Introduction
Human T-cell leukemia virus type 1 (HTLV-1) is the
etiologic agent of adult T-cell leukemia (ATL), an aggressive
neoplasia of CD4+ T cells (1,2). The
HTLV-1 transcriptional transactivator protein Tax transactivates
the expression of several cellular genes in addition to the viral
long term repeat (LTR) (3–5). The cellular genes activated by the Tax
protein include several involved in cell growth which suggests that
it is the expression of this protein that deregulates T-cell growth
during HTLV-1 infection and that the constitutive expression of Tax
is correlated with immortalization in T cells and the
transformation of other cell types (6–9). Tax
corresponds to a 40-kDa transforming protein (10,11)
from the pathogenic retrovirus HTLV-1 that induces the expression
of various family members of the transcription factor AP-1, such as
c-Jun, Jun-D, c-Fos and Fra-1, at the level of RNA expression in T
cells (12,13). The Jun-N-terminal kinase (JNK) is
the only member of the MAP kinases to phosphorylate c-Jun, the main
component of AP-1 complexes, and also has ATF-2 and Elk-1 as
substrates (14,15). In mammalian cells, three
mitogen-activated protein kinase (MAPK) families have been clearly
characterized: namely classical MAPK (also known as ERK), c-Jun
N-terminal kinase/stress-activated protein kinase (JNK/SAPK) and
p38 kinase. The JNK group of MAPKs is activated in response to the
treatment of cells with inflammatory cytokines and through exposure
to environmental stress. JNK activation is mediated by a protein
kinase cascade composed of a MAPK kinase and a MAPK kinase kinase
(14–16). JNK and p38 kinases were initially
proposed to mediate apoptosis in neuronal cells (17) and phosphorylation of c-Jun is
necessary for neuronal cell death (18). The use of kinase inhibitors and the
overexpression of dominant-negative mutant forms of MAPKs have
demonstrated the involvement of JNK and/or p38 kinase in apoptosis
induced in non-neuronal cells by various stimuli, including
estrogen, cisplatin, UV-B radiation and singlet oxygen (19,20).
Retinoids are comprised of a group of compounds
including retinoic acid (RA), vitamin A (retinol) and a series of
natural and synthetic derivatives with A-like activity (21–24).
Retinoids, including retinol and its analogues, are critical for a
variety of biologic functions. Retinol is metabolized in retinal
pigment epithelial cells to 11-cis-retinal and is required for
normal ocular function (25);
retinol and retroretinoids have also been implicated in maintaining
and regulating immune function (26,27).
Retinol is also essential for epithelial differentiation (28) and the control of embryonic
development (29). The vast
majority of retinoid-driven biologic effects are mediated by RAs,
the active metabolites of retinol. All-trans retinoic acid
(ATRA) and 9-cis-RA both play an important role in the regulation
of gene transcription that ultimately results in the regulation of
cell division, growth, differentiation and proliferation (30,31).
RAs exert their biologic activity by binding to nuclear receptors
that regulate the transcriptional activity of a variety of target
genes, which are implicated in the inhibition of cell growth, the
induction of cell differentiation and the induction of apoptosis in
a variety of tumor cell lines (32,33).
Much of our understanding of the signaling events
initiated following drug treatment has been based upon model cell
systems. One of the most useful and widely studied of these T cell
models has been the human Jurkat leukemia T cell line (34). Several studies have shown that
Jurkat cells behave similar to normal cells (35,36).
Likewise, other studies have shown a correlation between JNK and
the expression of interleukin (IL)-2, and interferon (IFN)-γ in
CD4+ T cells (37,38).
Using Jurkat cells as a model, we studied the effect of ATRA in
decreasing the ability of the Tax protein to induce Jurkat cell
proliferation as well as its correlation with the expression of JNK
and a panel of cytokines such as IL-2, IFN-γ, IL-4 and IL-10.
Materials and methods
Reagents
ATRA was obtained from Sigma Chemical Co. (St.
Louis, MO, USA). The protease inhibitors phenylmethylsulfonyl
fluoride (PMSF), leupeptin, pepstatin, aprotinin and bestatin were
purchased from Roche (USA); T4 polynucleotide kinase and
poly(dI-dC)2 were obtained from Amersham Pharmacia Biotech
(Piscataway, NJ, USA). Tris-borate-EDTA buffer and
acrylamide-bisacrylamide (29:1) were obtained from Bio-Rad
(Richmond, CA, USA). Luciferase assay reagent, lysis buffer and the
pGL-2 luciferase vector were obtained from Promega (Madison, WI,
USA). TPA and ionomycin were purchased from Sigma Chemical Co.
Anti-JNK, p38 and ERK antibodies were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). MBP was purchased from
Stratagene (La Jolla, CA, USA).
Cell culture and ATRA treatment
Jurkat T cells and Jurkat T cells expressing the Tax
protein, were grown in RPMI-1640 containing 10% heat-inactivated
fetal bovine serum (FBS), 200 mM glutamine, nonessential amino
acids, penicillin and streptomycin sulfate. Before ATRA treatment,
cells were grown overnight (16–20 h) in medium containing 0.5%
heat-inactivated FBS and subsequently stimulated in the presence of
the same medium with low concentrations of serum. ATRA was diluted
in a culture medium containing 0.5% FBS, with the DMSO
concentration below 0.5%. Appropriate controls containing the same
amount of solvent were included in each experiment. Intermittent
passage in G418-containing medium was performed to ensure retention
of the plasmid.
Plasmid construction and preparation of
nuclear extracts
The plasmid expressing the wild-type Tax protein and
the Tax inducible cell line were a gift from Dr Warner Greene
(Gladstone Institute of Virology and Immunology, University of
California, San Francisco, CA, USA). The human IL-2
promoter-enhancer fragment (−500 to +60) was subcloned from plasmid
SV-IL-2-CAT into the luciferase vector pGL2 (35). The AP-1-luciferase reporter plasmid
driven by the rat prolactin minimal promoter (−36 to +37) under the
control of four copies of the human AP-1 site (36) was kindly provided by M. Rincón and
R. A. Flavell (Section of Immunobiology, Howard Hughes Medical
Institute, Yale University School of Medicine, New Haven, CT, USA).
Plasmids containing multimers of the recognition sites for NF-κB
and AP-1 were constructed and linked to the pLuc-prolactin minimal
promoter plasmid (35,36). The orientation for each element was
confirmed by the restriction enzyme cleavage. The tandem sequences
used to construct the different plasmid multimers were as follows:
i) four copies of the AP-1,
12-O-tetradecanoylphorbol-13-acetate (TPA),
5′-TCGATTGAGTCAGG-GTAA3′; ii) two copies of the NF-κB-binding site
of the human Ig κ light-chain enhancer 5′-GGGACTTTCC-3′; iii) IL-2
promoter construct bearing a −500 to +30 (35).
