Introduction
Cancer testis antigens (CTAs) are proteins that are
normally expressed only in the male germ cells. These proteins are
not expressed in other normal somatic tissues but are aberrantly
upregulated in a variety of cancers (1–3). CTAs
are divided into two categories, X-chromosome encoded (CT-X) and
non X-chromosome encoded. To date, 110 CTAs have been reported in
the literature of which, ~30 are encoded by multi-gene families on
the X-chromosome also called CT-X genes (1,4).
Amongst the CT-X is the MAGE family whose members are divided into
two classes. MAGE-A, -B and -C have been classified as class I and
the remaining MAGE genes are classified as class II (5). The non X-chromosome encoded CTAs are
distributed throughout the genome and do not form multi-gene
families. Several studies have evaluated the expression of CTAs to
show that these proteins are frequently expressed in variety of
cancers. Melanomas, lung cancers and ovarian cancers have the
highest frequency of CTA expression while leukemia, lymphomas,
renal, colon and pancreatic cancers have a lower frequency of CTA
expression. The frequency of expression of these proteins has also
been found to correlate with tumor grade and metastatic behavior.
An expression array study performed in lung cancer cell lines
showed that 30% of the overexpressed genes (6 out of 20) were CTAs
(5 MAGEA and NY-ESO-1) (6).
CTAs are known to be regulated epigenetically in
cancers (7–9). Derepression of these genes in cultured
cells after treatment with DNA methyl-transferase inhibitors like
5-aza-2′-deoxycytidine shows that demethylation is a key factor
governing the regulation of these genes (9–11).
Using an integrative epigenetic screening approach, we have
previously shown that CTAs are upregulated in non-small cell lung
cancer (NSCLC) and head and neck squamous cell carcinoma (HNSCC) by
promoter hypomethylation (10,11).
Recently, we showed that transcription factor BORIS (brother of the
regulator of imprinted sites) regulates 3 MAGEA genes,
MAGEA2, A3 and A4, by binding to their promoters and
enriching transcription activating histone modifications (12). Two earlier reports have also
implicated BORIS in the activation of MAGEA1 and
NY-ESO-1 genes (13,14).
Because of their exclusive expression in a wide
variety of human cancers and their ability to elicit cellular and
humoral immune responses, CTAs have been explored as potential
targets for cancer immunotherapy (4,15,16).
Two CTAs, MAGEA3 and NY-ESO-1, are being evaluated as targets for
cancer vaccines in multiple clinical trials. Although much
attention has been given to the development of cancer vaccines
based on these antigenic proteins, their physiological functions
remain unclear. Recent efforts to resolve their functional roles
provide evidence for both tumor suppressive and oncogenic roles for
these proteins. While some studies have shown that MAGEs are
involved in growth stimulation and apoptosis inhibition (17–21),
others have shown that MAGEs are pro-apoptotic (22,23).
In the present study, we investigated the role of
MAGEA4 in promoting cell growth and the possible mechanisms by
which it acts as a growth promoter. We showed that overexpression
of MAGEA4 prevents cell cycle arrest and also makes the cells
resistant to apoptosis. Further, we showed that overexpression of
MAGEA4 represses p53 targets, BAX and CDKN1A.
Materials and methods
Tissue samples and cells
Human tissue samples were collected after approval
from Johns Hopkins Medicine Institutional Review Board and informed
written consent from the patients. Spontaneously immortalized
normal oral keratinocytes (NOK-SI) were provided by Dr Silvio
Gutkind (National Institutes of Health, Bethesda, MD).
Transfection of human expression vectors
and anchorage-dependent growth assay
Full-length ORF cDNA of MAGEA4 in pCMV-SPORT6
vector was obtained from Invitrogen (Carlsbad, CA). NOK-SI cells
(21) were plated in 96-well plates
and transfected with either the MAGEA4 expression vector or
the empty vector using FuGene 6 transfection reagent per the
vendor’s instructions (Roche). Cell Counting kit-8 (CCK-8)
(Dojindo) absorbance was measured by the Spectramax M2e 96-well
fluorescence plate reader Molecular Devices (Sunnyvale, CA) after
transfection every 24 h for 72 h. Absorbance was also measured at 0
h (time of transfection) to ensure equal plating of cells.
