Introduction
Lung cancer is the most commonly diagnosed cancer
worldwide and is the leading cause of cancer-related mortality
among men and women (1).
Chemotherapy remains one of the primary modalities for treating
lung cancer. However, the clinical use of cytotoxic drugs is
limited due to intrinsic or acquired resistance and toxicity
(2). Despite the improvements in
cancer therapy over the last 30 years, the overall 5-year survival
rate is generally less than 15% (3). To improve the survival rate, intensive
efforts have been made to find new anticancer agents.
Evodiamine is one of the major bioactive components
derived from Wu-Chu-Yu, a long-standing Chinese herb. Evodiamine
has been reported to possess various biological activities,
including anti-inflammatory effects (4,5), and
to have inhibitory effects on adipogenesis (6) and gastrointestinal disorders (7). Evodiamine is effective for inducing
cell cycle arrest and apoptosis in human colon LoVo cells (8), cervical HeLa cells (9), melanoma A375-S2 cells (10), thyroid cancer cells (11), hepatocellular SMMC-7721 cells
(12) and prostate DU145 and PC3
cell lines (13). Moreover,
evodiamine inhibited tumor cell migration in murine Lewis lung
carcinoma (LLC) cells (14).
However, the effect of evodiamine on LLC cell growth and death
remains unclear.
The study of autophagic activity and its
contribution to the development of cancer has gained interest in
recent years. Autophagy is an evolutionary conserved eukaryotic
process in which organelles and bulk proteins are turned over by
lysosomal activity. The physiologic function of autophagy is to
maintain homeostasis by eliminating unnecessary proteins and
injured or aged organelles. These processes are regulated by a
group of autophagy-specific genes (Atgs) which function to sense
environmental stress, assemble double- or multi-membrane-bound
autophagic vacuoles to sequester the cytoplasmic materials (termed
autophagosome), and facilitate the transport of autophagosomes to
lysosomes for degradation (15).
Autophagy has different effects on cell survival depending on the
stage of cell development (16). It
has been reported that autophagy displays a suppression effect at
the initial stage, in which tumor growth is rapid, or may be
required to provide essential nutrients to the cells in advanced
stages of cancer (17). Thus,
autophagy is not only involved in cell death, it can also induce
recycling of proteins and organelles to sustain cell survival.
However, this self-defense mechanism can render cancer cells
insensitive to anticancer agents. This can be overturned by
inhibiting autophagy, whereby autophagy inhibition could potentiate
cisplatin-induced apoptosis in esophageal squamous carcinoma cells
(18), upregulate
anthocyanin-induced apoptosis in hepatocellular carcinoma cells
(19) and augment 5-fluorouracil
chemotherapy in human colon cells in vitro and in
vivo(20,21). Furthermore, evodiamine stimulated
autophagy in human cervical HeLa cells as a survival function and
an autophagy inhibitor enhanced the sensitivity of HeLa cells to
evodiamine (22). It is unclear
whether evodiamine can induce autophagy in LLC cells, or whether
evodiamine-induced autophagy in LLC cells is cytoprotective.
In this study, we examined the anticancer activity
of evodiamine in LLC cells. We demonstrated that evodiamine
inhibited proliferation of LLC cells and increased apoptosis in
vitro and in vivo. Evodiamine induced autophagy by
converting microtubule-associated protein 1 light chain 3 (LC3)-I
to LC3-II and expressions of Atgs. Inhibiting autophagy enhanced
evodiamine-induced apoptosis in vitro in a
caspase-independent manner and in vivo in a
caspase-dependent manner. Overall, evodiamine-induced autophagy may
play a cytoprotective role in LLC cells, and evodiamine combined
with an autophagy inhibitor therapy could attenuate the
chemoresistance of LLC cells.
Materials and methods
Animals
All animals used in our study were athymic mice, not
human or non-human primates. Animals were used to establish a
murine Lewis lung carcinoma xenograft model through subcutaneously
injecting cells and agents for the treatment were administered
intratumorally every 3 days for a total of 5 times. All efforts
were made to minimize suffering and to provide appropriate
conditions, including a sterile environment and room temperature at
24°C, sterile distilled water, eggs and apples.
Cell line and cell culture
The murine Lewis lung carcinoma cell line (LLC) was
obtained from the State Key Laboratory of Trauma, Burns and
Combined Injury, Research Institute of Surgery (Chongqing, China).
The cells were cultured in Dulbecco’s modified Eagle’s medium
(DMEM) containing 10% fetal bovine serum (FBS), 100 U/ml
penicillin, and 100 μg/ml streptomycin at 37°C in the presence of
95% air, 5% CO2 with medium change every 2 days. Cells
in the exponential growth phase were harvested for use.
Chemicals and antibodies
Evodiamine was purchased from Xi’an Guanyu Bio-Tech
Co., Ltd. (Xi’an, China) with a purity of 98% determined by HPLC.
