Introduction
Colorectal cancer (CRC) is the third leading cause
of cancer-related mortality worldwide (1) and has created new challenges to the
methods and tools of treatment. CRC is often the result of a
combination of environmental and genetic mutations, accompanied by
a variety of gene expression profile changes (2–4).
Genetic mutations, including point mutations, chromosomal
translocation and gene amplification, result in oncogenes and tumor
suppressor gene mutations involved in cancer development (2). Tumor cells are parts of tissues that
have lost normal regulation of their growth, resulting in clonal
dysplasia and the formation of neoplasm, following the impact of a
variety of carcinogenic factors. Inappropriate expression of
tumor-suppressor genes or oncogenes is regarded the principal cause
of tumorigenesis.
The biological behavior of cancer, including
carcinogenesis and functional heterogeneity, can be explained by
the cancer stem cell (CSC) hypothesis. According to this model,
CSCs, which exhibit stem-like features, are involved in tumor
formation, proliferation, differentiation, metastasis and
resistance to therapy (5–7). Through their ability of self-renewal
and unlimited proliferation, tumor stem cells maintain the vitality
of the tumor cell population, and through their increased movement
and migration, tumor stem cells make cell metastasis possible. With
a variety of drug-resistant molecules, tumor stem cells allow the
tumor to become non-sensitive to external physical and chemical
factors. At the same time, numerous studies have indicated that
some cytokines, protein-coding genes and its products are involved
in the maintenance of the biological characteristics of CSCs
(8–10). However, the underlying mechanisms
remain largely unknown.
microRNAs (miRNAs), approximately 18–24 nt in
length, are a class of non-coding single-stranded RNA molecules in
eukaryotes. miRNAs regulate their target gene expression through
post-transcriptional regulation, which includes mediating
degradation of target mRNA, inhibiting target mRNA translation
through complementary combining with the target mRNA
3′-untranslated region (UTR) completely/incompletely (11–14).
Similar to other transcription factors, miRNAs are adjustment
factors which can determine cell fate. miRNAs and other
post-transcriptional regulatory mechanisms are regarded the
mechanisms that control gene expression (11,15,16).
In recent years, the combination of computer
science, information technology, mathematical theory and gene chip
technology, has given rise to an interdisciplinary science,
bioinformatics. Bioinformatics includes biological data handling,
processing of the genetic and physical map, nucleotide and amino
acid sequence analysis, the discovery of new genes and protein
structure prediction. With bioinformatics we can increase our
knowledge of genes, and from our previous understanding of a single
gene, we can now examine the genes in the whole genome level
organizational structure and information structure, and examine the
mutual relationships between gene location, structure and
function.
However, the reasons underlying the generation,
development, recurrence and metastasis of CRC remain unclear and
the role of stem cells in the tumor biological processes has yet to
be fully elucidated. Using a combination of bio-chip and computer
bioinformatics, herein we report a comprehensive analysis of stem
cell properties in the CRC cell line.
Materials and methods
Cell culture and tissues
The SW1116 human CRC cell line was obtained from the
Shanghai Chinese Academy of Sciences (CAS). The CRC cells were
carefully cultured in RPMI-1640 medium containing 10% fetal bovine
serum, 50 U/ml penicillin and 50 μg/ml streptomycin. Cells were
cultured at 37°C, in a 5% CO2 atmosphere with 95%
humidity.
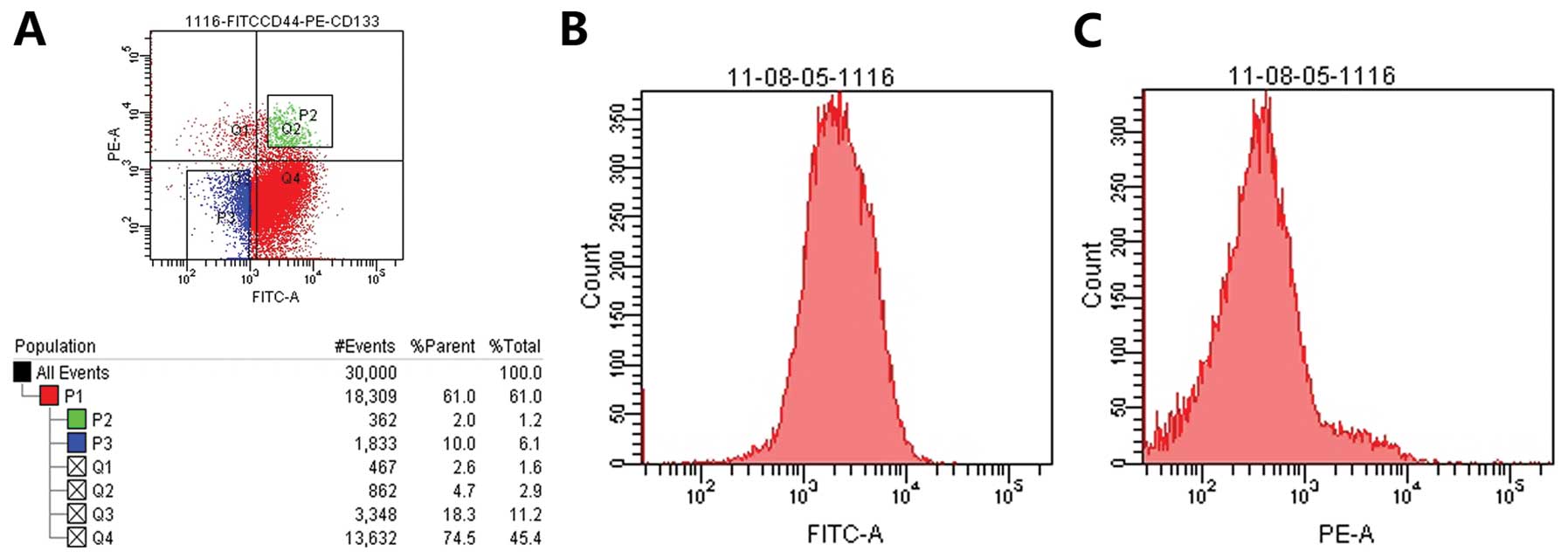
We separated
CD133+CD44+-positive and
CD133−CD44−-negative cells using flow
cytometry. CD133+CD44+-positive cells were
cultured in serum-free DMEM/F12 with EGF (10 ng/ml) and βEGF (10
ng/ml). CD133−CD44−-negative cells were
cultured in DMEM/F12 medium with fetal calf serum. Cells were
cultured at 37°C, in a 5% CO2 atmosphere with 95%
humidity (Fig. 1).