RNA extraction and quantitative
Reverse-Transcription PCR (qRT-PCR)
Total RNA was extracted using QIAzol and RNeasy mini
kit (Qiagen). RNA was reverse transcribed to cDNA using qScript
cDNA mix (Quanta Biosciences). Real-time PCR was performed using
the Fast SYBR green master mix on the ABI 7900HT real-time PCR
machine (Applied Biosystems). Primers used were: GAPDH, forward
5′-AGC CACATCGCTCAGACAC-3′ and reverse 5′-GCCCAATAC GACCAAATCC-3′;
MAGEA4, forward 5′-AGGAGAAGA TCTGCCTGTGG-3′ and reverse
5′-CAGCCTCCTGCTCCT CAGTA-3′.
Cell cycle assay
For cell cycle analysis, NOK-SI cells were plated in
100-mm dishes and transfected with the MAGEA4 expression vector or
empty vector using FuGene HD (Roche). To ensure contact inhibition
of growth, cells were allowed to remain confluent for 48 h before
being processed further. The cells were then harvested by
trypsinization, fixed in methanol and stained with 0.1 mg/ml
propidium iodide. DNA content was measured using flow cytometric
analysis performed using a FACScan flow cytometer (Becton
Dickinson).
Apoptosis assay
NOK-SI cells were transiently transfected with
MAGEA4 expression vector or the control empty vector.
Twenty-four hours post-transfection, the cells were treated with
2.5 mg/ml G418 for 2 days to induce apoptosis (24). At the end of 2 days, the cells were
harvested by trypsinization. Cell growth media were also collected