Evodiamine was dissolved in dimethylsulphoxide (DMSO) and diluted
with DMEM prior to use. The DMSO concentration in the cell culture
medium was <0.1%.
DMEM and FBS were obtained from Gibco BRL
(Gaithersburg, MD, USA). DMSO, MTT
(3-(4,5-dimethylthiazole-2-yl)-2, 5-diphenyltetrazolium bromide),
3-methyladenine (3-MA), monodansylcadaverine (MDC) and
4′,6-diamidino-2-phenylindole (DAPI) were purchased from
Sigma-Aldrich (St. Louis, MO, USA). Annexin V-FITC/PI apoptosis
detection kit was from KeyGen Biotech (Nanjing, China).
Chemiluminescence kit and BCA protein assay kit were from KangChen
Bio-tech (Shanghai, China). TUNEL apoptosis detection kit was
obtained from Roche Applied Science (Indianapolis, IN, USA).
Athymic mice were from the Chinese Academy of Sciences (Shanghai,
China). The following primary antibodies were used in this study:
LC3, Atg3, Atg4b, Atg5 and Atg7 (Sigma-Aldrich), caspase-3 (Fisher
Scientific, Hudson, NH, USA).
MTT assay and cell morphological
assay
A modified MTT assay was employed to quantify the
cell proliferation. LLC cells were seeded at a density of
1×104/well in 96-well plates. Following incubation with
agents of different concentrations for the indicated times, 20 μl
of 5 mg/ml MTT solution were added to each well followed by
incubation for 6 h. Then, 50 μl of 20% SDS solution were added in
each well followed by incubation overnight. The optical density was
determined with a microplate reader (BioTek ELx800, USA) at 570 nm
and 50% inhibition of cell growth (IC50) was evaluated.
The percentage of cell viability was calculated as follows: Cell
viability (%) =
A570(evodiamine)/A570(control) × 100%.
LLC cells at a density of 1×105/well were
seeded in 6-well plates and maintained overnight. The medium was
discarded and replaced with fresh medium or medium containing
evodiamine of different concentrations for 48 h. Cell morphology
was observed and representative images were captured under a light
microscope.
Flow cytometry
Apoptosis was measured by detecting
phosphatidylserine that was exposed on cell membranes using an
apoptosis detection kit. LLC cells treated with indicated agents at
indicated times were harvested and washed with PBS, then
resuspended in staining solution containing PI (50 μg/ml) and
Annexin V-FITC (25 μg/ml) followed by incubation for 15 min at room
temperature in the dark. Cells were resuspended in the binding
buffer and apoptotic cells were analyzed on a flow cytometry system
(FACScan; Becton-Dickinson, Heidelberg, Germany). Data were
analyzed using CellQuest software (Becton-Dickinson).
Analysis of autophagy
A fluorescent compound, MDC, has been proposed as a
special tracer for autophagic vacuoles. Treated and untreated cells
were harvested and washed with PBS, then resuspended in 0.05 mM MDC
and incubated at 37°C for 30 min. Following incubation, cells were
washed with PBS and cell number was counted under a light
microscope. Then, cells were lysed with lysis buffer (1 mM
Tris-HCl, 1% Triton X-100, pH 7.5) at room temperature in the dark.
Cell lysate was added into 96-well plates at a density of 100
μl/well and MDC (excitation wavelength 325 nm, emission filter 525
nm) fluorescence in cell lysate was determined with Optima FLUOstar
plate reader (BMG Labtech, Durham, NC, USA). The amount of
autophagic vacuoles in single cells was evaluated by obtaining the
ratio of MDC fluorescence to the amount of cells. Also, the
autophagic vacuoles of cells growing on cover-slips were labeled
with MDC at 37°C for 30 min and then cell nuclei were stained with
DAPI. Following incubation, cells were washed with PBS and
representative images were captured under a laser confocal scanning
microscope (Nikon Eclipse TE 300, Germany).
Western blot assay
Treated or untreated cells were harvested and washed
twice in ice-cold PBS, then lysed with lysis buffer. Cell lysate
was centrifuged at 13,000 g for 5 min at 4°C. The protein
concentration was determined by a BCA protein assay kit. Protein
was loaded onto SDS-PAGE gel and electro-transferred to
polyvinylidene fluoride membranes. After blocking with 5% non-fat
milk, the membranes were incubated with specific primary antibodies
followed by horseradish peroxidase conjugated secondary antibodies.
Finally, the membranes were visualized with an enhanced
chemiluminescence kit. Signal intensity was quantified by
densitometric analysis (Tanon, Shanghai, China).
In vivo antitumor effect
A total of 32 female athymic mice aged 4–6 weeks
were given ad libitum access to sterilized food and water.