Extraction of total cellular RNA
Total-RNA extraction was performed with TRIzol
(Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s
instructions. Extracted RNA samples were quantified by NanoDrop
1000 (Nanodrop, Wilmington, DE, USA). To remove any genomic DNA
contamination, the samples were treated by DNase (DNA-free kit;
Ambion, Austin, TX, USA).
miRNA chip analysis
The samples were analyzed with Human microRNA
OneArray® v3 which is produced by Phalanx Biotech Group
(Belmont, CA, USA). Human microRNA OneArray microarrays are made of
polydeoxynucleotide probes spotted onto a proprietary chemical
layer coated on top of a 1″×3″ (25×75 mm) standard format
microarray glass slide. Each probe is spotted onto the array in a
highly consistent manner using proprietary, non-contact spotting
technology. Each microarray contains 1711 unique human miRNA probes
and 189 experimental control probes. Each unique probe has 3
features, and probes contain 100% of Sanger miRBase v17 miRNA
content. We used ULS miRNA labeling kit (Kreatech, Durham, NC, USA)
to label target. We used miRNA OneArray Hyb Buffer V3 and miRNA
OneArray Hybridization Buffer II to complete the hybridization
process. All these steps were conducted in accordance with the
manufacturer’s instructions. We used an Axon 4000B scanner
(Molecular Devices, Sunnyvale, CA, USA) to scan miRNA chip, and
GenePix 4.1 data analysis software.
mRNA chip analysis
We analyzed samples with Illumina®
Whole-Genome Gene Expression Direct Hybridization Assay system
(direct hybridization assay) which integrates Illumina proprietary
BeadArray technology, a precise microarray scanning system (the
Illumina HiScan™ or iScan System or the Illumina BeadArray™
Reader), hybridization equipment and accessories, and standard,
off-the-shelf sample labeling protocols. First we amplified RNA
samples with Illumina® TotalPrep RNA Amplification kit.
The procedure consists of reverse transcription to synthesize first
strand cDNA, second strand cDNA synthesis, cDNA purification, in
vitro transcription to synthesize cRNA and cRNA purification.
All steps followed the instruction manual. The BeadArray chip
analysis and data analysis were performed by YiKe Co., Shanghai,
China. All analyses were conducted in accordance with the
manual.
Real-time polymerase chain reaction assay
for miRNA
Total-RNA was extracted from cells with TRIzol
(Invitrogen). Total-RNA was assessed by measuring the absorbance at
260 nm. cDNA was synthesized using ImProm-II reverse transcriptase.
With strand cDNA (0.5 μl), forward and reverse primers (both 0.5
μl) and SYBR green supermix (12.5 μl), real-time qPCR was
performed. Quantitative real-time PCR reaction was performed with
the 7000 Sequence Detection System (ABI). Relative expressions were
calculated using the formula 2−ΔΔCT values
(ΔCt=Ctgene-Ctcontrol). The primer sequences
and PCR conditions are summarized in Table I.
 | Table ISequences of primers and parameters
for reverse-transcription and real-time PCR. |
Table I
Sequences of primers and parameters
for reverse-transcription and real-time PCR.
| Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| MiR-29a |
ACACTCCAGCTGGGTAGCACCATCTGAAAT | Kit provides |
| MiR-29b |
ACACTCCAGCTGGGTAGCACCATTTGAAATC | Kit provides |
| MiR-449b |
ACACTCCAGCTGGGAGGCAGTGTATTGTTA | Kit provides |
| MiR-4524a |
ACACTCCAGCTGGGATAGCAGCATGAACCT | Kit provides |
| U6 |
CTCGCTTCGGCAGCACA |
AACGCTTCACGAATTTGCGT |
| NRAS |
TGAAACCTCAGCCAAGACCAGACA |
TGGCAATCCCATACAACCCTGAGT |
| FOS |
TGTCTGTGGCTTCCCTTGATCTGA |
TGGATGATGCTGGGAACAGGAAGT |
| WASF2 |
AGATGCTGCAGGACACCAAGGATA |
ACCAAAGTGGGTGGATACCCAGAA |
| COL5A1 |
TGCTCCAGGGATTCCTTCAAGGTT |
ATAGGAGAGCAGTTTCCCACGCTT |
| CDK6 |
TGCACAGTGTCACGAACAGACAGA |
TTAGATCGCGATGCACTACTCGGT |
| CCND1 |
AGAAGCTGTGCATCTACACCGACA |
TGATCTGTTTGTTCTCCTCCGCCT |
| E2F3 |
AGTTCATTCAGCTCCTGAGCCAGT |
CAGCCCATCCATTGGACGTTGTTT |
| GNG12 |
AGCACCAACAATATAGCCCAGGCA |
ACTCCTGGCATGTTCCTCACAGTA |
| GNA12 |
TCAAGAAGCACTTCCCGGACTTCA |
TTTCACAGCATGGAACACGAAGCG |
| β-actin |
ACCAACTGGGACGACATGGAGAAA |
TAGCACAGCCTGGATAGCAACGTA |
miRNA target gene prediction
The target gene prediction software TargetScan was
used to analyze differentially expressed miRNA (TargetScan human
V6).
miRNA-mRNA correlation analysis
Negative regulation underlines the miRNA-mRNA
relationship. We performed negative correlation analysis of
significant expression patterns. We focused on the intersection of
the 6444 miRNA target genes and the 2049 mRNA gene.
Gene ontology analysis of inversely
related target genes
We first mapped target genes to each node of the
gene ontology (GO) database. The analysis was carried out using the
software DAVID (http://david.abcc.ncifcrf.gov/), according to the
statistical test method (P-value) of significantly enriched
categories (P≤0.01 as a result of the final output).
Pathway analysis of inversely related
target genes
We first mapped target genes to the KEGG pathway
database. We used the software DAVID for analysis (http://david.abcc.ncifcrf.gov/). P≤0.05 was
regarded as statistically significant.
Network analysis of inversely related
target genes Functional regulatory network of miRNA target genes:
the miRNA-GO-network
The target gene cluster was classified in accordance
with the classification of the GO BP. miRNA-GO network was built.
The network reflects the target miRNA target gene function. Network
eigenvalue (degree) was calculated according to the location of
each miRNA function. The miRNAs with the highest eigenvalues which
regulate a number of gene functions and the sample status, are
always located in pivotal positions in the network.
miRNA-gene network
Using the Sanger miRNA database, we screened target
genes regulated by differentially expressed miRNAs (TargetScan).
Then we took the differentially expressed genes and the
intersection of target genes and differentially expressed genes,
which is differential target genes regulated by differential miRNA.
Classifying this part of differential target genes regulated by
differential miRNA with GO BP, we found that significantly
differential target genes belong to significant GO (P<0.01).
Using the relationship between miRNA and target gene, we built the
miRNA-gene network.
Network of significant, differentially
expressed target genes: miRNA-path network
With pathway analysis results, we built the pathway
network. The network reflects the relationship between miRNAs and
pathways. We calculated the specific degree according to the
location of each miRNA and pathways in the network. miRNAs and
pathways with the highest degree are always located in pivotal
positions in the network.
