Introduction
Lung cancer is one of the worst prognostic human
cancers, taking more lives than any other human cancer worldwide
(1). Progress in therapy for lung
cancer has made recent progress with the identification of
molecular markers such as EGFR and EML4-ALK (2–4).
Gefitinib (Tarceva) targeting EGFR and Crizotinib (Xalkori)
targeting EML4-ALK are leading examples of molecular
targeting in lung cancer therapeutics (2,4).
Patients with EGFR mutations have responded well to Tarceva.
However, many patients later relapse due to a secondary EGFR
mutation (T790M), decreasing the advantage of Tarceva treatment
with respect to patient prognosis and survival (5). In 2007, a new fusion protein,
EML4-ALK, was identified in a Japanese lung cancer patient
without EGFR or K-ras mutations (3). The incidence of this fusion protein is
greater in Asian patients without EGFR or K-ras
mutations (6). Encouragingly,
patients with EML4-ALK showed a dramatic response to an ALK
inhibitor (7) and in 2011, the FDA
approved Crizotinib for the treatment of non-small-cell lung cancer
(NSCLC) in patients with EML4-ALK fusion and aberrant ALK
expression. Overall mutation frequencies of EGFR and
EML4-ALK are ~25% and 1–7% (3,6),
respectively, which suggests that >65% of lung cancer patients
lack specific molecular targets for treatment. Identification of
new targets either for diagnosis or therapy is important in the
effort to reduce mortality by lung cancer.
NKX2-1 (TTF1 or TITF1 or
thyroid transcription factor-1) was originally identified in
thyroid and lung (8). Many efforts
have been made to identify characteristics of NKX2-1 not
only in thyroid but also in other organs, including the lung.
NKX2-1 was first thought to act as a lung oncogene (9,10), but
later study suggested that it may act as a tumor suppressor in lung
adenocarcinoma (11).
Notwithstanding the clear contribution of NKX2-1 to lung
development and homeostasis, it is not clear how to inhibit or
boost NKX2-1 for the treatment of lung cancer. This gene is not
clearly amplified, overexpressed, or mutated and as such no clear
agonistic or antagonistic molecular targets of NKX2-1 have been
developed for lung cancer.
In order to find potential lung cancer or general
thoracic malignancy-related markers associated with NKX2-1, we
screened other binding partners of NKX2-1, filtering out known type
II pneumocyte and clara cell markers. One of the interesting
binding partners of NKX2-1 is TSHR (12). NKX2-1 binds the promoter of
TSHR and promotes constitutive TSHR expression and
TSH/cAMP-induced negative regulation of TSHR (12,13).
Activating and inactivating mutations have been reported in many
thyroid-related diseases, such as hyper/hypothyroidism and thyroid
cancer (14,15). Most activating mutations are known
to be associated with autoimmune diseases of the thyroid (14,15).
Epigenetic changes in TSHR have been reported to cause a
significant down-regulation and decreased expression of TSHR
in thyroid cancer (16).
TSHR has not been studied in lung cancer or thoracic
malignancies except in a mutation screening of a lung metastasis
from thyroid cancer (17). No
TSHR mutation was found in the pulmonary metastasis of
thyroid cancer (17). As such, to
date it is not clear whether TSHR is genetically involved in
primary lung cancer development. Thus, we screened for TSHR
mutation, DNA copy number, and mRNA expression in 96 lung
adenocarcinoma and matched normal tissues. In addition to lung
adenocarcinoma, other thoracic malignancies including squamous cell
carcinoma (SCC) and malignant pleural mesothelioma (MPM) were
measured for TSHR protein expression by immunohistochemistry.
Materials and methods
Patient samples
The Committee on Human Research (CHR) of UCSF
reviewed and approved the application for the collection of tissue
samples from patients with thoracic malignancies (approval number:
10-03352). All samples were collected under the IRB approval and
written informed consents were obtained from all patients in this
study.