Transient transfection and luciferase
assays
Transfection of cells was conducted by
electroporation, using an Electro Cell Manipulator 600 (BTX, San
Diego, CA, USA) using 130 V/1700 μF capacitance. Briefly,
8×106 cells were transfected with 10 μg of luciferase
reporter plasmid and 5 μg of each expression plasmid, and the
mixture was incubated for 24 h. Transfected cells were cultured in
complete medium for 24 h and stimulated with ATRA or SP600125 for
another 8 h. Cells were harvested 32 h post-transfection, washed
twice in phosphate-buffered saline (PBS) and treated with lysis
buffer (luciferase assay; Promega) for 5–10 min on ice. Lysates
were spun down for 1 min, and the total supernatants were analyzed
using luciferase reagent and measured as a duplicate in a
luminometer (MicroLumat LB 96 P; Berthold) for 5 sec. Background
measurement was subtracted from each duplicate, and experimental
values were expressed either as recorded light units of luciferase
activity or as relative activity compared with extracts from
unstimulated cells (35,36).
Preparation of nuclear extracts
Nuclear extracts, were prepared as previously
described (34). The cells were
grown at 37°C in a humidified atmosphere of 10% CO2.
Cells (2×107), incubated with 0.1% DMSO (control) or 30
nM TPA, or 1 mM ionomycin, were collected and washed with ice-cold
PBS, washed again in buffer A [10 mM HEPES (pH 7.9), 15 mM KCl, 2
mM MgCl2, 6 mM DTT, 0.1 mM EDTA and 1 mM PMSF] and lysed
in buffer A with 0.2% Nonidet P-40. The pelleted nuclei were
re-suspended in buffer B [50 mM HEPES (pH 7.6), 50 mM KCl, 0.1 mM
EDTA, 1 mM DTT, 1 mM PMSF and 10% glycerol], in the presence of 0.3
M (NH4)2SO4 (pH 7.9) and rocked
for 30 min at 4°C. The broken nuclei were centrifuged for 10 min at
100,000 × g. A 125 μl aliquot of supernatant was transferred to a
second tube and additional
(NH4)2SO4 was added to a final
concentration of 1.5 M followed by a second centrifugation of
50,000 × g for 5 min. The supernatant was removed and the pellet
was re-suspended in 50 μl of buffer B and stored at 70°C. Protein
concentration was estimated using the Bio-Rad stain protein assay
kit with bovine albumin as the standard.
Western blot analysis
Jurkat cells (5×107) were seeded onto
6-well plates. Forty-eight hours after transfection, the cells were
collected and washed twice by cold PBS, and each well was treated
with 50 ml lysis buffer [2 mmol/l Tris-HCl (pH 7.4), 50 mmol/l
NaCl, 25 mmol/l EDTA, 50 mmol/l NaF, 1.5 mmol/l
Na3VO4, 1% Triton X-100 and 0.1% SDS], and
supplemented with protease inhibitors (1 mmol/l
phenylmethylsulfonyl fluoride, 10 mg/l pepstatin, 10 mg/l aprotinin
and 5 mg/l leupeptin) (all from Sigma Chemical Co.). Protein
concentrations were determined using the Bradford protein assay.
Equal amounts of protein (50 mg) were separated on a 15% SDS
polyacrylamide gel and transferred onto a nitrocellulose membrane
(Hybond C; Amersham, Freiburg, Germany). Membranes were blocked in
5% non-fat dry milk in TBS for 1 h at room temperature and probed
with rabbit anti-JNK-1 (SC-1648) antibody (dilution 1:500; Santa
Cruz Biotechnology, Inc.) overnight at 4°C. After washing 3 times
with TBS containing 0.1% Tween-20, membranes were incubated with
anti-rabbit IgG horseradish peroxidase (1:5,000; Santa Cruz
Biotechnology, Inc.) and developed by luminal mediated
chemiluminescence (Appylgen Technologies, Inc., China). To confirm
equal protein loading, membranes were reprobed with a 1:1,000
dilution of an anti-actin antibody (Santa Cruz Biotechnology,
Inc.). Densitometric analyses were performed using Scion Image
software (39,40).
Labeling of apoptotic cells with Annexin
V
Jurkat cells (500,000) were treated with RA as
indicated. Cells were washed with PBS and stained with Annexin
V-FITC (Pharmingen) and propidium iodide (PI) in binding buffer [10
mM HEPES (pH 7.4), 140 mM NaCl and 2.5 mM CaCl2] or 15
min at room temperature in the dark. Cells were subsequently
analyzed using flow cytometry (FACSCalibur) for apoptosis (FITC)
and viability using PI.
Cytokine (IL-2, IL-4, IL-6 and IL-10)
quantification in the culture supernatants
Jurkat leukemia T cells stably transfected with the
Tax protein were cultured under a condition identical to that
described for proliferation assay. ATRA was added at a final
10−7 M concentration 36 h after the initiation of the
culture in two independent pulses (every 12 h). After 60 h of
culture, supernatants were collected and the different cytokines
were measured in triplicate by using an enzyme-linked immunosorbent
assay (ELISA) system: IL-2, IL-4, IL-6 and IL-10 (R&D Systems,
Minneapolis, MN, USA) following the manufacturer’s protocols.
Results
We examined the effect of ATRA treatment on the
activity of one member of the MAP kinases in Jurkat leukemia and
Jurkat leukemia Tax-expressing protein.