Results
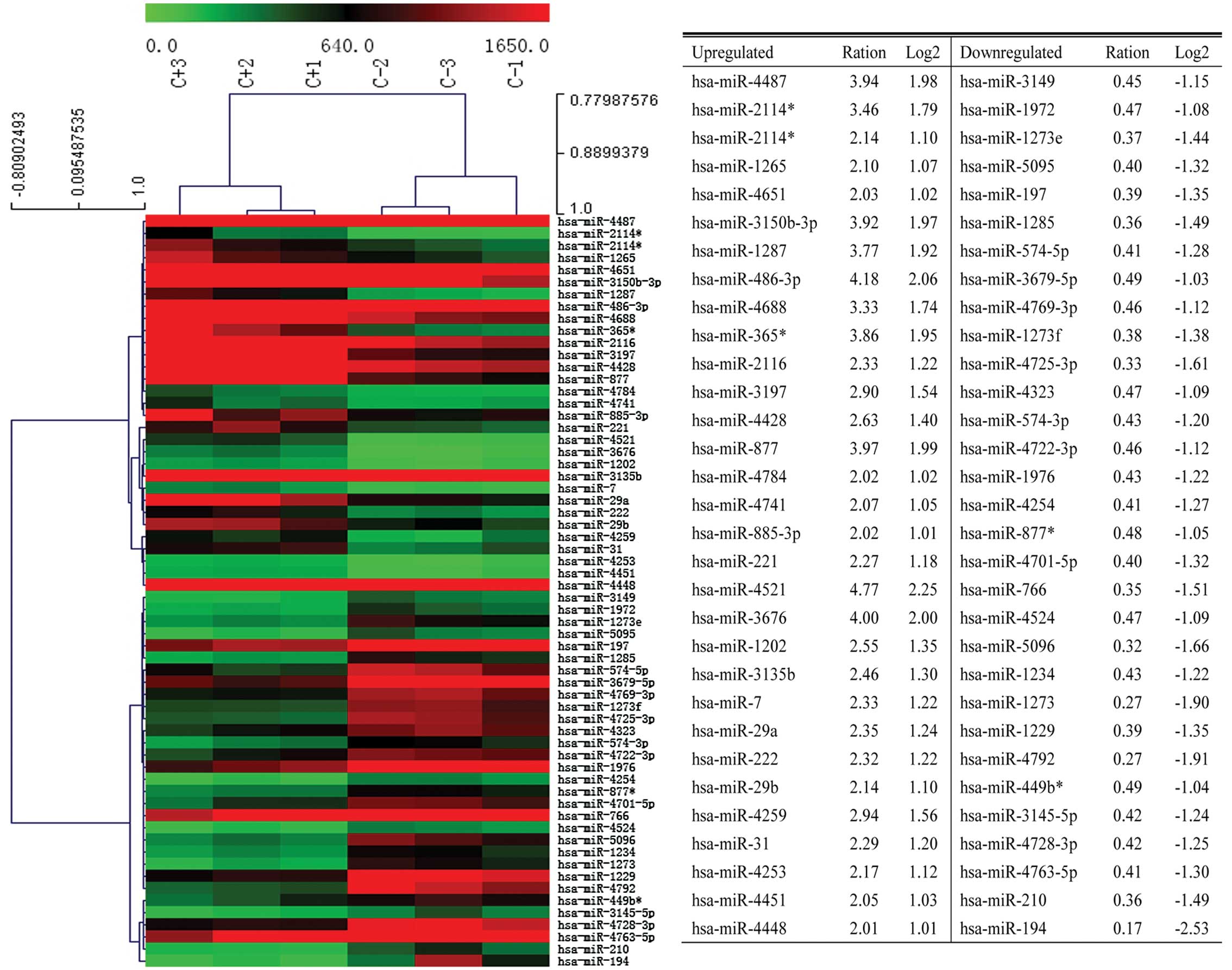
Differential miRNA expression profile in
colon CSCs and non-stem cells
We used an array-based miRNA chip to investigate the
differential miRNA expression profile in colon CSCs and non-stem
cells. A total of 1711 human miRNAs were examined. There are 31
miRNAs significantly upregulated and 31 miRNAs significantly
downregulated. Of these, miR-4521 was the most significantly
upregulated miRNA. Similarly, miR-194 was the most significantly
downregulated miRNA (Fig. 2).
Differential mRNA expression profile in
colon CSCs and non-stem cells
We used mRNA chip to investigate the differential
mRNA expression profile in colon CSCs and non-stem cells. We
examined 34694 mRNAs. There are a total of 2049 differentially
expressed mRNAs detected (Fig.
3).
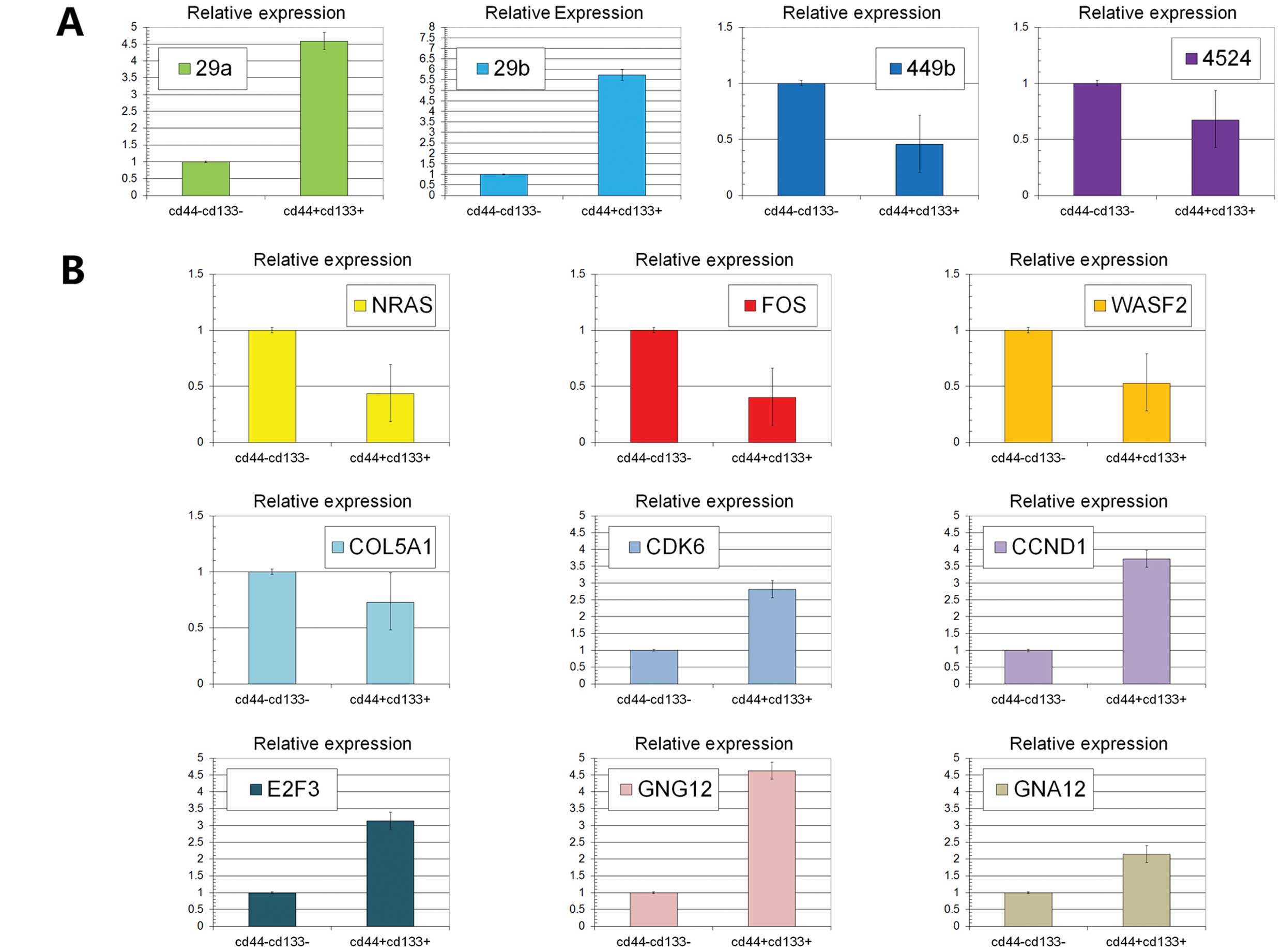 | Figure 3(A) The expression levels of miR29a,
miR29b, miR449b and miR4524 were detected using RT-PCR. We found
that miR29a and miR29b were upregulated while miR449b and miR4524
were downregulated in colon stem cells compared with non-stem cells
(P<0.05). (B) The expression levels of NRAS, FOS, WASF2, COL5A1,
CDK6, CCND1, E2F3, GNG12 and GNA12 were detected using RT-PCR.