Mice were subcutaneously administered 2×106 of LLC cells
in 0.1 ml of PBS. Two weeks later, when the tumor size reached ~100
mm3, mice were randomly divided into the DMSO, 3-MA,
evodiamine and evodiamine + 3-MA groups (n=8 per group). Evodiamine
(1 mg/kg) and/or 25 mg/kg 3-MA dissolved in 100 μl DMSO was
injected intratumorally every 3 days for a total of 5 times (Days
15, 18, 21, 24 and 27) and 100 μl DMSO were injected as the
control. The tumor volume and body weight were measured during the
experimental period, and tumor volume was determined by the
following formula: 0.5×L×W2 (L, length; W, width). The
animals were sacrificed after five times of measurement and tumors
were removed. A section of the tumor was fixed in 4%
paraformaldehyde, embedded in paraffin, cut into 5-μm sections and
mounted on slides for further analysis; another section was stored
at −70°C for western blot analysis.
Apoptosis in tumors
Apoptosis was measured by a TUNEL staining kit
according to the manufacturer’s instructions. The sections were
deparaffinized in xylene, and dehydrated in gradient ethanol. The
sections were permeabilized with 20 μg/ml protease for 30 min at
37°C followed by 50 μl of TUNEL reaction mixture for 60 min at
37°C, and further analyzed using a fluorescence microscope.
Statistical analysis
Data are presented as means ± standard deviation
(SD). Statistical analysis was performed with one-way analysis of
variance (ANOVA) or Student’s t-test for independent variables.
P<0.5 was considered to indicate statistically significant
differences.
Results
Evodiamine inhibits cell
proliferation