TSHR mutation screening
Ninety-six lung adenocarcinoma tissue samples
screened for our previous study (18) were used for TSHR mutation
screening. All coding regions of TSHR were screened using
PCR primers as previously described (19). Amplification was performed at
58–62°C using 20 ng of genomic DNA in 96 sample pairs, 10X PCR
buffer supplemented with 1.5 mM MgCl2, 10 pmol of each
primer, 50 mM of each dNTP, and 0.2 units of Taq polymerase
(Qiagen) in a total reaction volume of 25 μl. The PCR products were
cleaned up using 1 μl of SAP (1 U/μl), 0.1 μl of Exo1 enzyme (10
U/μl), 1 μl of 1X PCR buffer, 2.4 μl of 10X NEB buffer (type 3), 5
μl of the PCR product, and 15 μl of water. Bi-directional
sequencing was performed using the Taq dideoxy-terminator cycle
sequencing kit and an ABI 3730xl DNA analyzer (Applied Biosystems,
Foster City, CA, USA).
TSHR mRNA expression analysis in 96 lung
adenocarcinoma and matched normal tissues
RNA from 96 snap-frozen lung adenocarcinoma and
matched normal tissues was extracted with TRIzol (Invitrogen),
treated with DNase I (New England Biolabs), and purified with the
RNeasy RNA purification kit (Qiagen). cDNA was synthesized using a
reverse transcript RT III (Invitrogen). TSHR expression was
quantified by Taqman real-time PCR (ABI, 7900HT) using 96 lung
adenocarcinoma and matched normal tissues in triplicate
(TSHR expression probe: 4331182, ABI). GAPDH was
selected to normalize cDNA input. ΔCT 2 was calculated from an
expression difference between TSHR and GAPDH. ΔCT
values were then used to compare TSHR expression between
tumor and normal samples.
TSHR DNA copy number analysis in 96 lung
adenocarcinoma and matched normal tissues
DNA copy number analysis of TSHR was done in
the same 96 pairs of samples (TSHR DNA probe:
Hs01214599_cn). DNA copy number assay for TSHR was performed
using Taqman real-time PCR. All samples were analyzed in triplicate
and RNase P gene (VIC/Tamra probe) was used for internal control to
normalize DNA quantity. Taqman probes were labeled by
FAM/non-fluorescent quencher. Sample DNA (10 ng) were analyzed on a
384-well plate using 2X Taqman Universal PCR master mix with no
AmpErase UNG. Deletion or gain was determined by ΔCT calculation
and analyzed by software provided by ABI. Further data analysis was
also performed manually. After average ΔCT values of normal
controls were calculated, each ΔCT value of samples was divided by
this normal value. ΔΔCT values were then calculated and the
threshold level for copy number was set as <1.2 for deletion and
>2.6 for gain.
Immunohistochemistry (IHC)
Ten pairs of matched normal and lung adenocarcinoma
(AD) and squamous cell carcinoma (SCC), and five malignant pleural
mesothelioma (MPM) samples were stained with anti-TSHR antibody
(ab5492, Abcam). Sections (5 μm) were hydrated in xylene and graded
concentrations of ethanol, then steamed in citrate (Biogenex) for
20 min. Slides were then incubated at room temperature with a
blocking solution (10% goat serum in TBS and 1% BSA) for 1 h.
Blocking solution was then removed and the primary antibody was
applied. The slides were incubated at 4°C overnight. The slides
were incubated using reagents from the Invitrogen Histostain Plus
Broad Spectrum kit (85-9643) according to the protocol provided by
Invitrogen.