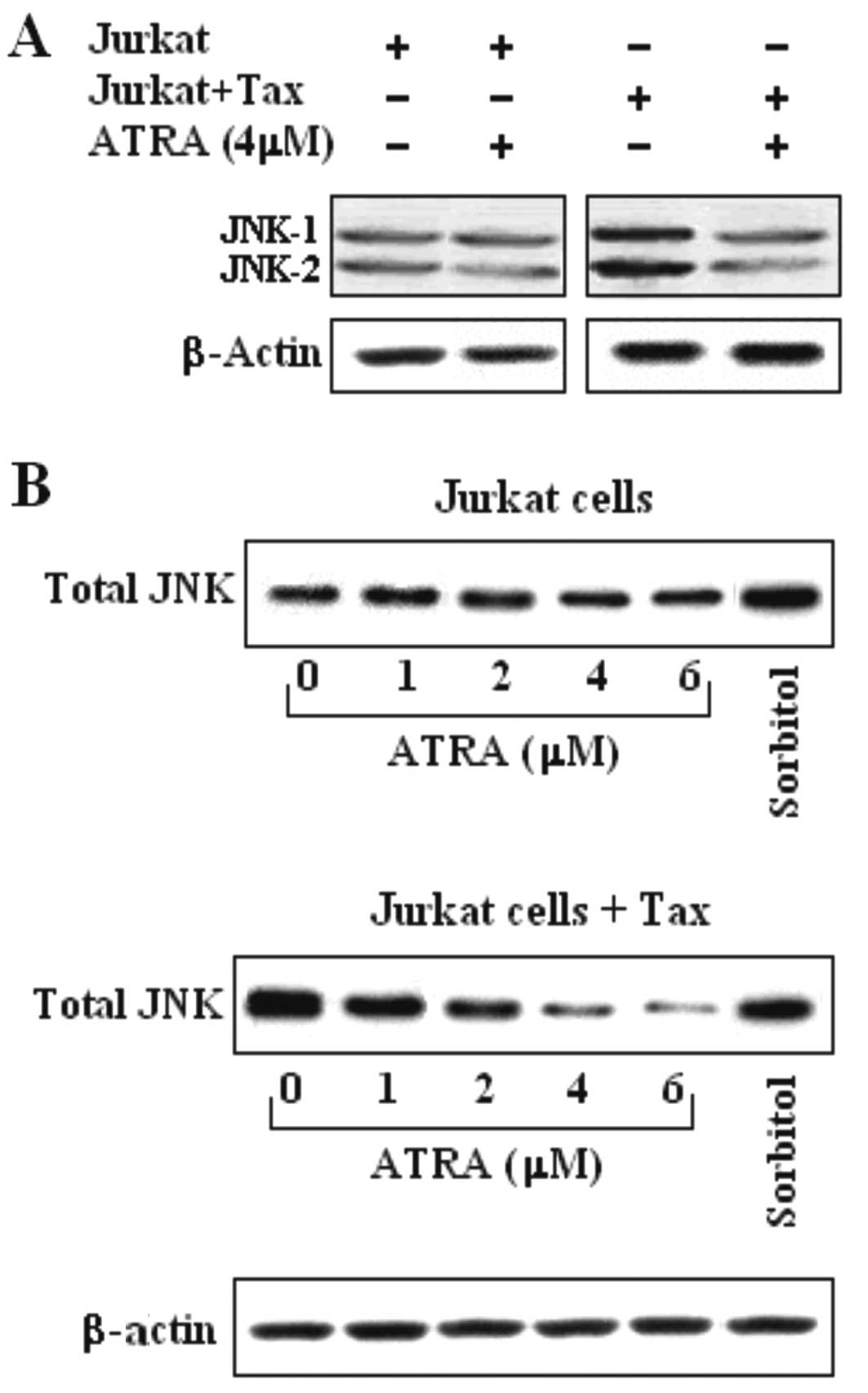
Inhibition of JNK activity by ATRA in
Jurkat-Tax expressing cells occurs in a dose-dependent manner
We examined the effect of ATRA (4 μM) treatment on
JNK kinase activity in Jurkat and Jurkat Tax-expressing cells.
JNK-1 and JNK-2 activity was detected in both Jurkat and Jurkat
Tax-expressing cells (Fig. 1A). The
strong inhibition of JNK by ATRA, in Jurkat Tax-expressing cells
occurred in a dose-dependent manner, ranging from 2 to 6 μM,
compared with the untreated Jurkat cells (Fig. 1B). However, the effect of ATRA in
the Jurkat cells was lower than that observed in the untransfected
Jurkat cells (Fig. 1B). The results
obtained in the Jurkat and Jurkat Tax-expressing cells after 24 h
of incubation are displayed in Fig.
1. Non-stimulated cells and cells treated with sorbitol were
used as the control.
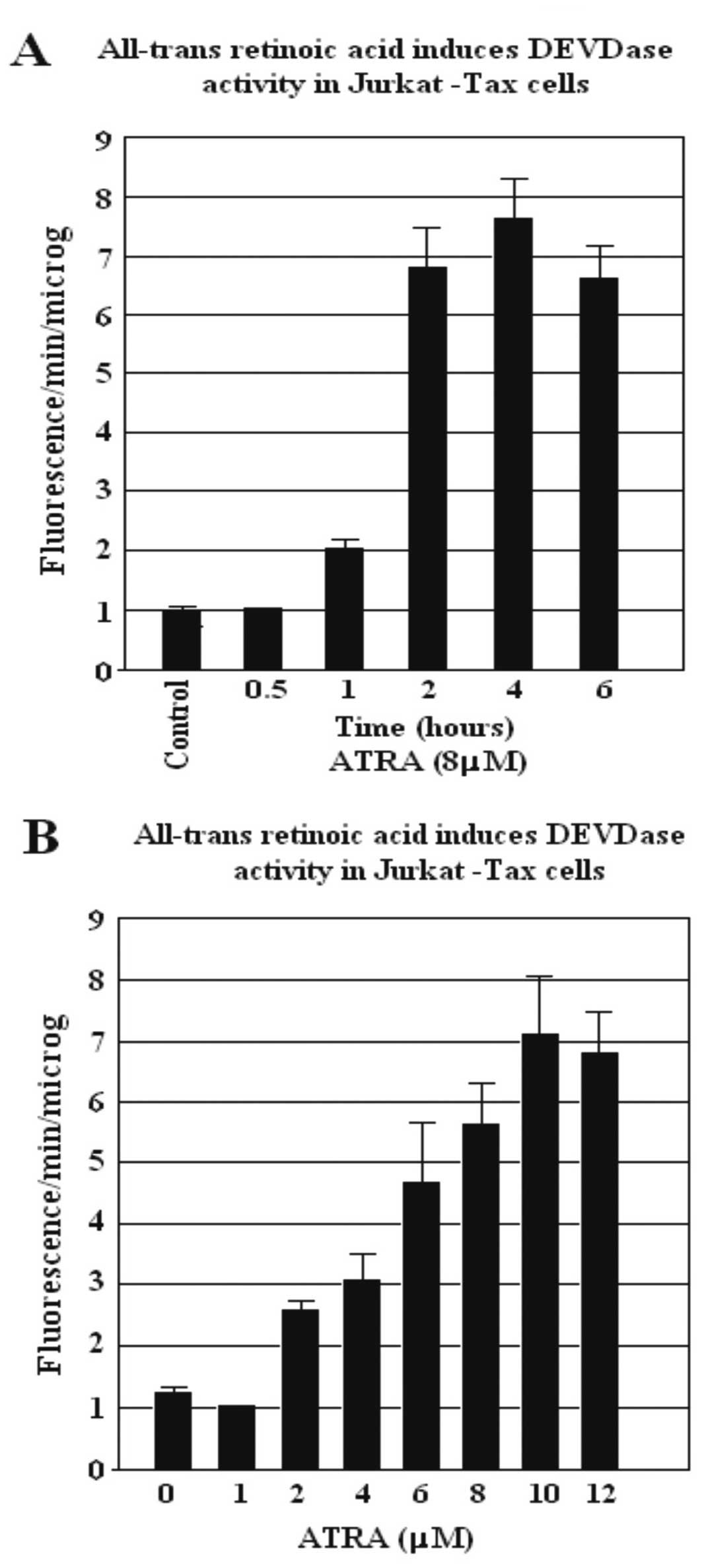
ATRA induces DEVDase activity Jurkat
Tax-expressing cells
We investigated the effect of ATRA on the ability to
induce caspase activity. Cells were pre-incubated in 10% FCS for 16
h and were later treated with 8 μM ATRA for different incubation
times (Fig. 2A) and with increasing
concentrations of ATRA (1–12 μM) for 4 h (Fig. 2B). Cytosol extracts were prepared at
the indicated periods of time, and caspase activity was
subsequently measured using DEVD-AFC as a substrate. We
demonstrated that the induction of DEVDase activity was clearly
dependent on the time of incubation and the concentration level of
ATRA. The increase in DEVDase activity was observed with
concentrations of ATRA >2 μM in the Jurkat Tax-expressing cells.