The effect of evodiamine on the growth of LLC cells
was measured by MTT assay. LLC cells were seeded and treated with
evodiamine (7.5–60 μM) for 1–3 days. After 24 h of treatment,
evodiamine modestly decreased the proliferation of LLC cells.
However, evodiamine (60 μM, IC50 = 113 μM) significantly
inhibited the growth of LLC cells at 48 h. Furthermore, the
viability of evodiamine-treated cells after 72-h treatment was
significantly decreased (60 μM, IC50 = 38 μM) (Fig. 1A). These results showed that
evodiamine inhibited the proliferation of LLC cells in a time- and
dose-dependent manner. Evodiamine-treated cells also displayed
noticeable morphological changes characterized by decreased cell
density and smaller cell conjunctions. Finally, cells became round,
floating and cell debris was found in the 60 μM evodiamine
treatment group (Fig. 1B).
Evodiamine induces caspase-independent
apoptosis
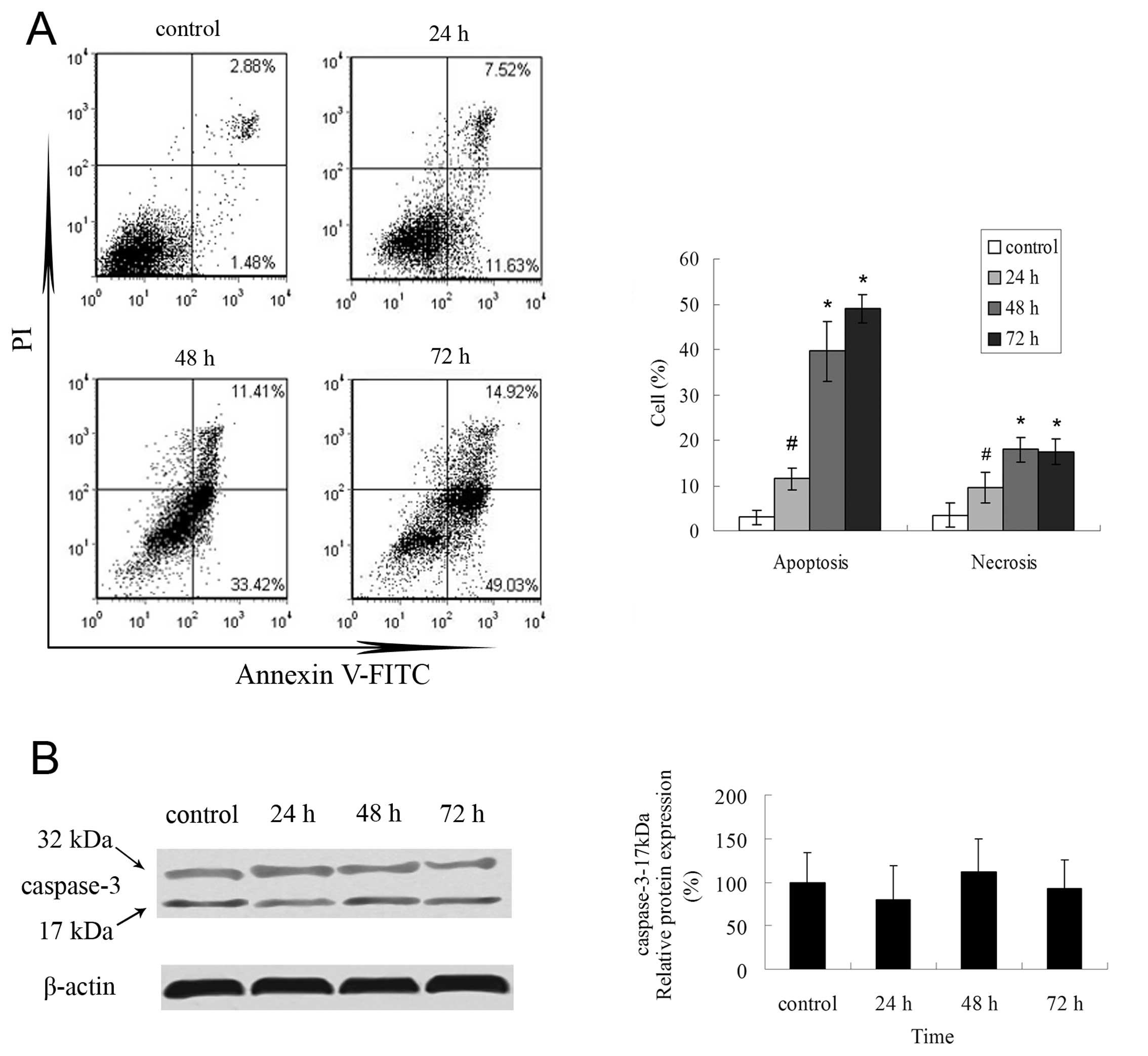
We further assessed whether evodiamine could induce
apoptosis of LLC cells by flow cytometry. By means of Annexin V
labeling to phosphatidylserine, evodiamine-treated LLC cells
underwent early apoptosis (Annexin V-FITC-positive and
PI-negative), which was increased to 11.5% in 24-h treatment and
persisted to 72 h (Fig. 2A).
Necrosis (Annexin V-FITC positive and PI-positive) was modestly
observed in a time-dependent manner (Fig. 2A). However, the apoptotic mechanism
was caspase-independent as evodiamine-treated LLC cells did not
display cleaved caspase-3 (17 kDa) (Fig. 2B).
Evodiamine promotes autophagy in LLC
cells
A fluorescent compound, MDC, was used to label