Results
Somatic TSHR mutation
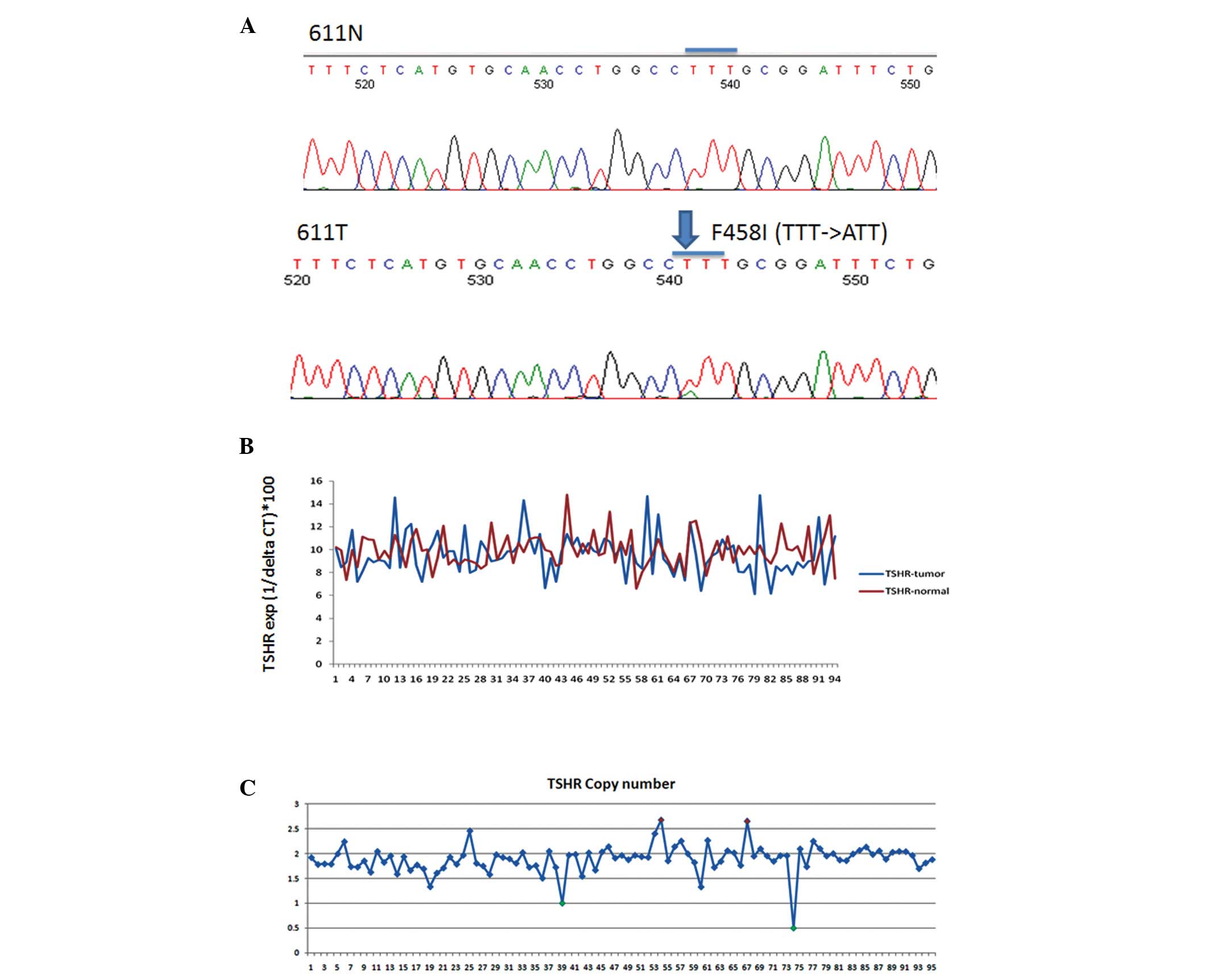
Out of 96 lung adenocarcinoma samples, one mutation
was identified in adenocarcinoma tissue (patient ID: 611) that did
not appear in matched normal tissue (Fig. 1A). A missense mutation at exon 10
(codon 458, TTT→ATT, F458I) was identified at the transmembrane
domain of TSHR (Fig. 1A).