NRAS, FOS, WASF2 and COL5A1 were downregulated in colon stem cells
(P<0.05). On the contrary, CDK6, CCND1, E2F3, GNG12 and GNA12
were upregulated (P<0.05). |
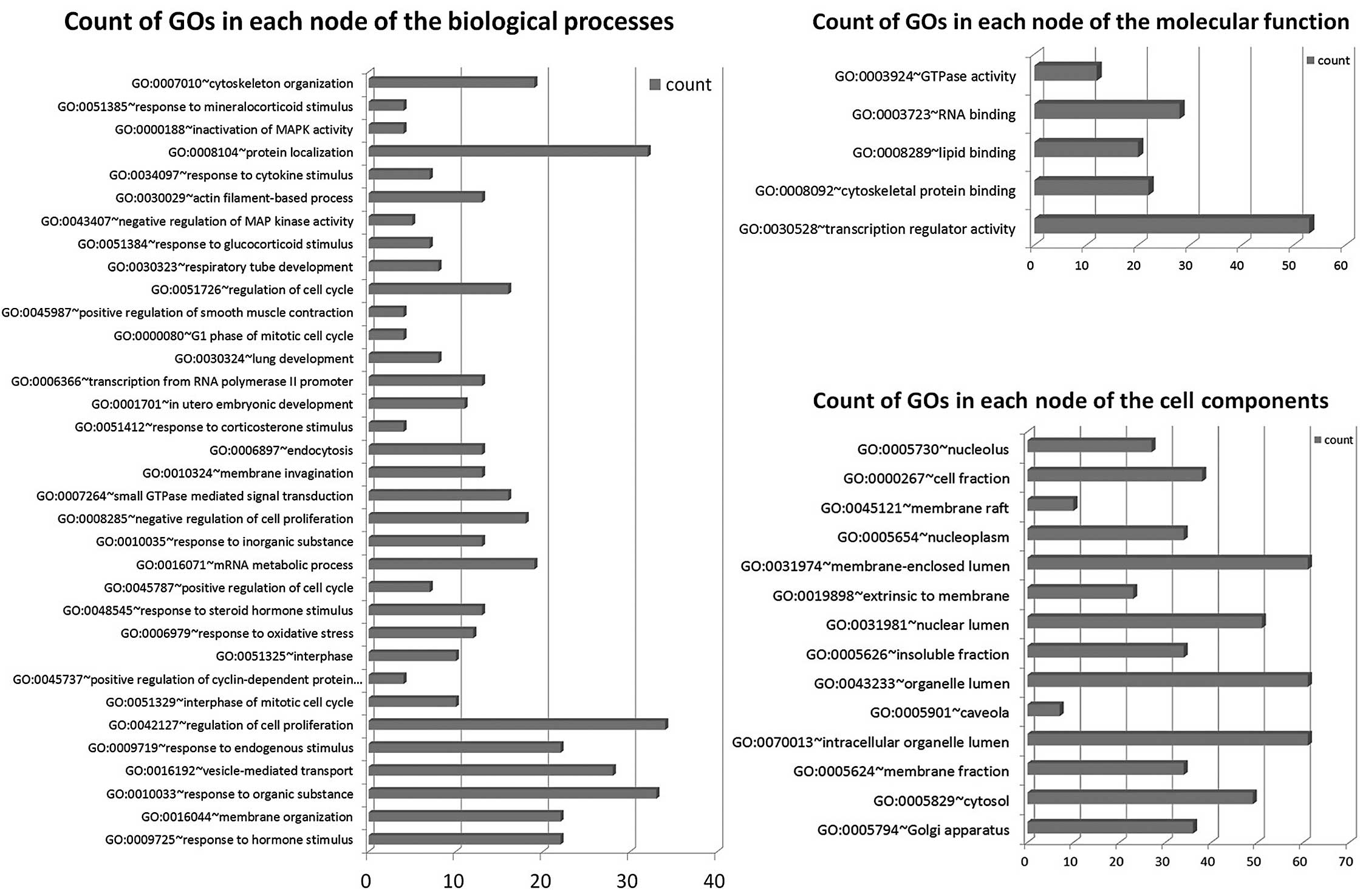
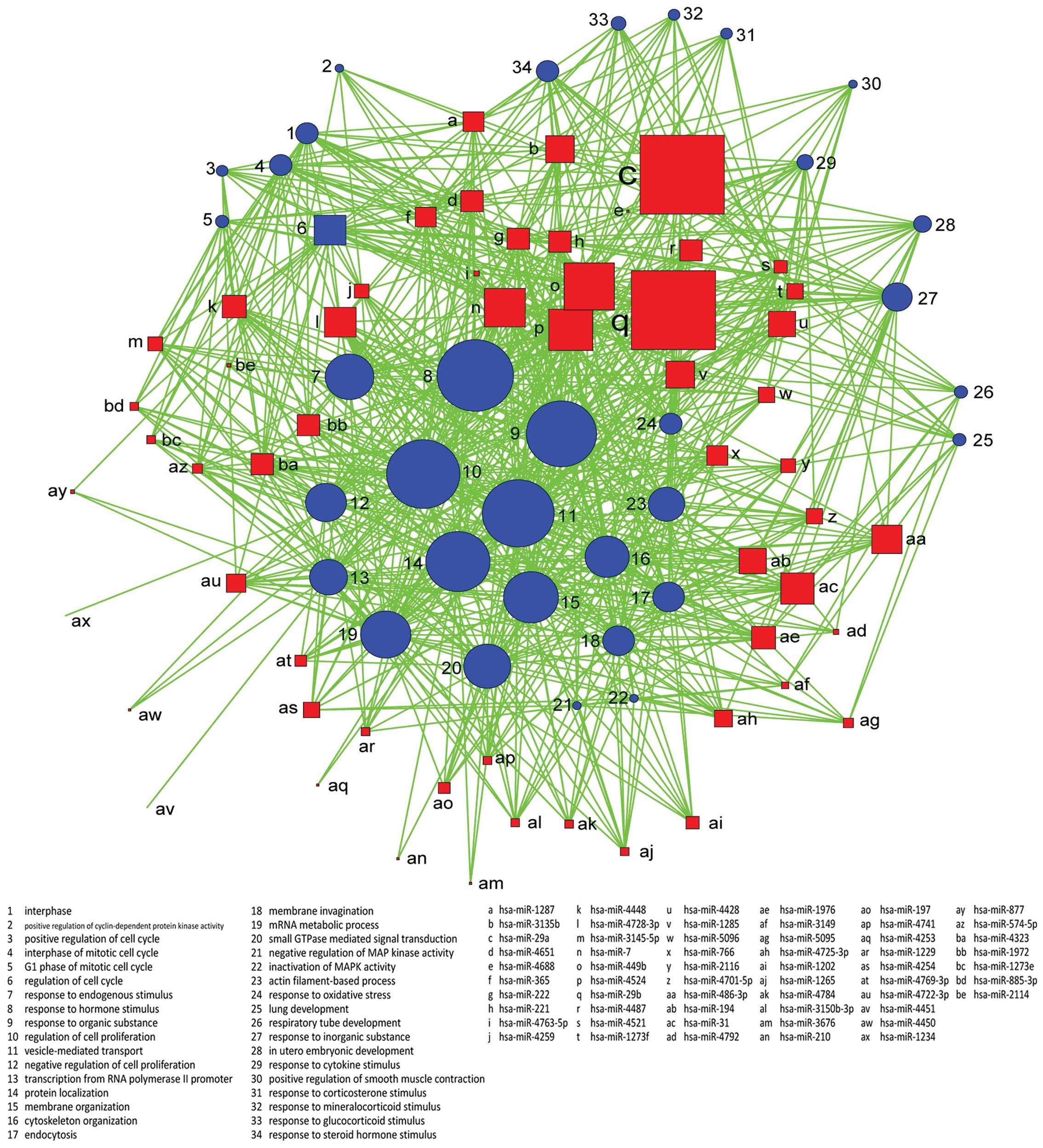
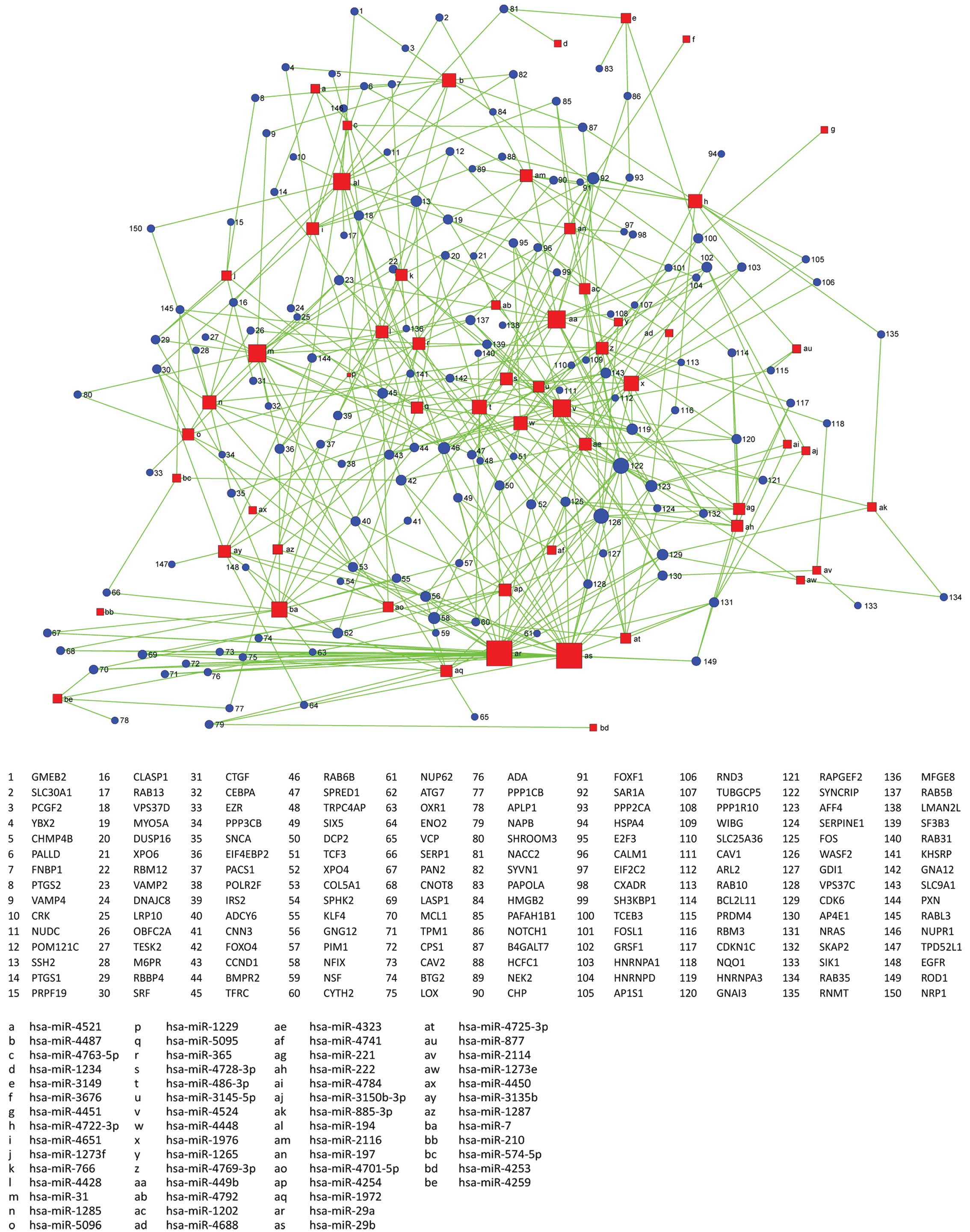
GOs are regulated by miRNAs
We found that GOs were significantly regulated by
differentially expressed genes in the biological process, molecular
function and cellular component. Results show differentially
expressed mRNAs regulated by differentially expressed miRNAs
involving a total of 34 significant GOs in the biological process,
5 GOs in the molecular function and 14 GOs in the cellular
component. The most differentially expressed GOs in the biological
process are hormone stimulus, membrane organization, response to
organic substance, vesicle-mediated transport, endogenous stimulus,
regulation of cell proliferation, interphase of mitotic cell cycle,
positive regulation of cyclin-dependent protein kinase activity and
regulation of cell cycle. The most differentially expressed GOs in
molecular function are transcription regulator activity,
cytoskeletal protein binding, lipid binding, RNA binding and GTPase
activity. The most differentially expressed GOs in cellular
component are golgi apparatus, cytosol, membrane fraction and
intracellular organelle lumen. Genes were classified in accordance
with the GO biological process, and we constructed the
miRNA-GO-network. miRNAs in different locations of the network have
a different degree. The miRNA which has the highest degree is
located in the central position of the network. From the network,
we find that hsa-miR-29a, hsa-miR-29b, hsa-miR-449b, hsa-miR-4524
and hsa-miR-7 are involved in more GOs related to the
characteristics of the sample. Regulation of cell proliferation,
vesicle-mediated transport, response to organic substance and
protein localization are important GOs represented by miRNA target
genes (Figs. 4 and 5).