to ensure collection of floating apoptotic cells. Cells were then
treated for the caspase-3 staining using the FITC active caspase-3
apoptosis kit (BD Biosciences) according to the manufacturer’s
instructions. The stained cells were subjected to flow cytometry
and data were analyzed using FlowJo software (Tree Star Inc.).
Results
MAGEA4 is differentially overexpressed in
primary HNSCC tissues
We have previously shown that MAGEA4 is a candidate
oncogene derepressed by promoter hypomethylation in HNSCC. In that
study, we performed bioinformatics analysis called Cancer Outlier
Profiling Analysis (COPA) on microarray data of 49 primary HNSCC
and 19 normal mucosal tissues retrieved from the Oncomine database
(www.oncomine.org) to show that MAGEA4 was
significantly overexpressed in tumors compared to normal tissues.
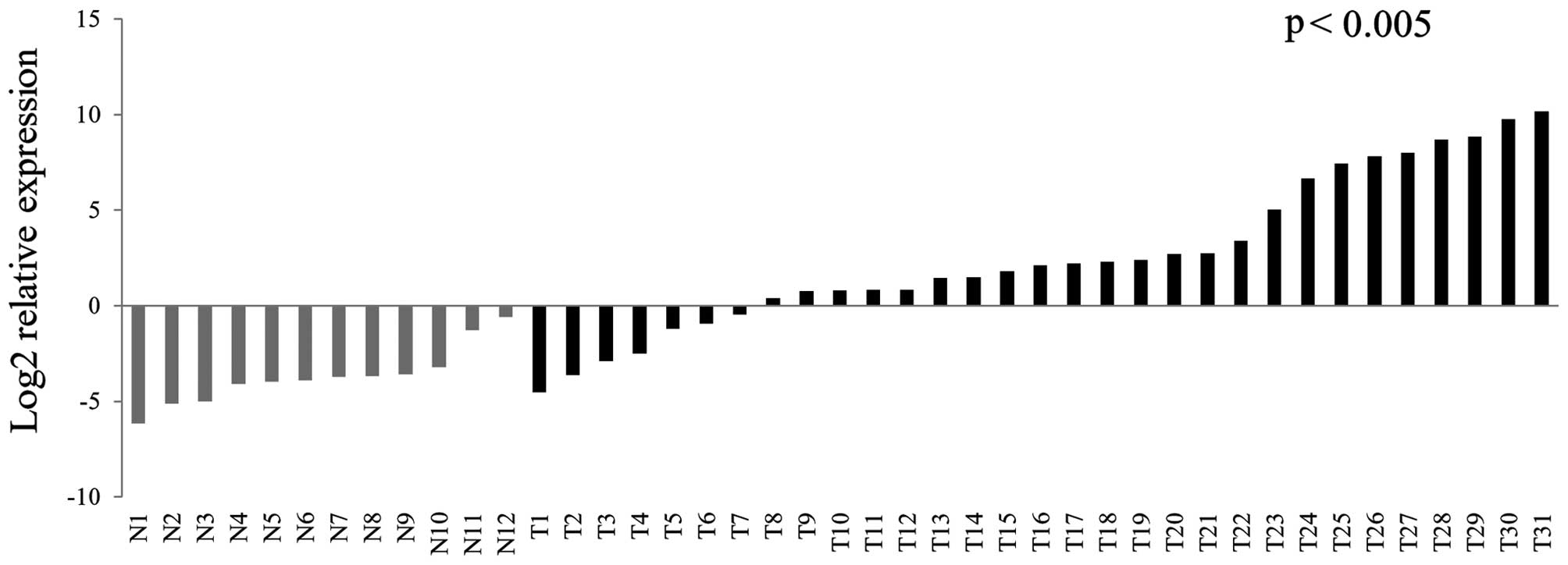
In the present study, we used qRT-PCR to analyze MAGEA4 expression
in a separate cohort of 31 HNSCC and 12 normal non-cancer upper
aerodigestive mucosal samples. We found significant upregulation of
MAGEA4 in the tumor samples compared to the normal samples
(p<0.005) (Fig. 1). Of 31 tumor
samples, 24 showed significant expression of MAGEA4.
MAGEA4 induces growth in NOK-SI cells by
preventing cell cycle arrest
To elucidate the function of MAGEA4 in HNSCC, we