The results suggest an overall observed TSHR mutation
frequency of 1% (one out of 96 AD). We investigated the prior
clinical history of the TSHR-mutated patient (patient ID:
611) to identify unique clinical insight. The patient was a
67-year-old (at the time of her second operation, during which the
specimen was collected) Caucasian woman with a history of coronary
artery disease, carotid stenosis, chronic obstructive pulmonary
disease (COPD) and a 50-pack-year smoking history who had presented
15 months earlier with a 3-year history of cough and 6-month
history of hoarseness. CT scan showed bilateral upper lobe lung
lesions, and fine needle aspiration suggested bilateral lung
adenocarcinoma. Her case was discussed at our institution’s
thoracic oncology tumor board, and the lesions were thought likely
to be two primary tumors. She first underwent left upper lobectomy,
and pathology showed stage IIB moderately differentiated
adenocarcinoma. Postoperatively, she received carboplatin and
gemcitabine for 2 cycles, then carboplatin and paclitaxel for 4
cycles. Imaging revealed stable disease, so she then underwent
right upper lobectomy, during which time our tumor and adjacent
lung specimens were collected. Pathology showed stage IB
adenocarcinoma with features of bronchioloalveolar carcinoma. This
patient had no signs or symptoms associated with thyroid cancer or
diseases (Table I). A systematic
review of English-language manuscripts available in the PubMed
database yielded no prior report that TSHR is associated
with COPD or coronary artery disease.
 | Table IPast medical and surgical history of
case 611 with TSHR mutation. |
Table I
Past medical and surgical history of
case 611 with TSHR mutation.
| History | Age |
|---|
| Coronary artery
disease, with history of myocardial infarction, status post
percutaneous coronary intervention with stent placement | 66 |
| Chronic obstructive
pulmonary disease (COPD) | |
| Bilateral severe
carotid stenosis, status post bilateral carotid
endarterectomies | 66 |
| Peripheral vascular
disease | |
| Hypertension | |
| Hyperlipidemia | |
| Hiatal hernia | |
| Gastroesophageal
reflux disease | |
| Diverticulosis | |
| Primary
hyperparathyroidism, status post parathyroidectomy | 33 |
| Nephrolithiasis,
status post bilateral nephrolithotomies | 33 |
| Pertussis, causing
right pneumothorax, status post right thoracotomy | 24 |
| Bilateral
cataracts, status post bilateral cataract repair | 62 |
TSHR mRNA expression and copy number
To determine if TSHR is amplified or
overexpressed in lung AD samples, we measured TSHR gene
expression in 96 AD and matched normal tissues using a real-time
quantitative PCR (Fig. 1B). There
was no clear up-regulation of TSHR in tumor samples compared
to the matched normal tissues. TSHR DNA copy number was then
checked, and only two potential amplifications of TSHR in 96
AD samples were found (Fig. 1C),
which suggests that TSHR amplification might not be a major
genetic event in lung cancer. For deletion, only additional two
samples showed clear deletions in 96 AD samples.
