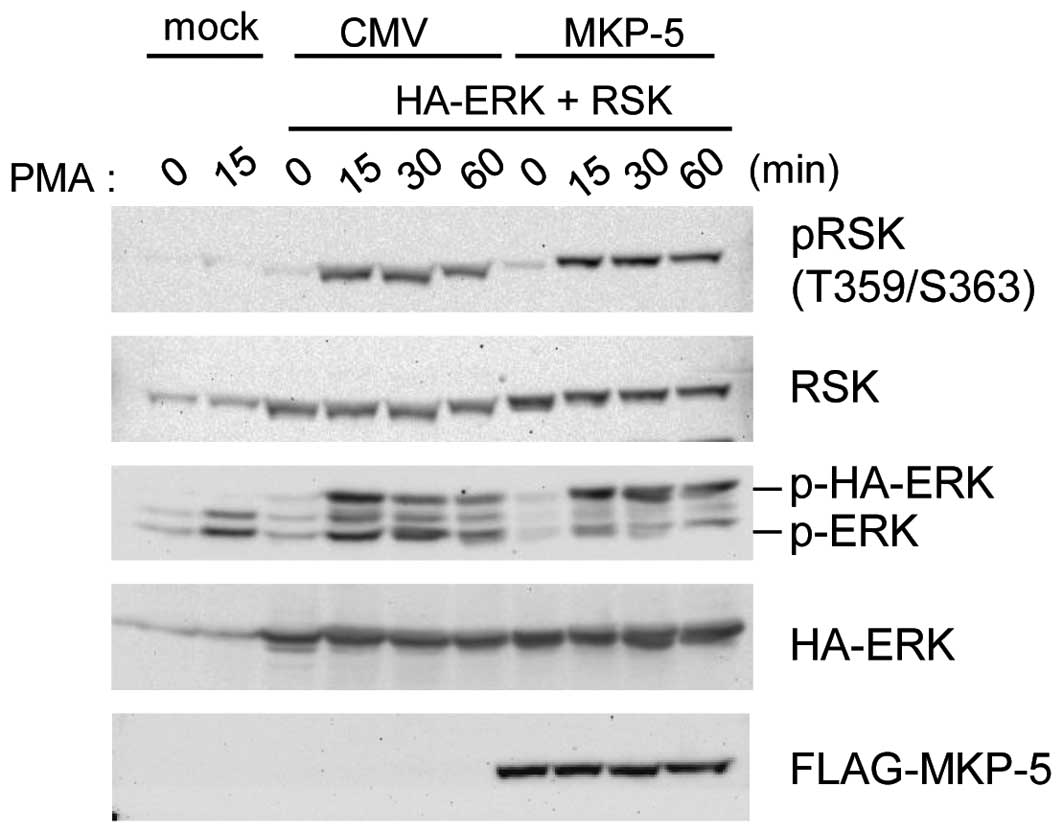
Phosphorylated ERK in MKP-5 co-expressing
cells is not active
When FLAG-MKP-5 was co-expressed, phospho-HA-ERK2
accumulated (Fig. 2) and was
retained in the cytoplasm (Fig.
1B). Thus it is possible that ERK substrates in the cytoplasm
are highly phosphorylated. We examined this possibility by
comparing phosphorylation levels of Thr-359 and Ser-363 of RSK1, a
cytoplasmic ERK substrate, in the presence or absence of
FLAG-MKP-5. As shown in Fig. 3,
sustained HA-ERK2 phosphorylation was detected in
FLAG-MKP-5-expressing cells, while enhanced RSK1 phosphorylation at
Thr-359 and Ser-363 was not observed. This data suggests that when
bound to MKP-5, phospho-ERK does not phosphorylate RSK1 as much as
non-bound phospho-ERK.