The presence of Annexin V-positive (apoptotic) cells was evident
after 1 h of incubation with ATRA (8 μM), and the number reached a
maximum after 4 h (Fig. 2A). The
apoptotic cells, dependent on ATRA concentration, as determined by
Annexin V labeling, were evident following 2 μM of ATRA treatment
and the number of apoptotic cells reached a maximum level following
10 μM ATRA treatment (Fig. 2B).
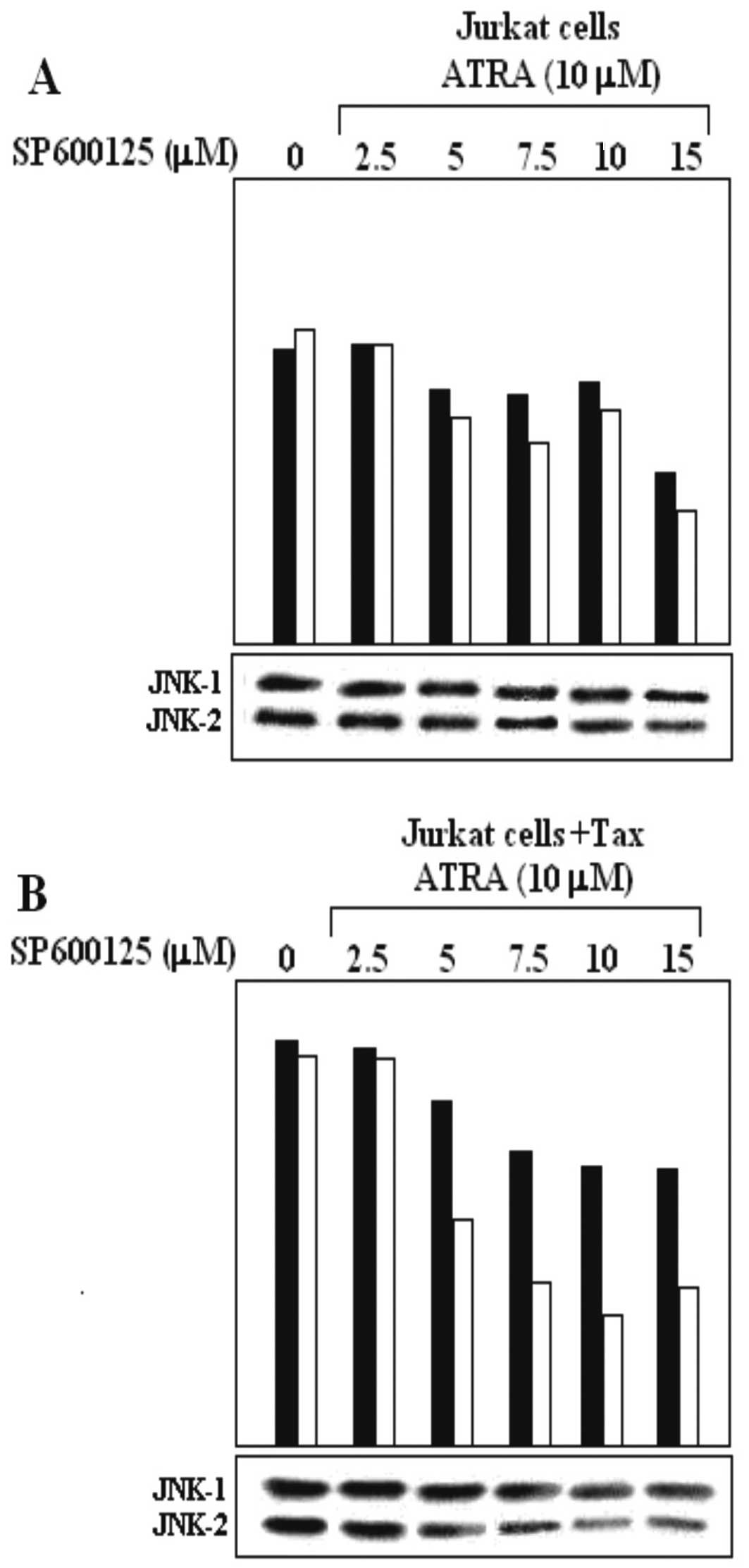
SP600125 and ATRA affect Tax upregulation
of JNK in Jurkat Tax-expessing cells
We demonstrated that ATRA, at concentration levels
of 2–6 μM, was able to decrease JNK expression in Jurkat and Jurkat
Tax-expressing cells (Fig. 1B). To
further study the inhibitory effect of ATRA on the expression of
JNK, we performed a combination assay using ATRA and SP600125 in
untransfected Jurkat cells (Fig.