TSHR protein expression by
immunohistochemistry
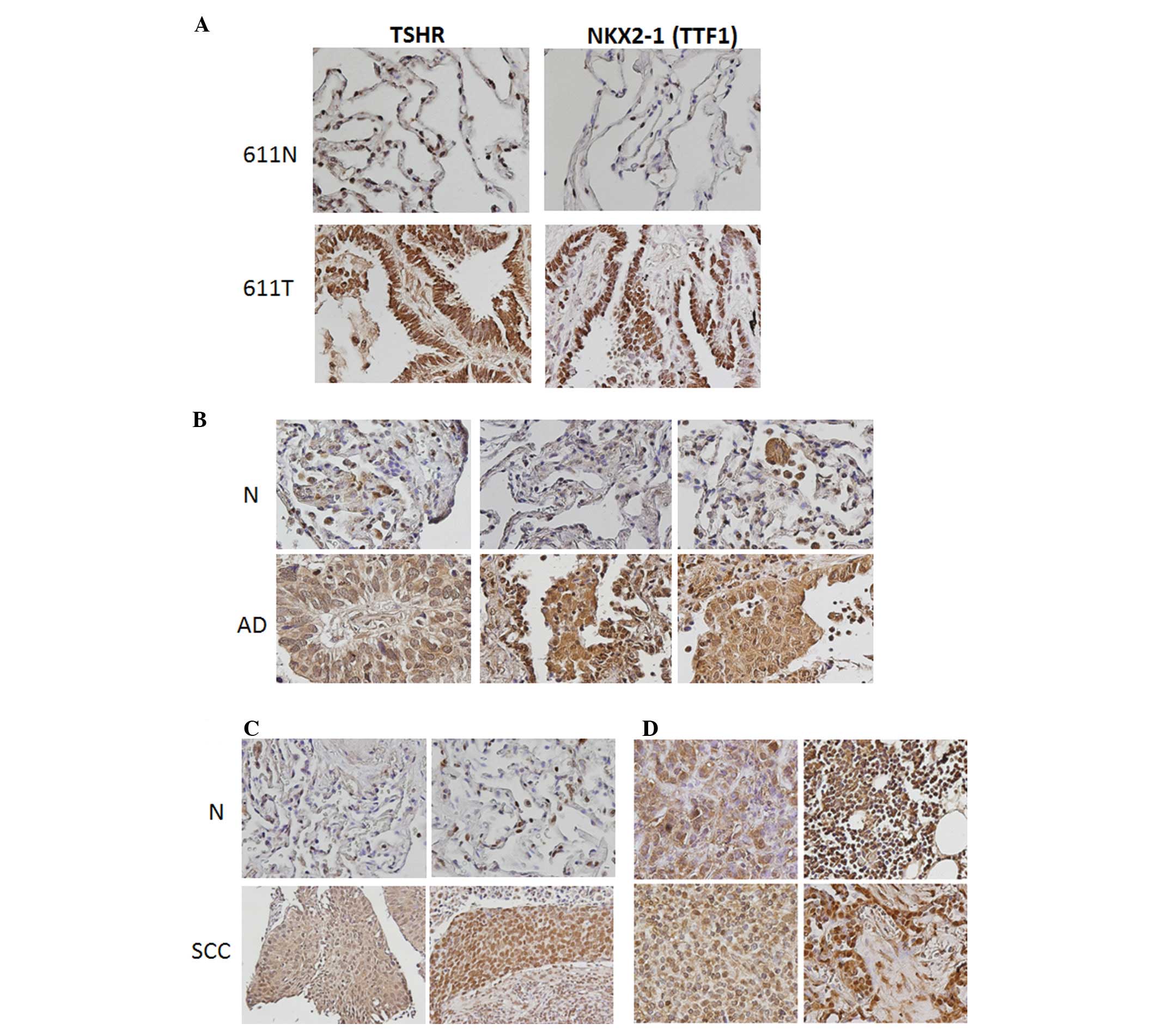
Next, we checked TSHR protein expression in tumor
and matched normal samples of the patient harboring the TSHR
mutation. Both TSHR and NKX2-1 (TTF1) were generally overexpressed
in tumor samples compared to normal tissues (Fig. 2A). Additional samples of three
different subtypes of thoracic malignancies, adenocarcinoma (AD),
squamous cell carcinoma (SCC), and malignant pleural mesothelioma
(MPM), were analyzed for TSHR expression by immunohistochemistry
(IHC) (Fig. 2B–D). High TSHR
protein expression was found in AD and SCC while low or mild
expression was detected in matched normal samples (Fig. 2B and C). For MPM, only tumor samples
were stained for TSHR and high TSHR expression was detected
(Fig. 2D). Although overall mRNA
expression of TSHR was low and not significantly different
in normal and tumor tissues (Fig.