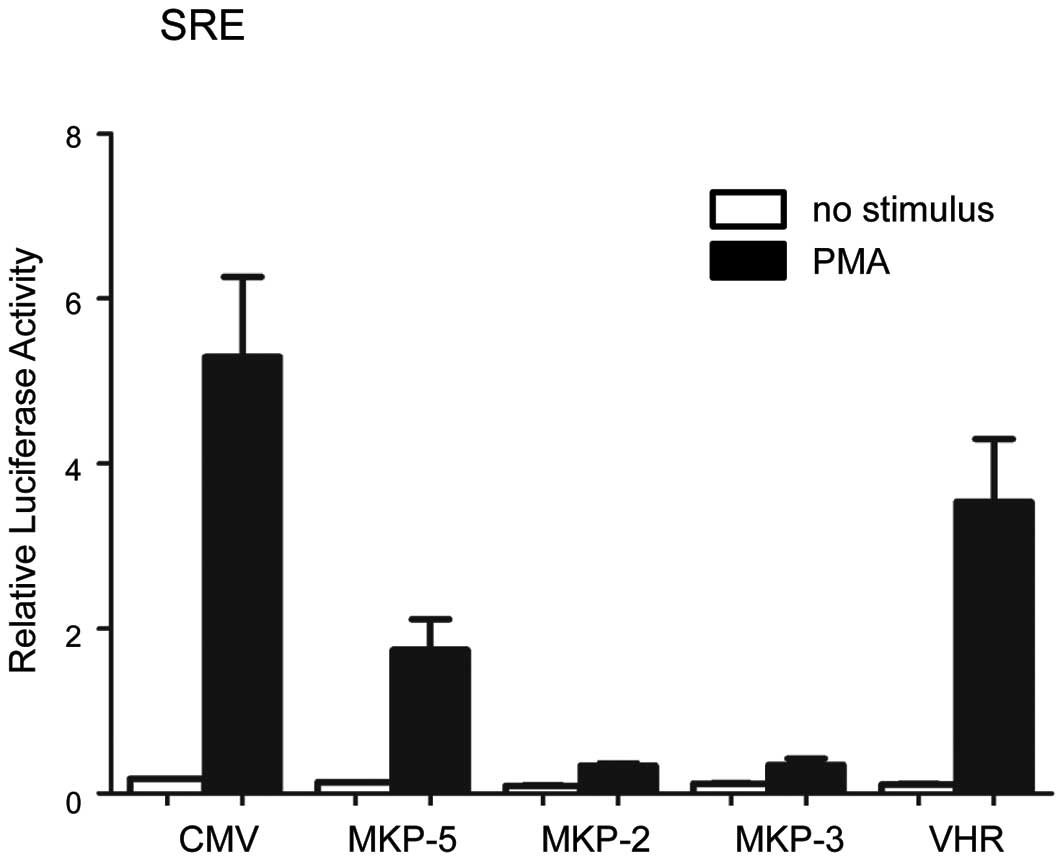
MKP-5 prevents ERK-dependent
transcriptional activation
The effect of MKP-5 on ERK-dependent transcriptional
regulation was analyzed using SRE (serum responsive element)-driven
luciferase reporter constructs (Fig.
4). MKP-2 and MKP-3, which dephosphorylate ERK effectively
(3), inhibited SRE-dependent
transcription almost completely. Under the same conditions, MKP-5
suppressed SRE-dependent transcription by 67%, whereas VHR, a
dual-specificity phosphatase that negatively regulates ERK
signaling (16) showed 33%
inhibition. These results show that MKP-5, which was believed to be
a specific regulator of gene expression through the JNK and p38
pathway, also downregulates ERK-dependent gene expression.