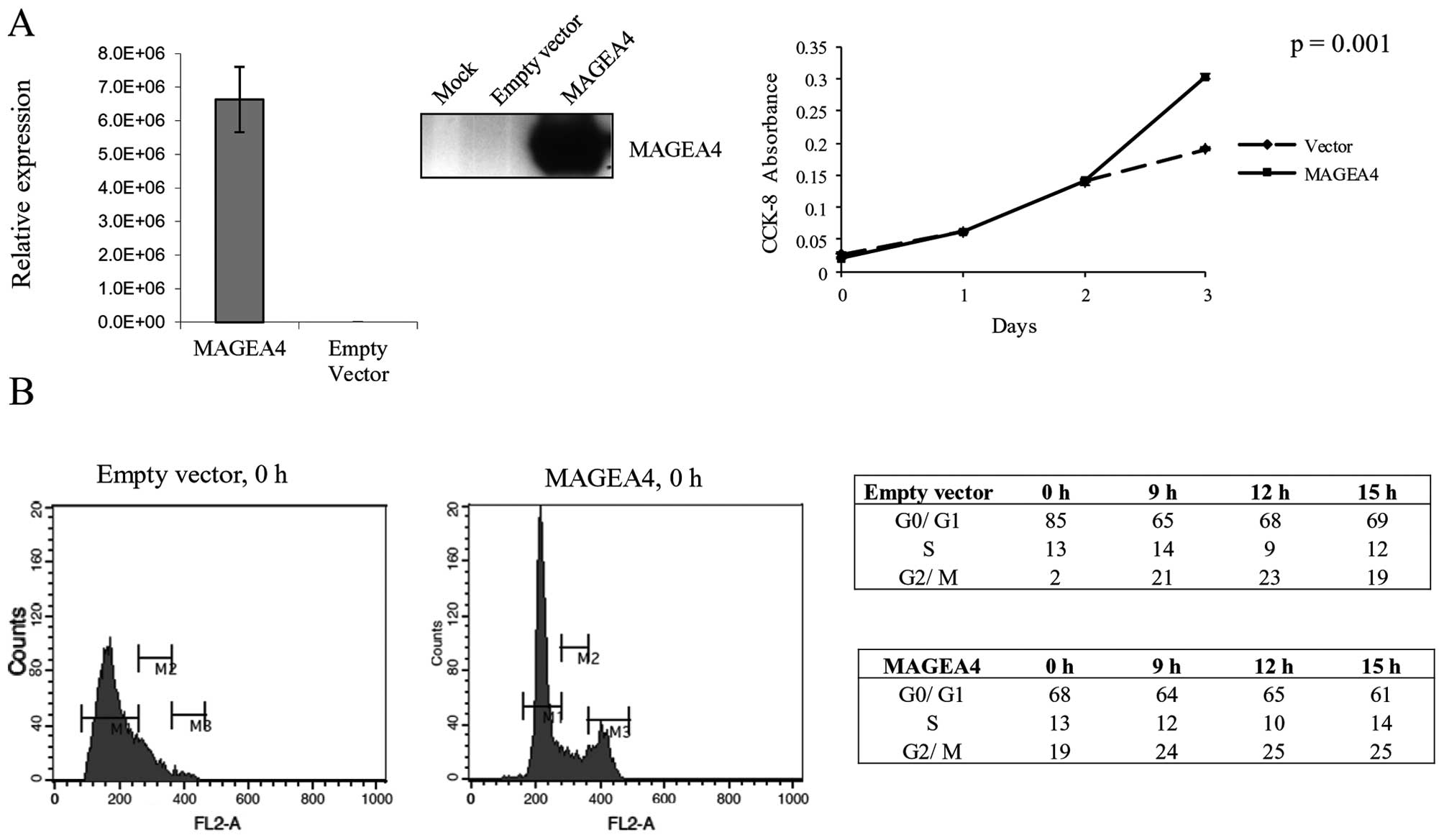
investigated the effect of overexpressing MAGEA4 in a spontaneously
transformed normal oral keratinocyte cell line (NOK-SI) (Fig. 2A). Transient overexpression of
MAGEA4 in NOK-SI cells resulted in 37% increase in cell
proliferation at 72-h post-transfection (Fig. 2A). We next investigated if the
growth stimulation caused by MAGEA4 is a result of changes in the
cell cycle. We transiently overexpressed MAGEA4 in NOK-SI cells and
allowed them to go into cell cycle arrest by contact inhibition. We
allowed the cells to remain confluent for 48 h to ensure growth
arrest by contact inhibition. The cells were then stained with
propidium iodide and analyzed by flow cytometry for cellular DNA
content. The analysis showed that a significantly smaller
percentage of cells overexpressing MAGEA4 were arrested in G1 phase
(68%) compared to cells transfected with the empty vector (85%)
(Fig. 2B). This shows that MAGEA4
is able to inhibit the growth arrest caused by contact inhibition
and allows growth under conditions of high confluency. A follow-up
by flow cytometry analysis at 9, 12 and 15 h after release from
density arrest, showed that MAGEA4 overexpressing and control cells
progressed through the cell cycle similarly (Fig. 2B).
MAGEA4 inhibits apoptosis and suppresses
p53 target genes
To gain further understanding of the mechanism of