1B), the protein level of TSHR is meaningfully high in all
three subtypes of thoracic malignancies, AD (Fig. 2B), SCC (C) and MPM (D).
Discussion
Lung cancer is the leading cause of cancer death in
men and women in the US and world-wide (1). Five-year survival has stubbornly
remained at a dismal 15–17% for a decade, making it one of the most
lethal cancers. Chronic respiratory diseases are another major
cause of deaths. Worldwide, COPD (Chronic Obstructive Pulmonary
Disease) is responsible for 3 million deaths annually (20,21).
COPD is relatively irreversible and the rate of incidence is
increasing, such that COPD is expected to be the 3rd or 4th leading
cause of death by 2030 (20,21) as
a result of increases in smoking and air pollution. However, as
only 15–20% of smokers are estimated to develop either lung cancer
or COPD, there must exist additional major genetic mechanisms
driving the development of these diseases (22–24).
Up to half of lung cancer patients suffer from COPD and ~1% of COPD
patients develop lung cancer per year (24,25).
The prognosis of lung cancer patients with COPD is worse than that
of patients without COPD (24).
Although several genetic and epigenetic mechanisms have been
proposed as playing a role in lung cancer development in COPD
patients, (22–26) the exact mechanisms by which some
patients with COPD later develop lung cancer remains unclear.
We screened for mutations in TSHR based on
its putative relationship with NKX2-1, a potential tumor suppressor
and type II pneumocyte marker in lung (11). The mutation frequency of TSHR
in lung cancer in our study is ~1%, similar to EML4-ALK, a
fusion protein estimated to play a role in 1–7% of lung
adenocarcinoma (3,5). When analyzing clinical characteristic
of patient 611 with TSHR mutation, we found that this
patient had a complicated prior medical history significant for
carotid stenosis, coronary artery disease and COPD. It was reported
that COPD is frequent in higher grade and non-BAC adenocarcinoma
(24,27). Thus, it is possible that THSR may be
involved in the development of a BAC-type lung adenocarcinoma
arising from a patient population with pre-existing COPD, although
further systematic study with large patient number will be required
to validate this.
TSHR has been reported to be mutated in many
thyroid-related diseases (14).
Activating TSHR mutations have been reported in congenital
hyperthyroidism, toxic multinodular goiter and hot nodules.
Inactivating TSHR mutations have been reported in congenital
hypothyroidism, multinodular goiter and cold nodules (14). Although several studies have
reported TSHR somatic mutations in thyroid cancer (15), epigenetic changes (methylation)
mainly seem to cause reduced expression of TSHR in thyroid
cancer (14,16). The novel mutation identified in this
study is located at codon 458 (exon 10), close to a proven
functional M453T TSHR transmembrane domain mutation, where
most activating TSHR mutations occur (14,15).
It was reported that an activating germline M453T TSHR
mutation caused non-autoimmune hyperthyroidism (28,29).
We hypothesized that the identified TSHR
mutation at the transmembrane domain would cause an overexpression
of TSHR or its binding partner, NKX2-1. We quantified TSHR
mRNA levels in both lung adenocarcinoma and matched normal tissues
and did not find a statistically significant change (Fig. 1B). At the DNA level, two samples
showed potential copy number amplifications while two other samples
showed clear deletions (Fig. 1C).
Next, we checked TSHR protein level by immunohistochemistry. A
significant overexpression of both TSHR and NKX2-1 in patient 611’s
tumor samples was observed (Fig.
2A). We also checked other subtypes of thoracic malignancies,
AD, SCC and MPM, for TSHR protein level. As shown in Fig. 2B–D, high TSHR expression was
identified in tumor samples but not in the matched normal tissues.