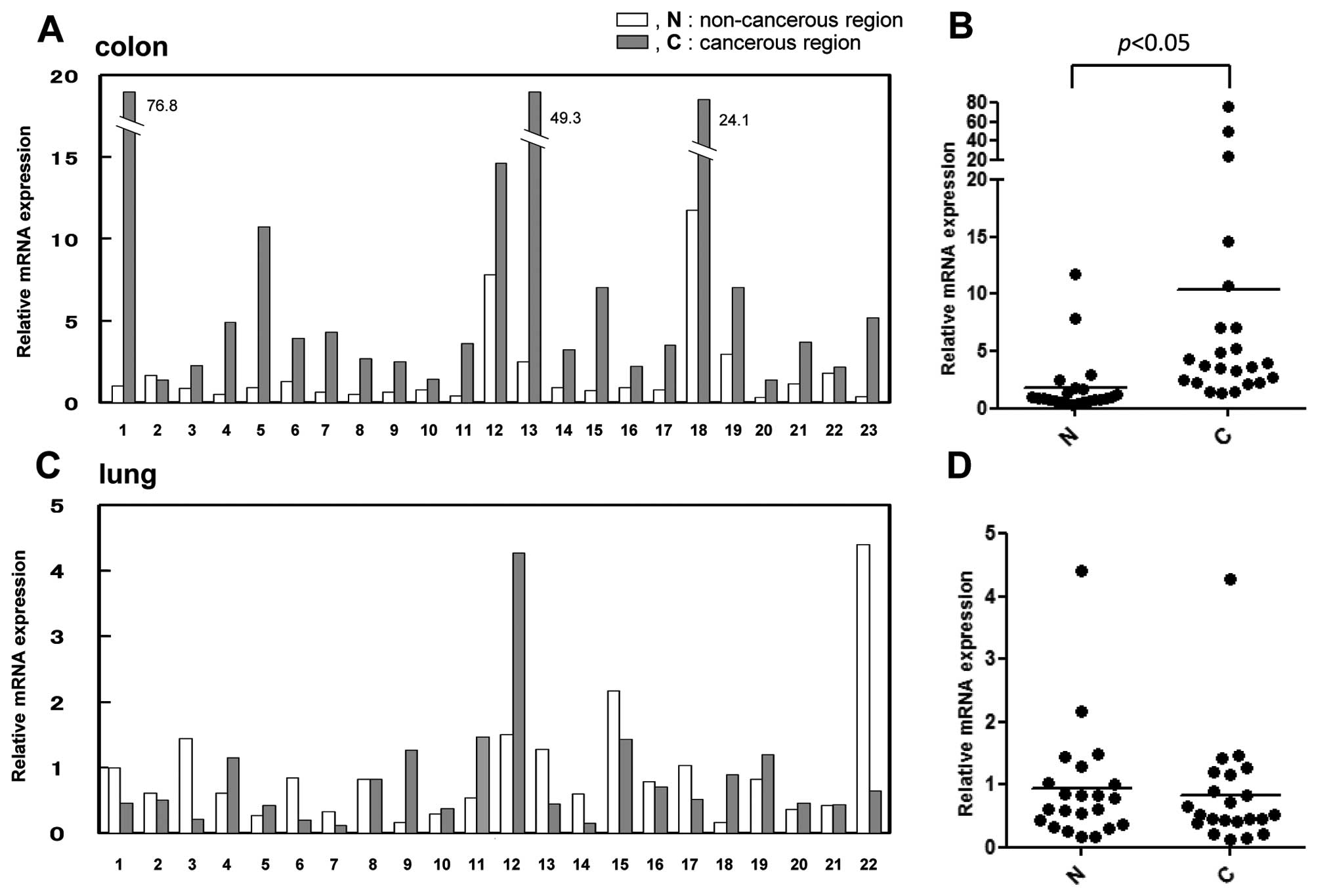
Upregulation of the MKP-5 gene in colon
carcinoma
Previous data revealed that in addition to the
significant negative regulation of the JNK and p38 pathways by
phosphatase activity, MKP-5 negatively regulates the ERK pathway as
a scaffold protein. This data suggested that, as a whole, the
function of MKP-5 on the three MAPK pathways is similar to that of
MKP-1 (JNK = p38 > ERK), which is regarded as a tumor regulator
according to the cancer type (5,6). To
obtain further insight on the role of MKP-5 in tumor formation, we
examined the expression levels of the MKP-5 gene in colon
carcinoma (Fig. 5A) and lung
carcinoma (Fig. 5C). The expression
levels of MKP-5 in colon cancerous tissues increased by
5-fold compared to the non-cancerous region of the same samples
(Fig. 5B). In contrast,
MKP-5 gene expression in lung cancer tissues was relatively
similar compared to that of non-cancerous regions (Fig. 5D). In breast cancer and glioblastoma
samples, there was no significant change in the expression between
tumor and normal samples (data not shown). This data suggests that
upregulation of MKP-5 was specific to colorectal carcinoma
tissues.
Discussion
In the present study, we found that in addition to
JNK and p38, MKP-5 interacts with ERK, and that MKP-5 and its
phosphatase-dead mutant enhanced and prolonged mitogen-stimulated
ERK phosphorylation. Immunohistological analysis showed that MKP-5
functions as a cytoplasmic anchor for ERK. We also analyzed the
physiological consequence of blocking nuclear translocation of
phospho-ERK, and found that despite the enhanced levels of
phospho-ERK, phosphorylation of the cytoplasmic target RSK was not
enhanced. Importantly, MKP-5 suppressed SRE-driven gene expression.