3A) and Jurkat cells transfected with a plasmid expressing the
Tax protein (Fig. 3B). The effect
of increasing concentrations of SP600125 on ATRA-mediated
inhibition of JNK expression in Jurkat Tax-expressing cells was
further investigated. Jurkat and Jurkat Tax-expressing cells were
pre-incubated with increasing concentrations of SP600125 (2.5, 5.0,
7.5, 10 and 15 μM) for 120 min. Then, 10 μM ATRA was added and
cells were incubated for an additional 180 min. Cell extracts were
prepared and subsequently assayed using a western blot assay for
JNK-1 and JNK-2 protein expression. We increased the ATRA
concentration to 10 μM, to reach a major inhibitory effect of JNK-1
expression. The combination treatment of ATRA with SP600125 further
decreased the JNK expression protein. Both ATRA and SP600125
treatments reduced the activity of JNK-1 and JNK-2 (Fig. 3A and B). ATRA-mediated inhibition of
JNK-1/JNK-2 expression in untransfected Jurkat cells was lower
(Fig. 3A) compared to that observed
in Jurkat cells transfected with the Tax protein expression plasmid
(Fig. 3B). The expression of JNK-1
was almost completely inhibited with increasing concentrations of
SP600125 (15 μM) and ATRA (10 μM) (Fig.
3B).
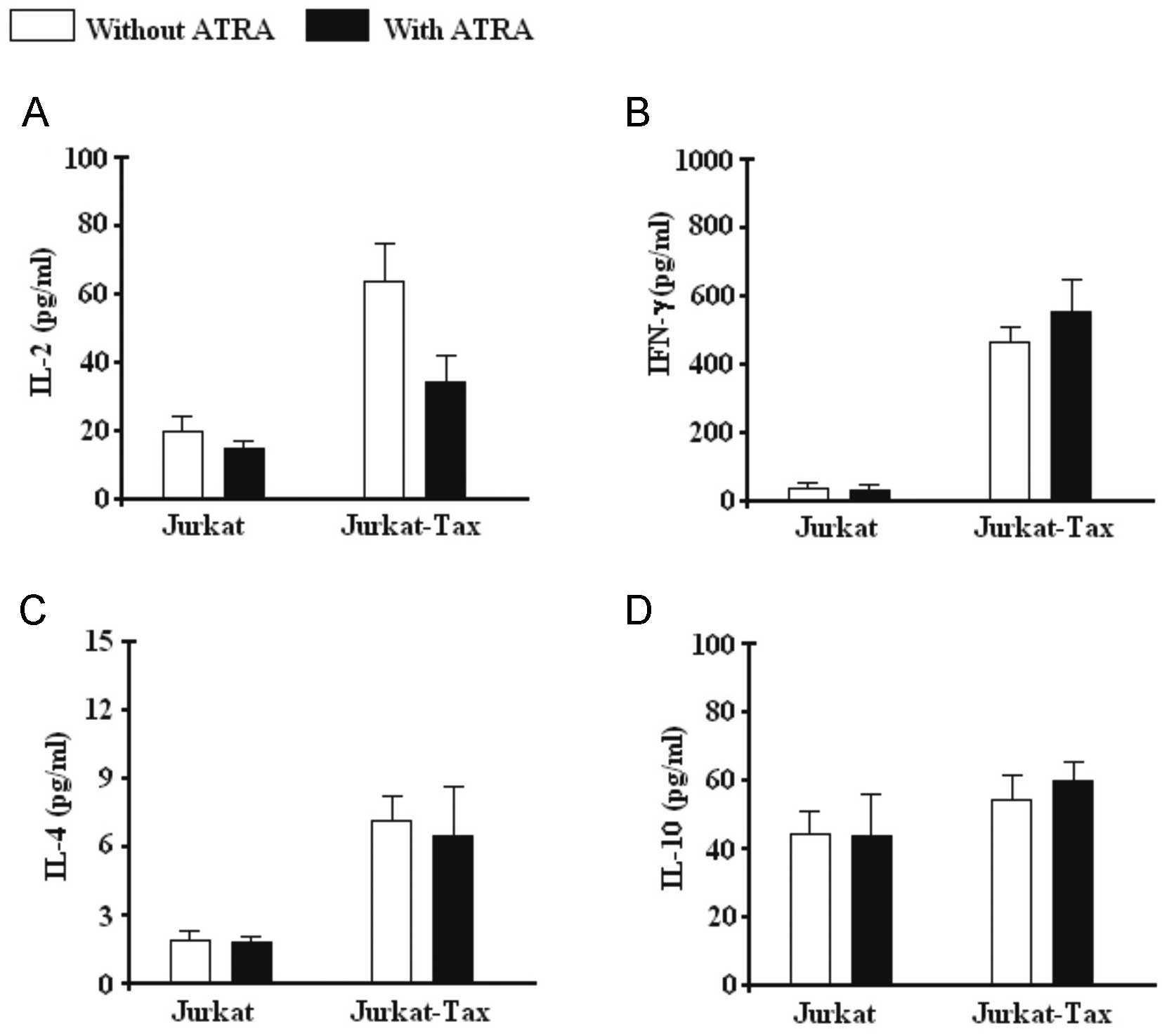
Regulation of cytokine production by ATRA
in Jurkat and Jurkat Tax-expressing cells
To further understand the effect of ATRA, cytokine
induction and secretion experiments were performed in Jurkat and
Jurkat Tax-expressing cells for IL-2, IL-4, IFN-γ and IL-10. A
strong induction of IL-2 (Fig. 4A),
IFN-γ (Fig. 4B) and IL-4 (Fig. 4C) production in Jurkat-Tax cells,
compared with Jurkat cells were observed, but not for IL-10
(Fig. 4D). The mean secretion of
IL-2 was statistically decreased in Jurkat Tax-expressing cells,
but not for IFN-γ, IL-4 and IL-10 when ATRA was added to the Jurkat
Tax-expressing stimulated cell culture.