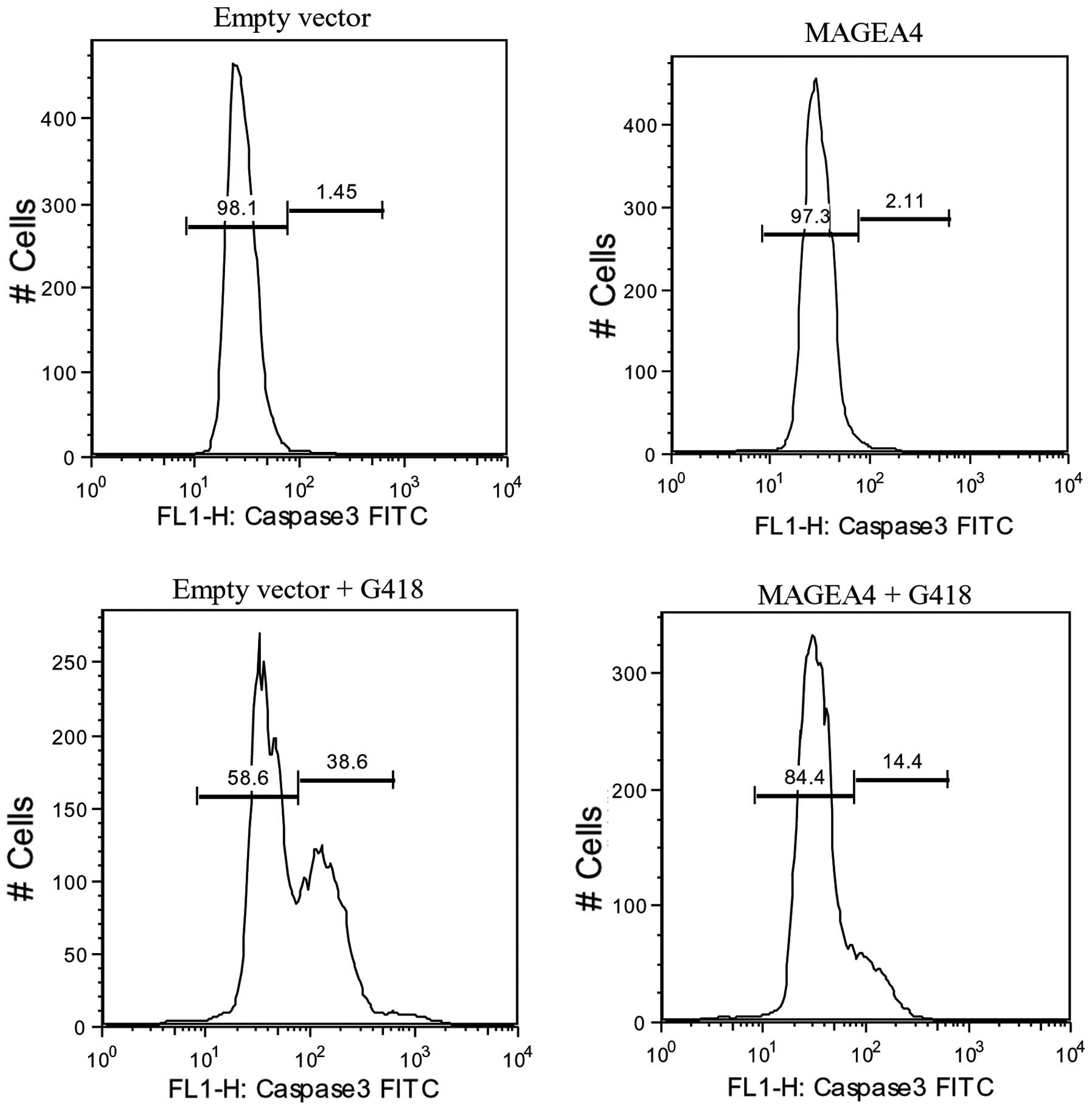
growth promotion by MAGEA4 in NOK-SI cells, we analyzed the effect
of MAGEA4 overexpression on apoptosis. We transiently transfected
NOK-SI cells with MAGEA4 expression vector or the control
empty vector and then induced apoptosis using G418. After 48 h of
treatment with G418, a significantly smaller percentage of
MAGEA4 expressing cells (14%) were positive for active
caspase-3 compared to the control cells (39%) indicating that
MAGEA4 expression can block apoptosis induced by G418 (Fig. 3). Control cells and cells
overexpressing MAGEA4 not treated with G418, were negative
for active caspase-3 (Fig. 3).
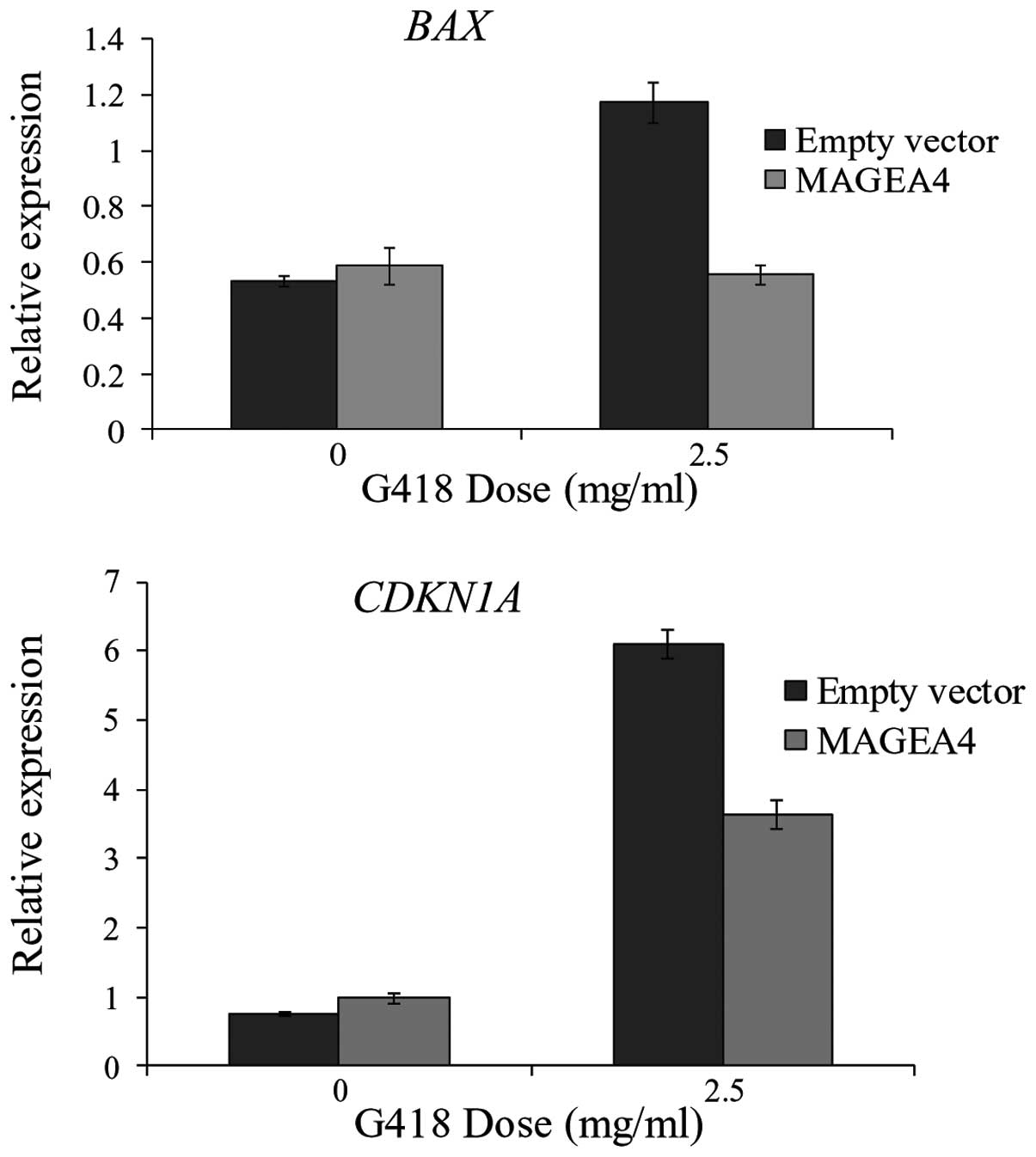
Monte et al (18) have shown that MAGEA2 recruits HDAC3
to p53 transcription sites to downregulate apoptotic activators,
BAX and CDKN1A. We reasoned that the inhibition of
apoptosis caused by MAGEA4 could be a result of downregulation of
these p53 target genes. The expression levels of both BAX
and CDKN1A were similar in MAGEA4 overexpressing and
control cells in absence of G418 (Fig.
4). After treatment with G418, the expression of both
BAX and CDKN1A increased in the control cells.
However, expression of these 2 genes remained significantly lower
in the MAGEA4 overexpressing cells compared to the control cells
(Fig. 4). This suggests that MAGEA4