autophagic vacuoles and changes in autophagic activity. LLC cells
exhibited an increase in MDC fluorescence within 1.5–6 h after
evodiamine treatment, while the peak of autophagy activity was
observed in 1.5-h treatment (Fig.
3A). In addition, evodiamine-treated LLC cells displayed a
greater number of distinct spots within the cytoplasm or
perinuclear regions compared to control (Fig. 3B).
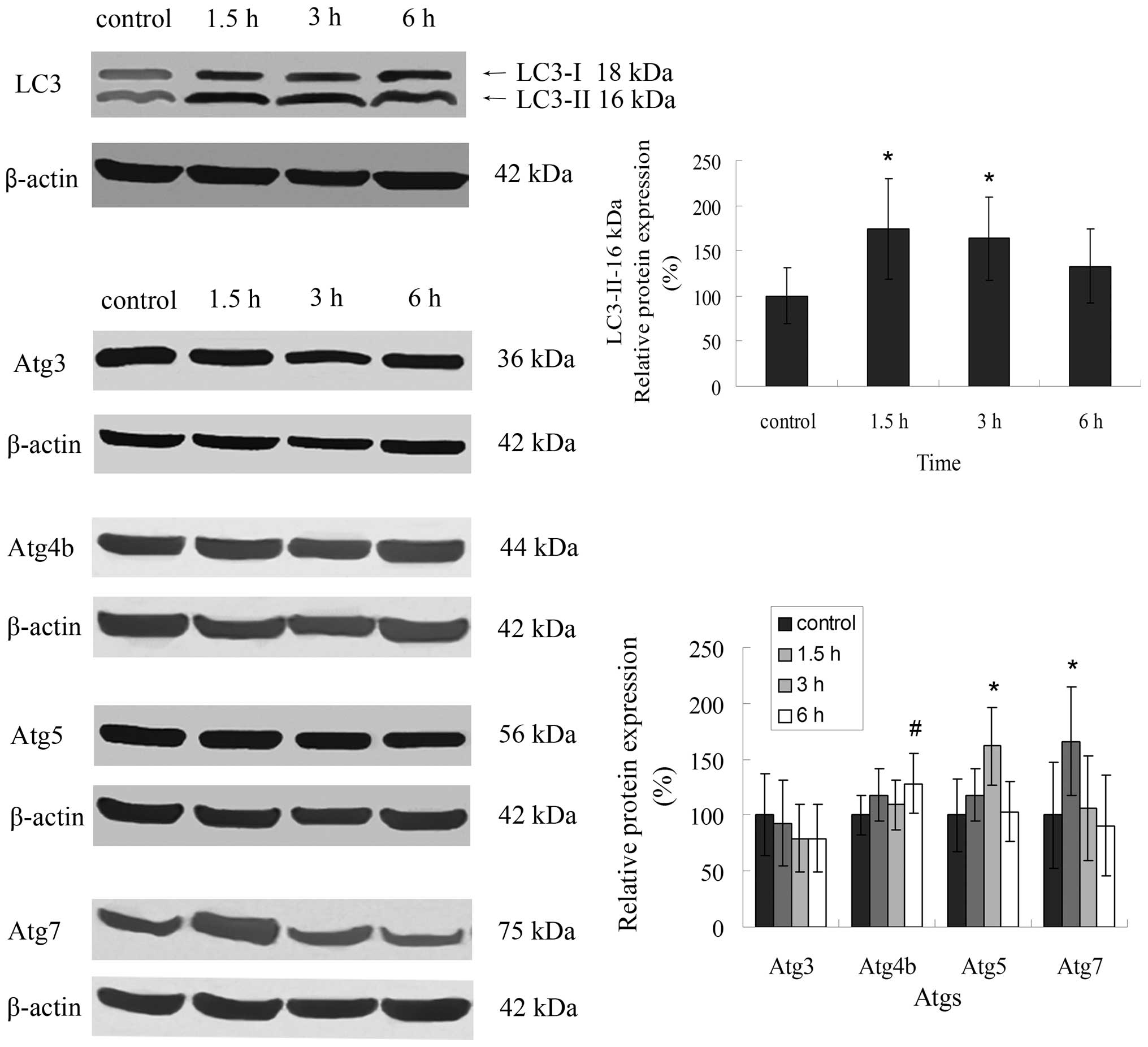
LC3 is involved in evodiamine-induced
autophagy
LC3 is widely used as a molecular marker protein for
autophagy. Evodiamine-treated LLC cells markedly upregulated LC3
expression with concomitant conversion of LC3-I to LC3-II, and the
peak of expression was at 1.5-h treatment, which was consistent
with our previous results (Fig. 4).
LC3 conversion as well as autophagosome formation are regulated by
Atgs including Atg3, Atg4b, Atg5 and Atg7. Accordingly, evodiamine
treatment increased the expression of Atg4b, Atg5 and Atg7 in LLC
cells. Evodiamine had no effects on Atg3 expression (Fig. 4).
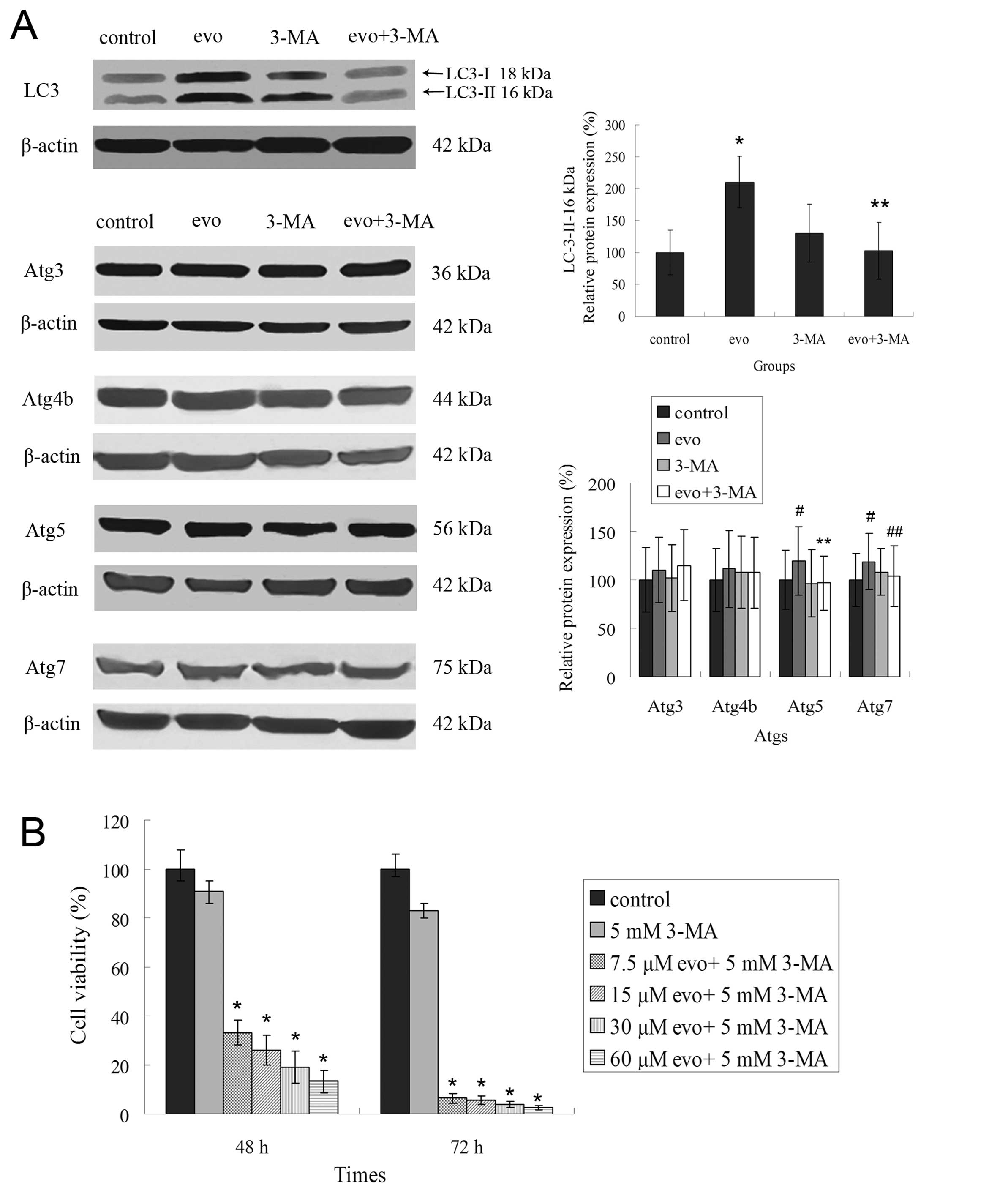
Inhibition of autophagy enhances
evodiamine-induced cell death
Evodiamine-induced autophagy has previously been
detected in LLC cells. To clarify the relevance of autophagy in the
cytotoxicity of evodiamine, we used an autophagy inhibitor, 3-MA,
that inhibits the activity of class III phosphatidylinositol 3
kinase (PI3K). There was a 1.98-fold increase in the incorporation
of MDC in evodiamine-treated LLC cells compared to control-treated
groups. The addition of 3-MA together with evodiamine decreased MDC
incorporation by 1.2-fold (Fig.