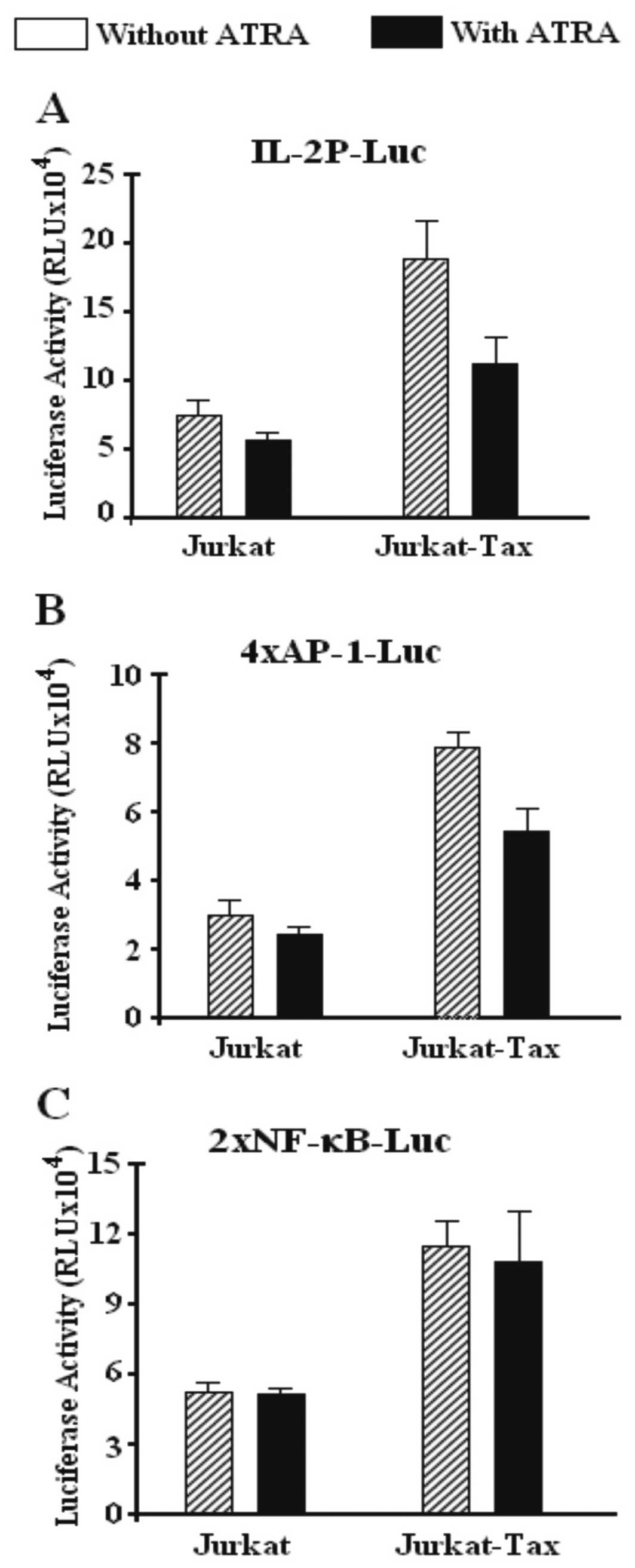
Tax transfection strongly enhances
transcriptional activity of three luciferase reporter constructs
expressing the IL-2 promoter, AP-1 and NF-κB reporter
construct
Jurkat and Jurkat T cells expressing the Tax protein
were transiently co-transfected with luciferase plasmid reporter
constructs containing the inducible region of the IL-2
enhancer/promoter (−500 to +60) (Fig.
5A) or an AP-1 (Fig. 5B), or an
NF-κB (Fig. 5C) reporter construct.
Tax expression was required to induce high transcription of these
constructs. The effect observed after Tax expression was 3-to
4-fold higher in all three constructs, the IL-2 promoter, the AP-1-
and the NF-κB-driven transcription, compared with untransfected
Jurkat cells which showed a moderate transcriptional activity.
However, after ATRA treatment the activity of the IL-2 promoter
(Fig. 5A) and AP-1 reporter
construct (Fig. 5B) was strongly
affected decreasing the transcriptional activity of both the IL-2
promoter and the AP-1 reporter, but failed to affect and slightly
decreased the transcriptional activity of the NF-κB reporter
construct promoter (Fig. 5C).
Noteworthy, NF-κB-driven transcription in the Tax-transfected and
untransfected Jurkat cells was not affected by ATRA (Fig. 5C). Apparently, NF-κB was not a
target for ATRA, since no activity was noted in Jurkat cells
transfected or not with the Tax-expression plasmid.
Discussion
In the present study, the effect of ATRA alone or in
combination with SP600125 on human Jurkat leukemia T cells,
transfected or not with a Tax expression plasmid was studied. We
demonstrated that ATRA in concentration levels between 4 to 10 μM,
caused a significant and sustained decreasing effect on JNK
expression in Jurkat Tax-expressing cells. A slight effect in all
the assays performed was observed in untransfected Jurkat cells.
Although the action of retinoids on human cells has been previously
studied (26–29), conclusive results have not been
obtained. JNK inactivation in Tax cells correlated with the
induction of apoptosis, as determined by the measurement of DEVDase
activity in the cells treated with ATRA. In addition, the
inhibitory effect of ATRA was further increased by simultaneous
stimulation with SP600125, a strong inhibitor of JNK. Several in
vitro studies have shown that JNK signaling plays an important
role in enhancing cell viability and cell cycle progression of
several cancer cell types (17,18,39).
Extensive studies on several other cell lines have shown that the
inhibition of JNK by SP600125 leads to G2/M cell cycle arrest
(19,20,40).
To further analyze the molecular mechanism of ATRA-mediated
suppression of the Tax protein in Jurkat cells we studied its
effects on the expression of IL-2, IFN-γ, IL-10 and IL-4, before
and after ATRA treatment. The effect of retinoids in specific
cytokine induction has been widely investigated in different
cellular lineages with contradictory results (26–28,41).
ATRA inhibition of JNK may be explained by a putative regulation of
the events mediated by IL-2 and/or IFN-γ. To address this issue,
IL-2, IL-4, IL-10 and IFN-γ were measured in the supernatant of
Jurkat cells with or without ATRA treatment (8 μM). Tax expression
strongly activated IL-2 and IFN-γ production but failed to
substantially increase the activity of IL-4 and IL-10. However,
ATRA treatment was able to moderately decrease the production of
IL-2 and failed to decrease the production of IFN-γ. Only a slight
Tax-mediated IL-4 and IL-10 increase was observed in all tested
cells.
To further understand the effect of ATRA on
Tax-expressing cells, we performed a luciferase assay with reporter
constructs for IL-2 promoter-Luc, AP-1-Luc and NF-κB-Luc. The
results showed that the addition of ATRA reduced the activity of
the IL-2 promoter and AP-1 reporter construct, but failed to reduce
the activity of the NF-κB reporter construct, suggesting that the
activity of ATRA may occur through AP-1. Previous studies have
demonstrated that the Tax response of the IL-2R α chain promoter
and the HIV LTR is mediated by the activation of NF-κB (42,43).
Tax has been shown to increase the expression of NF-κB proteins and
to prolong its localization to the nucleus where it is
transcriptionally active (44,45).
Tax also interacts specifically with IκB proteins (46).
In this context, IL-2 promoters have both NF-κB and
AP-1 binding sites (47) which are
important for promoter activity (48,49).