These observations indicate that MKP-5 inhibits ERK-dependent gene
expression by preventing nuclear translocation of phospho-ERK and
preventing phospho-ERK from further phosphorylating and activating
RSK1.
Other protein phosphatases also regulate ERK
localization (2). Phosphotyrosine
phosphatases, such as PTP-SL, STEP and He-PTP, retain ERK in a
dephosphorylated form in the cytoplasm by tyrosine
dephosphorylation. MKP-3/DUSP6 also functions in cytoplasmic
retention of dephosphorylated ERK2. Therefore, these phosphatases
may function to dephosphorylate and regulate ERK. In contrast,
MKP-5 may function as a scaffold protein and not as a phosphatase
to anchor ERK in the cytoplasm.
Temporal and spatial control of MAPK signaling is
regulated by protein scaffolds (17,18).
Two proteins, PEA-15 and SEF, have been identified as cytoplasmic
anchors for ERK2 (18). Both can
retain ERK2 in the cytoplasm in the active state, indicating that
they may act to restrict ERK2 activity to cytoplasmic targets. In
contrast, our data indicates that MKP-5 does not lead to enhanced
RSK1 phosphorylation. Therefore, the MKP-5 interaction may inhibit
the kinase activity of ERK, or MKP-5 may occupy the ERK substrate
recognition site. In our study, MKP-5 induced enhanced and
prolonged phosphorylation of ERK. As PEA-15 induces prolonged ERK
phosphorylation (19), this
activity may be a common characteristic of ERK cytoplasmic scaffold
proteins (18). It is likely that
cytoplasmic retention prevents phospho-ERK from dephosphorylation
by nuclear phosphatases, such as MKP-1 and MKP-2.
To our knowledge, this is the first report
demonstrating the analysis of MKP-5 expression in malignant
tissues. We examined its expression levels in colon carcinoma, lung
carcinoma, breast cancer and glioblastomas. Overexpression of the
MKP-5 gene was observed in colon carcinoma, but no
significant up- or downregulation was observed in lung carcinoma,
breast cancer and glioblastomas. It is unclear whether this
overexpression in colon carcinoma is a cause of or consequence of
cell transformation. With respect to the stress-activated MAP
kinases, their role in cancer can be complex, but JNK and p38 may
act as tumor suppressors in vivo(7). Since MKP-5 is a specific JNK and p38
phosphatase, this overexpression may direct cell transformation by
abrogating JNK and p38 activity. On the other hand, it is also
accepted that the ERK pathway is associated with the ability of
cancer cells to grow. In this study, we found that MKP-5 negatively
regulated ERK-dependent gene expression. MKP-5 gene
expression may be elevated as a negative feedback mechanism to
counteract ERK activation. Importantly, data suggested that
anti-inflammation activity by vitamin D and curcumin is mediated by
MKP-5, which is a potential mechanism for prostate cancer
prevention (20,21). In the mouse model of colon cancer,
which was induced by azoxymethane and dextran sulfate sodium, ERK
phosphorylation was significantly increased by about 10-fold
(22). The vitamin D analog
suppressed this ERK phosphorylation and progression to cancer. We
believe that the present findings on the novel function of MKP-5
will aid in the utilization of MKP-5 as a molecular target for
cancer prevention and therapy.
Evidence presented in our study strongly suggest
that MKP-5, a JNK and p38 phosphatase, inhibits ERK-dependent gene
expression by blocking nuclear accumulation of phospho-ERK and
suppressing activation of phospho-ERK as a kinase. These findings
suggest that deregulation of MKP-5 expression may affect the
cross-talk between stress signaling and ERK signaling in these
cancer tissues, and is associated with the malignant phenotype of
colon tumors.
Acknowledgements
We thank Dr K. Yamashita (Kanazawa University) for
the anti-FLAG antibody. We thank Dr M. Karin (University of
California) for pSRα-HA-ERK2. This study was supported in part by
grants-in-aid for Scientific Research (Challenging Exploratory
Research, B and C) provided by the Japan Society for the Promotion
of Science (to K.-I.S., Masami Sato and H.S.).
References
|
1
|
Chang L and Karin M: Mammlian MAP kinase
signalling cascades. Nature. 410:37–40. 2001.
|
|
2
|
Boutros T, Chevet E and Metrakos P:
Mitogen-activated protein (MAP) kinase phosphatase regulation:
roles in cell growth, death, and cancer. Pharmacol Rev. 60:261–310.