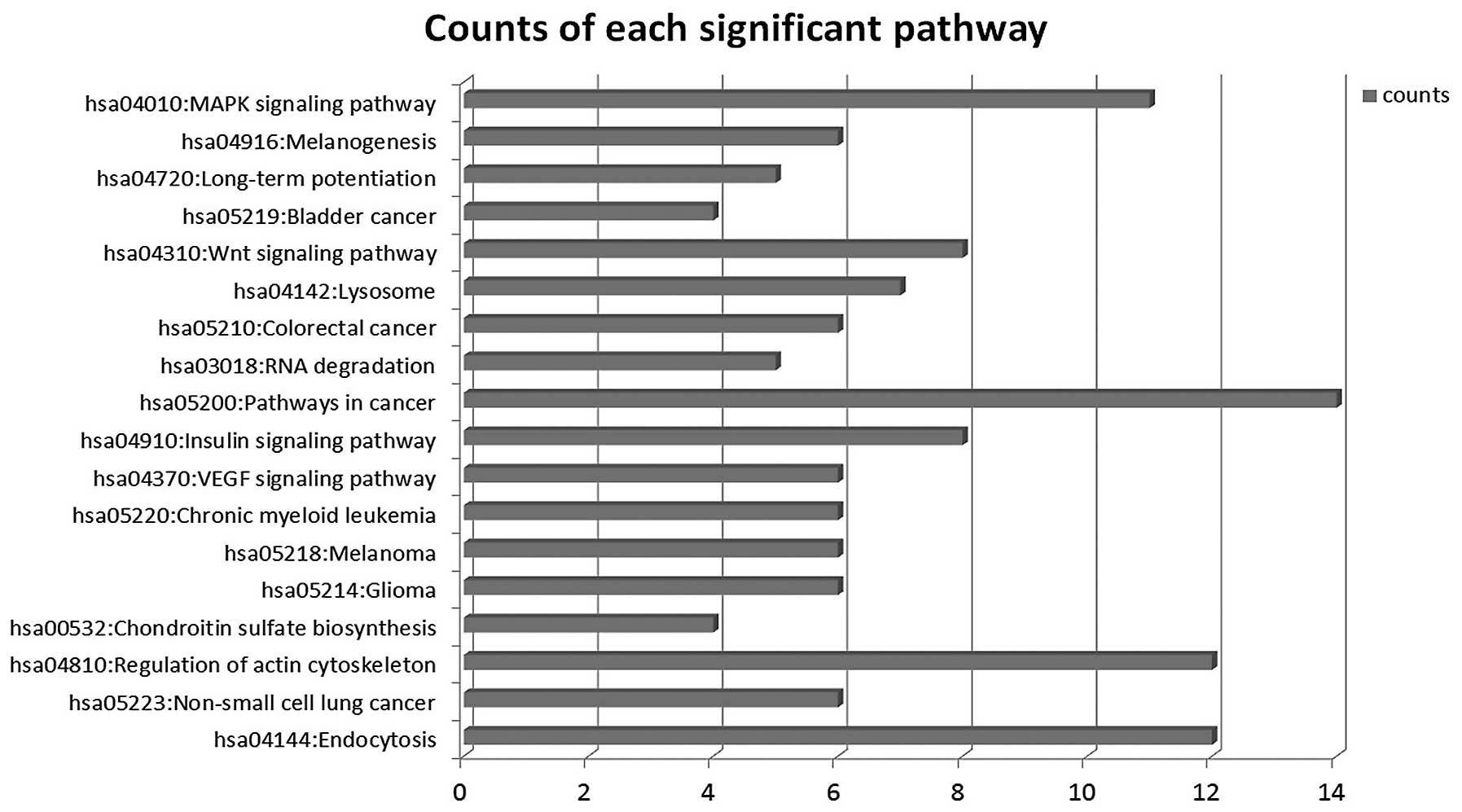
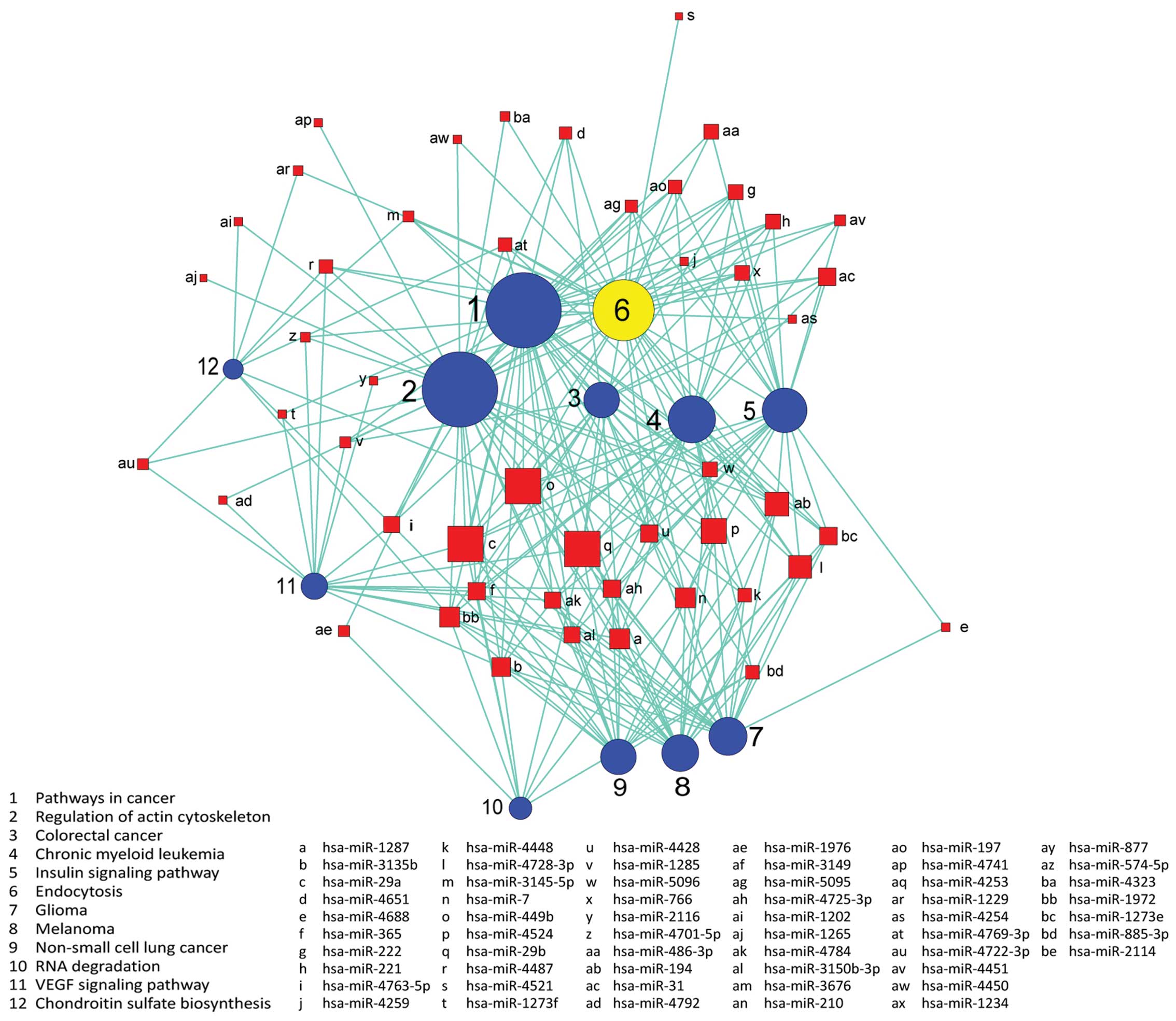
Signaling pathways are regulated by
miRNAs
We used the KEGG database to analyze target genes.