5A). Furthermore, combination treatment with 3-MA and the
evodiamine decreased the number of spots compared to evodiamine
alone group (Fig. 5B). Then,
molecular changes were detected during the following experiment.
The combination treatment also decreased the expression of LC3 and
the conversion of LC3-I to LC3-II, which were sharply increased by
60 μM evodiamine for 1.5 h. Moreover, upregulated expressions of
Atg5 and Atg7 induced by evodiamine were decreased by 3-MA, but had
no effect on Atg4 (Fig. 6A).
Autophagy has previously been reported to increase
cell survival. Furthermore, the inhibition of autophagy contributes
to enhanced cytotoxicity of chemotherapy in cancer cells. We
examined the effects of evodiamine-induced autophagy inhibition on
the cell death of LLC cells. Evodiamine induced cell death of LLC
cells in a concentration- or time-dependent manner as assessed by
MTT assay (Fig. 1A). Combination
treatment with 3-MA and evodiamine greatly enhanced cell death.
Cell viabilities were decreased to <35% at 48-h treatment and
10% at 72-h treatment. A low concentration of 3-MA did not induce
cell death at 48 or 72 h post-treatment (Fig. 6B). These observations suggest that
3-MA inhibited evodiamine-induced autophagy, but enhanced
evodiamine-induced cell death.
3-MA augments evodiamine-induced
apoptosis in LLC cells
We next evaluated the effects of 3-MA and evodiamine
combinational treatment on cell apoptosis. Evodiamine alone was
able to induce apoptosis, while 3-MA treatment was able to enhance
evodiamine-induced cell apoptosis. Necrosis was also moderately
increased in the combinatorial treatment (Fig. 7A). Furthermore, consistent with the
evodiamine alone group, the combinatorial treatment had no effect
on the active form of caspase-3 (17 kDa) (Fig. 7B).
In vivo effects of evodiamine and
3-MA
To determine the effects of evodiamine and 3-MA
in vivo, mice were subcutaneously administered LLC cells,
and were then treated with DMSO, 3-MA, evodiamine and evodiamine +
3-MA, respectively. In the final measurement, the tumor volume in
the evodiamine alone group was smaller than in the control group.
Furthermore, decreased tumor volume was greatest with the
combinatorial treatment using evodiamine and 3-MA (Fig. 8A and B). The tumor growth curve
revealed that treatment with evodiamine alone suppressed tumor
growth with further inhibitory effects using the combinatorial
treatment (Fig. 8C). Notably, the
treatment of 3-MA alone also inhibited tumor growth that was
comparable to the evodiamine treatment. No changes in body weight
or death were noted during the time of study (Fig. 8D).
 | Figure 8Anticancer effect of evodiamine and
3-MA combination against LLC xenografts. Mice were subcutaneously
administered 2×106 LLC cells in 0.1 ml of PBS. Two weeks
later, mice were randomly divided into the DMSO, 3-MA, evodiamine
and evodiamine + 3-MA groups. Agents were injected intratumorally
every 3 days for a total of 5 times (Days 15, 18, 21, 24 and 27)
and the tumor volume and body weight of these mice were measured.
The tumor volume was determined by the following formula:
0.5×L×W2 (L, length; W, width). (A) Representative
images of tumor-bearing mice and tumors. (B) The tumor volume of
the final measurement. (C and D) The tumor growth curve and body
weight curve. Data are expressed as means ± SD, (n=8).