2008.
|
|
3
|
Owens DM and Keyse SM: Differential
regulation of MAP kinase signaling by dual-specificity protein
phosphatases. Oncogene. 26:3203–3213. 2007.
|
|
4
|
Keyse SM: Dual-specificity MAP kinase
phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 27:253–261.
2008.
|
|
5
|
Haagenson KK and Wu GS: Miotogen activated
protein kinase phosphatases and cancer. Cancer Biol Ther.
9:337–340. 2010.
|
|
6
|
Bermudez O, Pages G and Gimond C: The
dual-specificity MAP kinase phosphatases: critical roles in
development and cancer. Am J Physiol Cell Physiol. 299:C189–C202.
2010.
|
|
7
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009.
|
|
8
|
Tanoue T, Moriguchi T and Nishida E:
Molecular cloning and characterization of a novel dual specificity
phosphatase, MKP-5. J Biol Chem. 274:19949–19956. 1999.
|
|
9
|
Theodosiou A, Smith A, Gillieron C, et al:
MKP5, a new member of the MKP kinase phosphatase family, which
selectively dephosphorylates stress-activated kinases. Oncogene.
18:6981–6988. 1999.
|
|
10
|
Masuda K, Shima H, Watanabe M and Kikuchi
K: MKP-7, a novel mitogen-activated protein kinase phosphatase,
functions as a shuttle protein. J Biol Chem. 276:39002–39011.
2001.
|
|
11
|
Masuda K, Shima H, Kikuchi K, et al:
Expression and comparative chromosomal mapping of MKP-5 genes
DUSP10/Dusp10. Cytogenet Cell Genet. 90:71–74. 2000.
|
|
12
|
Bar-Shira A, Rashi-Elkeles S, Zlochover L,
et al: ATM-dependent activation of the gene encoding MAP kinase
phosphatase 5 by radiomimetic DNA damage. Oncogene. 21:849–855.
2002.
|
|
13
|
Gobert RP, Joubert L, Curchod ML, et al:
Convergent functional genomics of oligodendrocyte differentiation
identifies multiple autoinhibitory signaling circuits. Mol Cell
Biol. 29:1538–1553. 2009.
|
|
14
|
Katagiri C, Masuda K, Urano T, et al:
Phosphorylation of Ser-446 determines stability of MKP-7. J Biol
Chem. 280:14716–14722. 2005.
|
|
15
|
Katagiri C, Masuda K, Nomura M, et al:
DUSP13B/TMDP inhibits stress-activated MAPKs and suppresses
AP-1-dependent gene expression. Mol Cell Biochem. 352:155–162.
2011.
|
|
16
|
Todd JL, Tanner KG and Denu JM:
Extracellular regulated kinases (ERK) 1 and ERK2 are authentic
substrates for the dual-specificity protein-tyrosine phosphatase
VHR. J Biol Chem. 274:13271–13280. 1999.
|
|
17
|
Brown MD and Sacks DB: Protein scaffolds
in MAP kinase signaling. Cell Signal. 21:462–469. 2009.
|
|
18
|
Ebisuya M, Kondoh K and Nishida E: The
duration, magnitude and compartmentalization of ERK MAPK kinase
activity: mechanisms for providing signaling specificity. J Cell
Sci. 118:2997–3002. 2005.
|
|
19
|
Formstecher E, Ramos JW, Fauquet M, et al:
PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev
Cell. 1:239–250. 2001.
|
|
20
|
Nonn L, Peng L, Feldman D and Peehl DM:
Inhibition of p38 by vitamin D reduces interleukin-6 production in
normal prostate cells via mitogen-activated protein kinase
phosphatase 5: Implication for prostate cancer prevention by
vitamin D. Cancer Res. 66:4516–4524. 2006.
|
|
21
|
Nonn L, Duong D and Peelh DM:
Chemopreventive anti-inflammatory activities of curcumin and other
phytochemicals mediated by MAP kinase phosphatase-5 in prostate
cells. Carcinogenesis. 28:1188–1196. 2007.
|
|
22
|
Fichera A, Little N and Bissonnette MA:
Vitamin D analogue inhibits colonic carcinogenesis in the AOM/DSS
model. Surg Res. 142:239–245. 2007.
|