The AP-1 transcription factor is composed of Jun and Fos proteins
that function as transcriptional regulators in a heterodimeric
complex (49). The formation of
heterodimers is performed by the JNK kinase, which phosphorylates
cJun allowing it to bind to c-Fos and form the complex AP-1
(50). Thus, the modulation of JNK
may be responsible for decreasing Tax activity observed in Jurkat
cells treated with ATRA. Tax-expressing cells prior to treatment
resulted in an increase of IL-2 transcription and expression.
However, our results demonstrated that cells treated with ATRA
demonstrated a decrease in both the transcription and expression of
IL-2. Using reporter constructs for AP-1 and NF-κB, two nuclear
factors presented in the IL-2 promoter, revealed that ATRA
specifically downregulated AP-1, but not NF-κB response element
activities, compared with the untreated cells. These results
strongly correlate with the decrease of JNK-1 and JNK-2 expression,
suggesting that the Tax protein may regulate the development and
function of Tax-expressing cells involved in cell-mediated
response, partly by inhibiting factors required for the
transcription of the IL-2 gene such as AP-1, a target of JNK
activity.
Acknowledgements
We gratefully acknowledge Dr Warner Greene
(Gladstone Institute of Virology and Immunology, University of
California, San Francisco, CA, USA) for providing the plasmid
expressing the wild-type Tax protein and the Tax inducible cell
line. This study was supported by the Biomedical Experimental
Laboratory, Faculty of Sciences, University of Tarapaca, Arica,
Chile.
References
|
1
|
Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn
PA, Minna JD and Gallo RC: Detection and isolation of type C
retrovirus particles from fresh and cultured lymphocytes of a
patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci USA.
77:7415–7419. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hinuma V, Nagata K, Hanaoka M, Nakai M,
Matsumoto T, Kinoshita K, Shirakawa S and Miyoshi I: Adult T-cell
leukemia antigen in ATL cell line and detection of antibodies to
the antigen in human sera. Proc Natl Acad Sci USA. 78:6476–6480.
1981. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cann AJ, Rosenblatt JD, Wachsman W, Shah
NP and Chen IS: Identification of the gene responsible for human
T-cell leukaemia virus transcriptional regulation. Nature.
318:571–574. 1985. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Felber BK, Paskalis H, Kleinman-Ewing C,
Wong-Staal F and Pavlakis GN: The pX protein of HTLV-I is a
transcriptional activator of its long terminal repeats. Science.
229:675–679. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sodroski J, Rosen C, Goh WC and Haseltine
W: A transcriptional activator protein encoded by the x-lor region
of the human T-cell leukemia virus. Science. 228:1430–1434. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Grassmann R, Berchtold S, Radant I, Alt M,
Fleckenstein B, Sodroski JG, Haseltine WA and Ramstedt U: Role of
human T-cell leukemia virus type 1 X region proteins in
immortalization of primary human lymphocytes in culture. J Virol.
66:4570–4575. 1992.PubMed/NCBI
|
|
7
|
Siekevitz M, Feinberg MB, Holbrook N,
Wong-Staal F and Greene WC: Activation of interleukin 2 and
interleukin 2 receptor (Tac) promoter expression by the trans
activator (tat) gene product of human T-cell leukemia virus, type
I. Proc Natl Acad Sci USA. 84:5389–5393. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ballard DW, Bohnlein E, Lowenthal JW, Wano
Y, Franza BR and Greene WC: HTLV-I tax induces cellular proteins
that activate the kappa B element in the IL-2 receptor alpha gene.
Science. 241:1652–1655. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Leung KY and Nabel GJ: HTLV-1
transactivator induces interleukin-2 receptor expression through an
NF-kappa B-like factor. Nature. 333:776–778. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Franchini G: Molecular mechanisms of human
T-cell leukemia/lymphotropic virus type I infection. Blood.
86:3619–3639. 1995.PubMed/NCBI
|
|
11
|
Franklin A and Nyborg JK: Mechanisms of
Tax regulation of human T cell leukemia virus type I gene
expression. J Biomed Sci. 2:17–29. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bergers G, Graninger P, Braselmann S,
Wrighton C and Busslinger M: Transcriptional activation of the
fra-1 gene by AP-1 is mediated by regulatory sequences in the first
intron. Mol Cell Biol. 15:3748–3758. 1995.PubMed/NCBI
|
|
13
|
Angel P and Karin M: The role of Jun, Fos
and the AP-1 complex in cell-proliferation and transformation.
Biochim Biophys Acta. 1072:129–157. 1991.PubMed/NCBI
|
|
14
|
Fuchs SY, Xie B, Adler V, Fried VA, Davis
RJ and Ronai Z: c-Jun NH2-terminal kinases target the
ubiquitination of their associated transcription factors. J Biol
Chem. 272:32163–32168. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yao M, Nguyen TV and Pike CJ:
Beta-amyloid-induced neuronal apoptosis involves c-Jun N-terminal
kinase-dependent downregulation of Bcl-w. J Neurosci. 25:1149–1158.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Widmann C, Gibson S, Jarpe MB and Johnson
GL: Mitogen-activated protein kinase: conservation of a
three-kinase module from yeast to human. Physiol Rev. 79:143–180.
1999.PubMed/NCBI
|
|
17
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Behrens A, Sibilia M and Wagner EF:
Amino-terminal phosphorylation of c-Jun regulates stress-induced
apoptosis and cellular proliferation. Nat Genet. 21:326–329. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang CC and Shapiro DJ: Activation of the
p38 mitogen-activated protein kinase pathway by estrogen or by
4-hydroxytamoxifen is coupled to estrogen receptor-induced
apoptosis. J Biol Chem. 275:479–486. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu R, Mandlekar S, Tan TH and Kong AN:
Activation of p38 and c-Jun N-terminal kinase pathways and
induction of apoptosis by chelerythrine do not require inhibition
of protein kinase C. J Biol Chem. 275:9612–9619. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brockes J: Developmental biology. Reading
the retinoid signals. Nature. 345:766–768. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maden M: The retinoic acid supergun
affair. Curr Biol. 4:281–284. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
O’Connell MJ, Chua R, Hoyos B, Buck J,
Chen Y, Derguini F and Hammerling UJ: Retro-retinoids in regulated
cell growth and death. Exp Med. 184:549–555. 1996.PubMed/NCBI
|
|
24
|
Chen Y, Buck J and Derguini F:
Anhydroretinol induces oxidative stress and cell death. Cancer Res.
59:3985–3990. 1999.PubMed/NCBI
|
|
25
|
Duester G: Families of retinoid
dehydrogenases regulating vitamin A function: production of visual
pigment and retinoic acid. Eur J Biochem. 267:4315–4324. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yan JP, Garrus JE, Giebler HA, Stargell LA
and Nyborg JK: Molecular interactions between the coactivator CBP
and the human T-cell leukemia virus Tax protein. J Mol Biol.