*P<0.01 vs. the control group, **P<0.01
vs. the evodiamine treatment group. |
Evodiamine and 3-MA combinatorial
treatment enhances caspase-dependent apoptosis in vivo
Our results indicated that 3-MA inhibited
evodiamine-induced autophagy and enhanced caspase-independent
apoptosis in vitro. Moreover, we found that evodiamine
combined with 3-MA significantly inhibited tumor growth in LLC
xenografts. Histological staining showed that evodiamine as well as
the combinatorial treatment led to shrinkage of the cell nucleus,
pyknosis and karyorrhexis. No effect on cell morphology was
observed with the treatment of 3-MA alone. Increased number of
TUNEL-positive cells was observed in all treated groups compared to
the control. Furthermore, this increasing effect of evodiamine and
3-MA combination treatment was the greatest (Fig. 9A). To determine the molecular
mechanism of apoptosis, the expression of cleaved caspase-3 (17
kDa) in tumor tissue was examined. Contrary to the in vitro
study, both the evodiamine and 3-MA alone groups weakly increased
the protein level of cleaved caspase-3 (17 kDa), while evodiamine
and 3-MA combination significantly induced increasing cleaved
caspase-3 protein levels (Fig. 9B).
Moreover, evodiamine slightly enhanced the conversion of LC3-I to
LC3-II which was slightly attenuated in the combinatorial treatment
(Fig. 9C).
Discussion
Lung cancer is the leading cause of cancer-related
mortality and effective treatment is needed. Evodiamine, a
bioactive component of Wu-Chu-Yu, is effective for treating
gastrointestinal disorders (7), and
possesses anti-inflammatory activity (4,5) and
inhibitory effects on adipogenesis (6). Evodiamine also possesses anticancer
properties by inhibiting the growth of various cancer cells
(8–13). In this study, we investigated its
anticancer effects against murine Lewis lung carcinoma cells. We
showed by flow cytometry and TUNEL staining that evodiamine induces
apoptosis of LLC cells in vitro and in vivo.
Targeting the apoptotic pathway is of great
therapeutic interest for novel targeted therapies to induce cancer
cell death or sensitize them to established cytotoxic agents
(23). The caspase-dependent
pathway is a classical apoptotic pathway. This includes the
extrinsic and intrinsic pathways for the activation of caspase-8
and -9, respectively, followed by the caspase-3 (24). Our study illustrates that
evodiamine-induced apoptosis in LLC cells was caspase-independent
in vitro. This indicates that other apoptotic mechanisms are
executed, such as those that are mediated by apoptosis-inducing
factor (AIF) and endonuclease G (endo G) (25). Whether this mechanism is involved in
evodiamine-induced apoptosis of LLC cells remains to be
determined.
During our study, evodiamine also induced autophagy
of LLC cells. Autophagy is a ubiquitous physiological process and
is induced in nutrient or growth factor deficient conditions.
Autophagosomes degrade cytoplasm and some organelles to provide
nutrients and energy for cell survival (26). Autophagosome-mediated degradation is
dependent upon two conjugation systems reminiscent of the
ubiquitination proteosomal pathway. Atg12 is conjugated to Atg5 by
the combined action of Atg7 and Atg10 (E1 and E2-like enzymes,
respectively). The final complex is formed by Atg5-Atg12
non-covalently associated with the coiled-coil protein Atg 16. This
system initiates the formation of the sequestering membrane
(27,28). The second ubiquitin-related system
leads to the conjugation of LC3 (the homologue of Atg8 in yeast) to
the lipid PE by Atg7, Atg4b and Atg3. The lapidated form of LC3 is
referred to as LC3-II and localizes to autophagosomal membranes.
LC3-II has been shown to be an autophagosomal marker in mammals
(29,30). We determined that evodiamine induces
autophagy in vitro and in vivo by detecting the
conversion of LC3-I to LC3-II as well as the protein expressions of
Atg4b, Atg5 and Atg7.
Autophagosome formation is also regulated by other
mechanisms. These include the association of class III PI3K with
Beclin 1 and recruitment of the Atg12-Atg5 complex (31). Atg12-Atg 5 conjugation is required
for the elongation of the isolation membrane and for localization
of conjugated LC3 (32). The
inhibitor, 3-MA, inhibits autophagy by blocking the activity of
class III PI3K without affecting protein synthesis or ATP levels
(33,34). We showed that 3-MA inhibits
autophagy of evodiamine-treated LLC cells, and decreases the
conversion of LC3-I to LC3-II and expression of Atg7 and Atg5.
Consistent with this result, conversion of LC3-I to LC3-II was also
attenuated by the combinatorial treatment, evodiamine and 3-MA,
in vivo.
Since autophagy is under the control of several
tumor-suppressor proteins, such as Beclin 1 (35) and PTEN (36), researchers hypothesized that the
autophagy process decrease is associated with tumor progression.