may inhibit apoptosis by reducing the response of p53 targets,
BAX and CDKN1A to apoptosis inducing agents.
Discussion
MAGE proteins are members of class I family of CTAs
which elicit both cellular and humoral immune responses and thus
have been explored as targets for therapeutic cancer vaccines. In
the present study, we evaluated the expression of MAGEA4 in
a cohort of HNSCC and normal mucosa and found that it is
significantly upregulated in tumors. Although it is well
established that CTAs are overexpressed in tumors, the biological
functions of these proteins remain poorly characterized.
Surprisingly, only a few studies have investigated the
physiological functions of CTAs in cancers. In order to define the
functions of MAGEA4 in HNSCC, we overexpressed MAGEA4 in normal
oral keratinocytes and found that it stimulates growth of these
cells by inhibiting cell cycle arrest and also making the cells
resistant to apoptosis. Of note, MAGEA4 overexpressing cells
treated with G418, an inducer of p53-dependent apoptosis, showed
lower levels of two p53 target genes, BAX and CDKN1A.
While BAX, a proapoptotic member of the Bcl-2 family proteins,
forms pores in the mitochondrial membrane and aids in the release
of cytochrome c into the cytoplasm, CDKN1A has also been reported
to enhance apoptosis in response to drug treatment (25,26).