281:395–400. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cantorna MT, Nashold FE, Chun TY and Hayes
CE: Vitamin A down-regulation of IFN-gamma synthesis in cloned
mouse Th1 lymphocytes depends on the CD28 costimulatory pathway. J
Immunol. 156:2674–2679. 1996.PubMed/NCBI
|
|
28
|
De Luca LM, Darwiche N, Celli G, et al:
Vitamin A in epithelial differentiation and skin carcinogenesis.
Nutr Rev. 52:S45–S52. 1994.PubMed/NCBI
|
|
29
|
Thaller C and Eichele G: Identification
and spatial distribution of retinoids in the developing chick limb
bud. Nature. 327:625–628. 1987. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Saari JC: Retinoids in photosensitive
systems. The Retinoids: Biology, Chemistry, and Medicine. Sporn MB,
Roberts AB and Goodman DS: 2nd edition. Raven; New York, NY: pp.
351–386. 1994
|
|
31
|
Sporn MB and Roberts AB: Role of retinoids
in differentiation and carcinogenesis. J Natl Cancer Inst.
73:1381–1387. 1984.PubMed/NCBI
|
|
32
|
Martin SJ, Bradley JG and Cotter TG: HL-60
cells induced to differentiate towards neutrophils subsequently die
via apoptosis. Clin Exp Immunol. 79:448–453. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Oridate N, Lotan D, Xu XC, Hong WK and
Lotan R: Differential induction of apoptosis by all-trans-retinoic
acid and N-(4-hydroxyphenyl) retinamide in human head and neck
squamous cell carcinoma cell lines. Clin Cancer Res. 2:855–863.
1996.PubMed/NCBI
|
|
34
|
Sigal HN and Dumont FJ: Cyclosporin A,
FK-506, and rapamycin: pharmacologic probes of lymphocyte signal
transduction. Ann Rev Immunol. 10:519–560. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Parra E, Varga M, Hedlund G, Kalland T and
Dohlsten M: Costimulation by B7-1 and LFA-3 targets distinct
nuclear factors that bind to the interleukin-2 promoter: B7-1
negatively regulates LFA-3-induced NF-AT binding. Mol Cell Bio.
17:1314–1323. 1997.PubMed/NCBI
|
|
36
|
Parra E, McGuire K, Hedlund G and Dohlsten
M: Over-expression of RelA and c-Jun substitutes for B7-1
costimulation by targeting the CD28RE within the IL-2 promoter. J
Immunol. 160:5374–5381. 1998.PubMed/NCBI
|
|
37
|
Smeets RL, Fleuren WM, He X, Vink PM,
Wijnands F, Gorecka M, Klop H, Bauerschmidt S, et al: Molecular
pathway profiling of T lymphocyte signal transduction pathways; Th1
and Th2 genomic fingerprints are defined by TCR and CD28-mediated
signalling. BMC Immunol. 13:122012. View Article : Google Scholar
|
|
38
|
Rincón M, Conze D, Weiss L, Diehl NL,
Fortner KA, Yang D, Flavell RA, Enslen H, Whitmarsh A and Davis RJ:
Conference highlight: do T cells care about the mitogen-activated
protein kinase signalling pathways? Immunol Cell Biol. 78:166–175.
2000.PubMed/NCBI
|
|
39
|
Parra E, Ortega A and Saenz L:
Downregulation of Egr-1 by siRNA inhibits growth of human prostate
carcinoma cell line PC-3. Oncol Rep. 22:1513–1518. 2009.PubMed/NCBI
|
|
40
|
Parra E: Activation of MAP kinase family
members triggered by TPA or ionomycin occurs via the protein
phosphatase 4 pathway in Jurkat leukemia T cells. Mol Med Rep.
5:773–778. 2012.PubMed/NCBI
|
|
41
|
Allende LM, Corell A, Madroño A, Góngora
R, Rodríguez-Gallego C, López-Goyanes A, Rosal M and Arnaiz-Villena
A: Retinol (vitamin A) is a cofactor in CD3-induced human
T-lymphocyte activation. Immunology. 90:388–396. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kanno T, Franzoso G and Siebenlist U:
Human T-cell leukemia virus type I Tax-protein-mediated activation
of NF-kappa B from p100 (NF-kappa B2)-inhibited cytoplasmic
reservoirs. Proc Natl Acad Sci USA. 91:12634–12638. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Maggirwar SB, Harhaj E and Sun SC:
Activation of NF-kappa B/Rel by Tax involves degradation of I kappa
B alpha and is blocked by a proteasome inhibitor. Oncogene.
11:993–998. 1995.PubMed/NCBI
|
|
44
|
Suzuki T, Hirai H, Murakami T and Yoshida
M: Tax protein of HTLV-I destabilizes the complexes of NF-kappa B
and I kappa B-alpha and induces nuclear translocation of NF-kappa B
for transcriptional activation. Oncogene. 10:1199–1207.
1995.PubMed/NCBI
|
|
45
|
Suzuki T, Hirai H and Yoshida M: Tax
protein of HTLV-I interacts with the Rel homology domain of
NF-kappa B p65 and a c-Rel protein bound to the NF-kappa B binding
site and activates transcription. Oncogene. 9:3099–3105.
1994.PubMed/NCBI
|
|
46
|
McGuire KL, Curtiss VC, Larson EL and
Haseltine WA: Influence of human T-cell leukemia virus type I tax
and rex on interleukin-2 gene expression. J Virol. 67:1590–1599.
1993.PubMed/NCBI
|
|
47
|
Visse E, Inostroza J, Cabello G and Parra
E: The MAP kinases are differently utilized by CD28 and CD2
adhesion pathways in superantigen-activated Jurkat T cells. Biol
Res. 36:263–278. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Olsson C, Michaelsson E, Parra E,
Pettersson U, Lando P and Dohlsten M: Biased dependency of CD80
versus CD86 in the induction of transcription factors regulating
the IL-2 promoter. Int Immunol. 10:499–506. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lee W, Mitchell P and Tjian R: Purified
transcriptional factor AP-1 interacts with TPA-inducible enhancer
elements. Cell. 49:741–752. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bannister A, Oehler T, Wilhelm D, Angel P
and Koumanidis T: Stimulation of c-Jun activity by CBP: c-Jun
residues Ser63/73 are required for CBP induced stimulation in vivo
and CBP binding in vitro. Oncogene. 11:2509–2514. 1995.PubMed/NCBI
|