The target genes regulated by differentially expressed miRNAs
involving a total of 18 significant pathways, containing 47 genes.
These pathways include pathways in cancer, endocytosis, regulation
of actin cytoskeleton, the VEGF signaling pathway, the insulin
signaling pathway, colorectal cancer and RNA degradation. Seen from
the significant pathway relationship network, the main signal
pathways are CRC, chronic myeloid leukemia, glioma, regulation of
actin cytoskeleton, endocytosis and the VEGF signaling pathway, of
which regulation of actin cytoskeleton and pathways in cancer have
the largest degree. miRNA-29a, miRNA-449b, miRNA-29b, miRNA-4524,
miRNA-194 are at the key position in the miRNA-pathway-network.
This is essentially the same as the result of the miRNA-GO-network
(Figs. 6 and 7).
Regulation network of differentially
expressed miRNAs
There are 62 miRNAs, 34 GOs, 18 pathways that have
significant differences between colon CSCs and non-stem cells.
Therefore, regulatory networks of differentially expressed miRNAs
displaying the miRNAs with the highest degree affected the
surrounding genes hsa-mi-29a, hsa-miR-29b, hsa-miR-4524,
hsa-miR-449b, and hsa-miR-31 confirming previous results. Details
are shown in Fig. 8.
Discussion
Cancer microRNA (miRNA) expression profiling has
been widely reported. Tumors of various organs in the body have
corresponding miRNA expression profile changes. Therefore, miRNAs,
as regulators of gene expression, are involved in the tumor
development process, acting as oncogenes or tumor suppressor genes
(17–20). Changes in miRNA expression profile
are found at various stages of colon tumor development. Li et
al reported that overexpression of miR-203 can significantly
decrease cell proliferation and survival, and induce cell apoptosis
in the p53-mutated colorectal cancer (CRC) cells (21). Strillacci et al demonstrated
that downregulation of the miR-101 level could represent one of the
leading causes of COX-2 overexpression in CRC cells (22). Li et al also reported that
miR-181b can suppress proliferation of U87 glioma stem cells.
Overexpression of miR-181b can reduce chemoresistance to
temozolomide in U87 glioma stem cells (23). Therefore, these data show that
miRNAs are related to the characteristics of tumor cells. Li et
al recently found expression of breast cancer resistance
protein BCRP/ABCG2 regulatory miRNAs (hsa-miR-328, -519c and -520h)
in stem-like ABCG2+ cancer cells (24). Zhang et al identified a colon
cancer stem cell (CSC) miRNA signature comprising a total of 19
differentially expressed miRNAs, such as miR-429, miR-155, and
miR-320d, in the HT29 adenocarcinoma cell line (25). The above show that the special
phenotype and biological characteristics of CSCs are results of
miRNA regulation. However, differences in expression profiles of
miRNA between colon CSCs and non-stem cells and the relationship
between differential miRNA expression and the function of stem
cells are rarely reported. Therefore, in our study, expression
profiling of 1711 miRNAs based on OneArray microarray platform and
34694 mRNAs based on Illumina Whole-Genome system of samples of
stem cells and non-stem cells of the SW1116 human CRC cell line was
carried out to further explore the characteristics of CRC. The
array data were confirmed using RT-PCR. We also classified colon
stem cells with the profiling of miRNA. By defining miRNAs that are
related to the characteristics of colon CSCs we will be able to
further clarify the regulatory pathway and gain insight into the
features of colon CSCs.
The results, by microarray analysis in the SW1116
CRC cell line, showed 62 miRNAs and 2049 mRNAs differentially
expressed in colon stem cells compared to non-stem cells. Among
these differentially expressed miRNAs, 31 miRNAs represented
overexpression in colon stem cells, whereas the remaining 31 miRNAs
demonstrated underexpression. Among these miRNAs, overexpression of
mir-29a, mir-29b and underexpression of mir-449b, mir4524 were
confirmed by quantitative RT-PCR assay, proving the chip results.
This miRNA expression profiling indicates characteristics of colon
CSCs. Thus, miRNA regulation is intricately related to distinctive
features and the biological performance of colon CSCs.
Gene cluster controlling the cell cycle gene cluster
is extremely important for the maintenance of stem cell growth and
proliferation characteristics. GO analysis showed that there is a
noticeable change (PPP1CB, CCND1, CDKN1C and CDK6) in cell
cycle-related (GO: 0051329, interphase of mitotic cell cycle; GO:
0045787, positive regulation of cell cycle; GO: 0000080, G1 phase
of mitotic cell cycle; GO: 0051726, regulation of cell cycle) gene
cluster. This shows that the change of cell cycle of stem cells is
key to maintaining its important characteristics, and this
distinguishes stem cells from non-stem cells. This feature gives
stem cells their characteristics and their ability to proliferate
and metastasize.
The change in the characterization of cell
differentiation is an important feature of tumor stem cells that
differentiates them from non-stem cells. Numerous studies have
shown that the change of cell differentiation plays a crucial role
in tumor occurrence, development and metastasis. We found that GO
0042127 (regulation of cell proliferation) related genes have
significantly altered. Thirty-four genes correspond to this GO,
including CAV1, KLF4, SERPINE1 and BMPR2. This reflects the need of
the stem cells to maintain the level of the high-density
proliferation, in order to progress to distant metastases, cell
proliferation-related gene expression was significantly
changed.