Supporting our results, Yang et al (20) have shown that suppression of the
MAGEA genes reduced the viability and also induced apoptosis
in three melanoma cell lines. Suppression of apoptosis by MAGE
proteins was p53-dependent MAGE knockdown-induced apoptosis in
wild-type HCT116 colon cancer cell line but not in
p53−/− cells. Inoculation of mice with mMAGEB siRNA
treated S91 melanoma cells, led to decreased tumor growth compared
to S91 cells treated with control siRNA. These results are also
supported by observations from another study that melanoma cells
expressing high levels of MAGEA proteins are resistant to
p53-dependent apoptosis. MAGEA2, specifically, interacts with p53
and recruits transcriptional repressors to p53 transcription sites
thus inhibiting the expression of p53 downstream targets (18). Knockdown of MAGEA and MAGEC2
expression in human mast cells and knockdown of murine MAGEB in
mouse mast cells decreases proliferation of these cells (19). Knockdown of these MAGE proteins also
led to increased apoptosis in these cells. Furthermore, treatment
with MAGE siRNA led to decreased tumor growth in a murine model of
mastocytosis. MAGEA3 has been found to specifically bind
procaspase-12 and render the cells refractory to endoplasmic
reticulum stress induced apoptosis (17). All the above studies provide
compelling evidence for an oncogenic role for MAGE proteins.
However, some contradictory studies have also emerged that support
a tumor suppressive role for MAGE proteins. One such study has
shown that overexpression of MAGEA4 in human embryonic kidney cells
(293 cells) leads to increased apoptosis while knockdown of MAGEA4
in a squamous cell lung cancer cell line H1703 and 293/MAGEA4 cells
reduces apoptosis suggesting a tumor suppressive role for MAGEA4
(22). Also, MAGEA4 specifically
binds to gankyrin, an oncogene overexpressed in hepatocellular
carcinomas, to suppress its oncogenic activity. It suppresses both
anchorage-independent growth as well as tumor formation of gankyrin
overexpressing cells in athymic nude mice (23).
The MAGEA proteins being highly homologous may be
expected to exhibit functional redundancy. However, emerging
evidence suggests the involvement of MAGE proteins in a wide
spectrum of cellular processes. Thus, in view of the differential
roles of MAGEA proteins, it is important to define the functions of
individual MAGE proteins in different cancers to better understand
their functional significance in tumorigenesis.
Acknowledgements
This study was based on a web database application
provided by Research Information Technology Systems (RITS) -
https://www.rits.onc.jhmi.edu/.
References
|
1
|
Simpson AJ, Caballero OL, Jungbluth A,
Chen YT and Old LJ: Cancer/testis antigens, gametogenesis and
cancer. Nat Rev Cancer. 5:615–625. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zendman AJ, Ruiter DJ and Van Muijen GN:
Cancer/testis-associated genes: identification, expression profile,
and putative function. J Cell Physiol. 194:272–288. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Koslowski M, Bell C, Seitz G, et al:
Frequent nonrandom activation of germ-line genes in human cancer.
Cancer Res. 64:5988–5993. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Caballero OL and Chen YT: Cancer/testis
(CT) antigens: potential targets for immunotherapy. Cancer Sci.
100:2014–2021. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Barker PA and Salehi A: The MAGE proteins:
emerging roles in cell cycle progression, apoptosis, and
neurogenetic disease. J Neurosci Res. 67:705–712. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sugita M, Geraci M, Gao B, et al: Combined
use of oligonucleotide and tissue microarrays identifies
cancer/testis antigens as biomarkers in lung carcinoma. Cancer Res.
62:3971–3979. 2002.PubMed/NCBI
|
|
7
|
De Smet C, De Backer O, Faraoni I, Lurquin
C, Brasseur F and Boon T: The activation of human gene MAGE-1 in
tumor cells is correlated with genome-wide demethylation. Proc Natl
Acad Sci USA. 93:7149–7153. 1996.PubMed/NCBI
|
|
8
|
De Smet C, Lurquin C, Lethe B, Martelange
V and Boon T: DNA methylation is the primary silencing mechanism
for a set of germ line- and tumor-specific genes with a CpG-rich
promoter. Mol Cell Biol. 19:7327–7335. 1999.PubMed/NCBI
|
|
9
|
Weber J, Salgaller M, Samid D, et al:
Expression of the MAGE-1 tumor antigen is up-regulated by the
demethylating agent 5-aza-2′-deoxycytidine. Cancer Res.