This indicates that TSHR protein expression, not mRNA expression,
may potentially be a useful lung tumor marker for either diagnostic
or therapeutic purposes. An anti-TSHR antibody-based ELISA assay
performed on blood specimens may provide a simple and reliable
method for earlier detection of lung cancer.
Taken together, we first identified a somatic
mutation of TSHR in a patient presenting with lung
adenocarcinoma with BAC features and a prior medical history
significant for COPD and coronary artery disease. Our observed
mutation frequency of TSHR is ~1% (1/96) in lung
adenocarcinoma patients, which suggests that mutation of
TSHR is not a major event in lung carcinogenesis generally.
It does not appear that TSHR contributes to lung cancer
development by classical genetic events such as mutation,
amplification, or overexpression. When considering a significant
overexpression of TSHR in tumor compared to matched normal samples,
TSHR protein, not DNA or mRNA, may be used for either diagnostic or
therapeutic purpose. Although further studies with larger numbers
of samples are required, it appears possible that a subgroup of
lung adenocarcinoma patients, such as those with BAC, COPD and/or
cardiovascular diseases, may be susceptible to a TSHR
mutation.
Acknowledgements
This work was supported by the Barbara Isackson Lung
Cancer Research Fund, The Eileen D. Ludwig Endowed Fund, and Kazan,
McClain, Abrams, Fernandez, Lyons, Greenwood, Harley & Oberman
Foundation Fund for Thoracic Oncology Research. I.J.K. is supported
by the UALC (Uniting Against Lung Cancer) foundation.
References
|
1
|
Edwards BK, Ward E, Kohler BA, et al:
Annual report to the nation on the status of cancer, 1975–2006,
featuring colorectal cancer trends and impact of interventions
(risk factors, screening, and treatment) to reduce future rates.
Cancer. 116:544–573. 2010.
|
|
2
|
Lynch TJ, Bell DW, Sordella R, et al:
Activating mutations in the epidermal growth factor receptor
underlying responsiveness of non-small-cell lung cancer to
gefitinib. N Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Soda M, Choi YL, Enomoto M, et al:
Identification of the transforming EML4-ALK fusion gene in
non-small-cell lung cancer. Nature. 448:561–566. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dahabreh IJ, Linardou H, Siannis F, et al:
Somatic EGFR mutation and gene copy gain as predictive biomarkers
for response to tyrosine kinase inhibitors in non-small cell lung
cancer. Clin Cancer Res. 16:291–303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pao W, Miller VA, Politi KA, et al:
Acquired resistance of lung adenocarcinomas to gefitinib or
erlotinib is associated with a second mutation in the EGFR kinase
domain. PLoS Med. 2:e732005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Horn L and Pao W: EML4-ALK: honing in on a
new target in non-small-cell lung cancer. J Clin Oncol.
27:4232–4235. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Koivunen JP, Mermel C, Zejnullahu K, et
al: EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in
lung cancer. Clin Cancer Res. 14:4275–4283. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guazzi S, Price M, De Felice M, et al:
Thyroid nuclear factor 1 (TTF-1) contains a homeodomain and
displays a novel DNA binding specificity. EMBO J. 9:3631–3639.
1990.PubMed/NCBI
|
|
9
|
Weir BA, Woo MS, Getz G, et al:
Characterizing the cancer genome in lung adenocarcinoma. Nature.
450:893–898. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kwei KA, Kim YH, Girard L, et al: Genomic
profiling identifies TITF1 as a lineage-specific oncogene amplified
in lung cancer. Oncogene. 27:3635–3640. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Winslow MM, Dayton TL, Verhaak RG, et al:
Suppression of lung adenocarcinoma progression by Nkx2-1. Nature.
473:101–104. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Civitareale D, Castelli MP, Falasca P, et
al: Thyroid transcription factor 1 activates the promoter of the
thyrotropin receptor gene. Mol Endocrinol. 7:1589–1595.