Many signal transduction pathways in stem cells also
changed. The most important change is the MAPK signaling pathway.
MAPK, downstream signaling molecules with serine and threonine
protein kinase activity in the Ras pathway, can format AP-1 acting
on the nucleus to activate specific genes in order to pass the
signal by activating the C-Fos, C-Jun transcription regulator. The
MAPK signal transduction pathway plays an important role in stress
responses such as inflammation and apoptosis. MAPK can promote
endothelial cell proliferation and angiogenesis. Tumor angiogenesis
can provide more nutrients to accelerate the growth of the tumor
and to promote the proliferation of cancer cells. The MAPK pathway
plays a unique role in the growth of the tumor stem cell and its
transfer characteristics (26). The
Wnt pathway is also involved. The majority of downstream target
genes of the Wnt pathway is involved in cell proliferation and
apoptosis genes. Playing a key role in the embryo during
development, the Wnt pathway occurs with a variety of human tumors,
especially in CRC. In colon CSCs, Wnt pathway-related genes (FOSL1,
CCND1, CHP, FZD9 FZD4, PPP3CB PPP2CA, TBL1X) change (27). In addition, the Jak-STAT signaling
pathway, ErbB signaling pathway, VEGF signaling pathway also showed
varying degrees of change in colon CSCs.
Changes in cytoskeletal proteins may also be
involved in stem cells. We found that the pathway of regulation of
actin cytoskeleton is involved in a significant change. Kasper
et al found that actin cytoskeleton is involved in
mesenchymal stem cell aging (28).
Cell-matrix adhesion is closely related to a variety of cellular
processes such as cell migration, cell differentiation, and cell
proliferation. Pathways such as focal adhesion, gap junction,
adherens junction and tight junction are also involved.
In conclusion, by analyzing the difference of miRNA
and mRNA expression in colon CSCs and non-stem cells, we found that
miRNAs play an important role in the expression of stem cell
characteristics. This study provides a new perspective on CRC
metastasis and recurrence and the findings of this study may
contribute to the treatment and diagnosis of CRC.
Acknowledgements
The authors are grateful for the support of the
Central Laboratory and Department of Pathology of Huashan
Hospital.
References
|
1
|
Jemal A, Murray T, Ward E, et al: Cancer
statistics. CA Cancer J Clin. 59:225–249. 2009.
|
|
2
|
Lengauer C, Kinzler K and Vogelstein B:
Genetic instabilities in human cancers. Nature. 396:643–649. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baylin S and Ohm J: Epigenetic gene
silencing in cancer - a mechanism for early oncogenic pathway
addiction? Nat Rev Cancer. 6:107–116. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Esteller M: Cancer epigenomics: DNA
methylomes and histone-modification maps. Nat Rev Genet. 8:286–298.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huang E and Wicha M: Colon cancer stem
cells: implications for prevention and therapy. Trends Mol Med.
14:503–509. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pardal R, Clarke M and Morrison S:
Applying the principles of stem-cell biology to cancer. Nat Rev
Cancer. 3:895–902. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tan B, Park C, Ailles L and Weissman I:
The cancer stem cell hypothesis: a work in progress. Lab Invest.
86:1203–1207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen Y, Hsu H, Chen Y, et al: Oct-4
expression maintained cancer stem-like properties in lung
cancer-derived CD133-positive cells. PLoS One. 3:e26372008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gangemi R, Griffero F, Marubbi D, et al:
SOX2 silencing in glioblastoma tumor-initiating cells causes stop
of proliferation and loss of tumorigenicity. Stem Cells. 27:40–48.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Todaro M, Alea M, Stefano DA, et al: Colon
cancer stem cells dictate tumor growth and resist cell death by
production of interleukin-4. Cell Stem Cell. 1:389–402. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gregory RI, Yan KP, Amuthan G, et al: The
microprocessor complex mediates the genesis of microRNAs. Nature.
432:235–240. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bernstein E, Caudy A, Hammond SM and
Hannon GJ: Role for a bidentate ribonuclease in the initiation step
of RNA interference. Nature. 409:363–366. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Meister G and Tuschl T: Mechanisms of gene
silencing by double-stranded RNA. Nature. 431:343–349. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Peters L and Meister G: Argonaute
proteins: mediators of RNA silencing. Mol Cell. 26:611–623. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Harfe BD, McManus M, Mansfield JH,
Hornstein E and Tabin CJ: The RNaseIII enzyme Dicer is required for
morphogenesis but not patterning of the vertebrate limb. Proc Natl
Acad Sci USA. 102:10898–10903. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Murchison EP, Stein P, Xuan ZY, et al:
Critical roles for Dicer in the female germline. Genes Dev.
21:682–693. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Y, Medvid R, Melton C, Jaenisch R and
Blelloch R: DGCR8 is essential for microRNA biogenesis and
silencing of embryonic stem cell self-renewal. Nat Genet.
39:380–385. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wienholds E, Koudijs MJ, Eeden F, Cuppen E
and Plasterk R: The microRNA-producing enzyme Dicer1 is essential
for zebrafish development. Nat Genet. 35:217–218. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li J, Chen YX, Zhao JF, Kong FR and Zhang
YD: miR-203 reverses chemoresistance in p53-mutated colon cancer
cells through downregulation of Akt2 expression. Cancer Lett.
304:52–59. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Strillacci A, Griffonia C, Sansone P, et
al: MiR-101 downregulation is involved in cyclooxygenase-2
overexpression in human colon cancer cells. Exp Cell Res.
315:1439–1447. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li P, Lu XM, Wang YY, et al: MiR181-b
suppresses proliferation of and reduces chemoresistance to
temozolomide in U87 glioma stem cells. J Biomed Res. 24:436–443.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li X, Pan YZ, Seigel GM, et al: Breast
cancer resistance protein BCRP/ABCG2 regulatory microRNAs
(hsa-miR-328, -519c and -520h) and their differential expression in
stem-like ABCG2+ cancer cells. Biochem Pharmacol.
81:783–792. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang HL, Li WH, Nan FF, et al: MicroRNA
expression profile of colon cancer stem-like cells in HT29
adenocarcinoma cell line. Biochem Biophys Res Commun. 404:273–278.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Seger R and Krebs EG: The MAPK signaling
cascade. FASEB J. 726–735. 1995.
|
|
27
|
Polakis P: Wnt signaling and cancer. Genes
Dev. 14:1837–1851. 2000.
|
|
28
|
Kasper G, Mao L, Geissler S, Draycheva A,
Trippens J, Kühnisch J, Tschirschmann M, Kaspar K, Perka C, Duda GN
and Klose J: Insights into mesenchymal stem cell aging: involvement
of antioxidant defense and actin cytoskeleton. Stem Cells.
27:1288–1297. 2009. View
Article : Google Scholar : PubMed/NCBI
|