54:1766–1771. 1994.PubMed/NCBI
|
|
10
|
Glazer CA, Smith IM, Ochs MF, et al:
Integrative discovery of epigenetically derepressed cancer testis
antigens in NSCLC. PloS One. 4:e81892009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Smith IM, Glazer CA, Mithani SK, et al:
Coordinated activation of candidate proto-oncogenes and cancer
testes antigens via promoter demethylation in head and neck cancer
and lung cancer. PloS One. 4:e49612009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bhan S, Negi SS, Shao C, et al: BORIS
binding to the promoters of cancer testis antigens, MAGEA2, MAGEA3,
and MAGEA4, is associated with their transcriptional activation in
lung cancer. Clin Cancer Res. 17:4267–4276. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vatolin S, Abdullaev Z, Pack SD, et al:
Conditional expression of the CTCF-paralogous transcriptional
factor BORIS in normal cells results in demethylation and
derepression of MAGE-A1 and reactivation of other cancer-testis
genes. Cancer Res. 65:7751–7762. 2005.
|
|
14
|
Hong JA, Kang Y, Abdullaev Z, et al:
Reciprocal binding of CTCF and BORIS to the NY-ESO-1 promoter
coincides with derepression of this cancer-testis gene in lung
cancer cells. Cancer Res. 65:7763–7774. 2005.PubMed/NCBI
|
|
15
|
Scanlan MJ, Gure AO, Jungbluth AA, Old LJ
and Chen YT: Cancer/testis antigens: an expanding family of targets
for cancer immunotherapy. Immunol Rev. 188:22–32. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ghafouri-Fard S and Modarressi MH:
Cancer-testis antigens: potential targets for cancer immunotherapy.
Arch Iran Med. 12:395–404. 2009.PubMed/NCBI
|
|
17
|
Morishima N, Nakanishi K, Takenouchi H,
Shibata T and Yasuhiko Y: An endoplasmic reticulum stress-specific
caspase cascade in apoptosis. Cytochrome c-independent activation
of caspase-9 by caspase-12. J Biol Chem. 277:34287–34294. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Monte M, Simonatto M, Peche LY, et al:
MAGE-A tumor antigens target p53 transactivation function through
histone deacetylase recruitment and confer resistance to
chemotherapeutic agents. Proc Natl Acad Sci USA. 103:11160–11165.
2006. View Article : Google Scholar
|
|
19
|
Yang B, O’Herrin S, Wu J, Reagan-Shaw S,
Ma Y, Nihal M and Longley BJ: Select cancer testes antigens of the
MAGE-A, -B, and -C families are expressed in mast cell lines and
promote cell viability in vitro and in vivo. J Invest Dermatol.
127:267–275. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang B, O’Herrin SM, Wu J, et al: MAGE-A,
mMage-B, and MAGE-C proteins form complexes with KAP1 and suppress
p53-dependent apoptosis in MAGE-positive cell lines. Cancer Res.
67:9954–9962. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Glazer CA, Smith IM, Bhan S, et al: The
role of MAGEA2 in head and neck cancer. Arch Otolaryngol Head Neck
Surg. 137:286–293. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Peikert T, Specks U, Farver C, Erzurum SC
and Comhair SA: Melanoma antigen A4 is expressed in non-small cell
lung cancers and promotes apoptosis. Cancer Res. 66:4693–4700.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nagao T, Higashitsuji H, Nonoguchi K, et
al: MAGE-A4 interacts with the liver oncoprotein gankyrin and
suppresses its tumorigenic activity. J Biol Chem. 278:10668–10674.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jin QH, Zhao B and Zhang XJ: Cytochrome c
release and endoplasmic reticulum stress are involved in
caspase-dependent apoptosis induced by G418. Cell Mol Life Sci.
61:1816–1825. 2004.PubMed/NCBI
|
|
25
|
Kuribayashi K and El Deiry WS: Regulation
of programmed cell death by the p53 pathway. Adv Exp Med Biol.
615:201–221. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lincet H, Poulain L, Remy JS, Deslandes E,
Duigou F, Gauduchon P and Staedel C: The p21(cip1/waf1)
cyclin-dependent kinase inhibitor enhances the cytotoxic effect of
cisplatin in human ovarian carcinoma cells. Cancer Lett. 161:17–26.
2000. View Article : Google Scholar : PubMed/NCBI
|