1993.PubMed/NCBI
|
|
13
|
Shimura H, Shimura Y, Ohmori M, et al:
Single strand DNA-binding proteins and thyroid transcription
factor-1 conjointly regulate thyrotropin receptor gene expression.
Mol Endocrinol. 9:527–539. 1995.
|
|
14
|
Davies TF, Yin X and Latif R: The genetics
of the thyroid stimulating hormone receptor: history and relevance.
Thyroid. 20:727–736. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hébrant A, van Staveren WC, Maenhaut C, et
al: Genetic hyperthyroidism: hyperthyroidism due to activating TSHR
mutations. Eur J Endocrinol. 164:1–9. 2011.PubMed/NCBI
|
|
16
|
Xing M, Usadel H, Cohen Y, et al:
Methylation of the thyroid-stimulating hormone receptor gene in
epithelial thyroid tumors: a marker of malignancy and a cause of
gene silencing. Cancer Res. 63:2316–2321. 2003.PubMed/NCBI
|
|
17
|
Führer D, Tannapfel A, Sabri O, et al: Two
somatic TSH receptor mutations in a patient with toxic
metastasising follicular thyroid carcinoma and non-functional lung
metastases. Endocr Relat Cancer. 10:591–600. 2003.PubMed/NCBI
|
|
18
|
Choi H, Kratz J, Pham P, et al:
Development of a practical fast mutation screening assay for human
lung adenocarcinoma. Int J Oncol. 40:1900–1906. 2012.PubMed/NCBI
|
|
19
|
Narumi S, Muroya K, Abe Y, et al: TSHR
mutations as a cause of congenital hypothyroidism in Japan: a
population-based genetic epidemiology study. J Clin Endocrinol
Metab. 94:1317–1323. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stevenson CS and Birrell MA: Moving
towards a new generation of animal models for asthma and COPD with
improved clinical relevance. Pharmacol Ther. 130:93–105. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Whitsett J, Wert S and Weaver T: Alveolar
surfactant homeostasis and the pathogenesis of pulmonary disease.
Annu Rev Med. 61:105–119. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Spitz MR, Wei Q, Dong Q, et al: Genetic
susceptibility to lung cancer: the role of DNA damage and repair.
Cancer Epidemiol Biomarkers Prev. 12:689–698. 2003.PubMed/NCBI
|
|
23
|
Cohen BH: Chronic obstructive pulmonary
disease: a challenge in genetic epidemiology. Am J Epidemiol.
112:274–288. 1980.PubMed/NCBI
|
|
24
|
Sekine Y, Katsura H, Koh E, et al: Early
detection of COPD is important for lung cancer surveillance. Eur
Respir J. Nov 16–2011.(Epub ahead of print).
|
|
25
|
Skillrud DM, Offord KP and Miller RD:
Higher risk of lung cancer in chronic obstructive pulmonary
disease. A prospective matched, controlled study. Ann Intern Med.
105:503–507. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wright JG and Christman JW: The role of
nuclear factor kappaB in the pathogenesis of pulmonary diseases:
implications for therapy. Am J Respir Med. 2:211–219. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brody JS and Spira A: State of the art.
Chronic obstructive pulmonary disease, inflammation, and lung
cancer. Proc Am Thorac Soc. 3:535–537. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kraemer S, Rothe K, Pfaeffle R, et al:
Activating TSH-receptor mutation (Met453Thr) as a cause of
adenomatous non-autoimmune hyperthyroidism in a 3-year-old boy. J
Pediatr Endocrinol Metab. 22:269–274. 2009.PubMed/NCBI
|
|
29
|
Supornsilchai V, Sahakitrungruang T,
Wongjitrat N, et al: Expanding clinical spectrum of non-autoimmune
hyperthyroidism due to an activating germline mutation, p. M453T,
in the thyrotropin receptor gene. Clin Endocrinol. 70:623–628.
2009. View Article : Google Scholar : PubMed/NCBI
|