However, autophagy can be induced by chemotherapy including
cisplatin (18), anthocyanin
(19), 5-fluorouracil (20,21),
dexamethasone (37), sulforaphane
(38), curcumin (39), oridonin (40), bafilomycin A1(41), and resveratrol (42). Although autophagy can serve as a
protective response and inhibit apoptosis, it can also enhance cell
death. In this study, evodiamine combined with 3-MA was much more
potent for suppressing cell proliferation than evodiamine alone
in vitro and in vivo. This indicated that
evodiamine-induced autophagy was protective against the
cytotoxicity of evodiamine on LLC cells. Autophagy inhibition
improved the sensitivity of LLC cells to evodiamine, and cell death
was increased. Furthermore, the apoptotic and autophagic pathways
are not mutually exclusive as they have been shown to act in
synergy (43). However, the exact
role and the relationship between autophagy and apoptosis in cancer
remain unknown. Our findings revealed that using both evodiamine
and 3-MA enhances cell apoptosis in vitro and in
vivo. Although in vitro treatment had no, in vivo
treatment increased caspase-3 activity. We presumed that the
apoptosis pathway induced by the combination treatment in
vitro is different from what occurs in vivo and may be
tumor microenvironment specific.
3-MA also blocks class I PI3K activity and
suppresses the invasion of HT1080 cells. This may be independent of
autophagic inhibition by decreasing type I and II PI3Ks and
possibly other molecules (44). The
PI3K/Akt signaling plays a central role in modulating cell survival
and apoptosis (45). It is possible
that 3-MA also affects this signaling pathway in vitro and
in vivo. In some previous studies, 3-MA was found to be
cytotoxic against cancer cells (42,46,47).
However, its cytotoxicity in LLC cells was modest in our
experiments.
Collectively, our study demonstrated that evodiamine
inhibits the growth of LLC cells by increasing apoptosis in a
caspase-independent manner in vitro and in a
caspase-dependent pathway in vivo. Meanwhile, evodiamine
enhanced the conversion of LC3-I to LC3-II, and upregulated the
expression of Atg4b, Atg5 and Atg7. This induced autophagy was
cytoprotective, as inhibition of evodiamine-induced autophagy by
3-MA could potentiate the cytotoxicity of evodiamine in
vitro and in vivo. Although evodiamine possesses
anticancer effects on multiple cancer cells, its anticancer
activity has yet to be fully elucidated. Our observations provide
additional insights into the therapeutic effects of evodiamine in
cancer.
Acknowledgements
This study was supported by Grants from the National
Natural Science Foundation of China (No. 30772253) and the Project
of State Key Laboratory of Trauma, Burns, and Combined Injury (No.
SKLZZ201005).
Abbreviations:
|
LLC
|
Lewis lung carcinoma
|
|
Atg
|
autophagy-specific gene
|
|
MDC
|
monodansylcadaverine
|
|
LC3
|
microtubule-associated protein 1 light
chain 3
|
|
3-MA
|
3-methyladenine
|
|
AIF
|
apoptosis-inducing factor
|
|
endo G
|
endonuclease G
|
|
PI3K
|
phosphatidylinositol 3 kinase
|
References
|
1
|
Jemal A, Siegel R, Ward E, et al: Cancer
statistics, 2008. CA Cancer J Clin. 58:71–96. 2008. View Article : Google Scholar
|
|
2
|
Petty RD, Nicolson MC, Kerr KM, et al:
Gene expression profiling in non-small cell lung cancer: from
molecular mechanisms to clinical application. Clin Cancer Res.
10:3237–3248. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Erridge SC, Moller H, Price A, et al:
International comparisons of survival from lung cancer: pitfalls
and warnings. Nat Clin Pract Oncol. 4:570–577. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang TY, Wu JB, Hwang TL, et al: A new
quinolone and other constituents from the fruits of Tetradium
ruticarpum: effects on neutrophil pro-inflammatory responses. Chem
Biodivers. 7:1828–1834. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu YN, Pan SL, Liao CH, et al: Evodiamine
represses hypoxia-induced inflammatory proteins expression and
hypoxia-inducible factor 1alpha accumulation in RAW264.7. Shock.
32:263–269. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang T, Wang Y and Yamashita H: Evodiamine
inhibits adipogenesis via the EGFR-PKCalpha-ERK signaling pathway.
FEBS Lett. 583:3655–3659. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu CL, Hung CR, Chang FY, et al: Effects
of evodiamine on gastrointestinal motility in male rats. Eur J
Pharmacol. 457:169–176. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang C, Fan X, Xu X, et al: Evodiamine
induces caspase-dependent apoptosis and S phase arrest in human
colon lovo cells. Anticancer Drugs. 21:766–776. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang J, Wu LJ, Tashino S, et al: Protein
tyrosine kinase pathway-derived ROS/NO productions contribute to
G2/M cell cycle arrest in evodiamine-treated human cervix carcinoma
HeLa cells. Free Radic Res. 44:792–802. 2010. View Article : Google Scholar
|
|
10
|
Wang C, Li S and Wang MW:
Evodiamine-induced human melanoma A375-S2 cell death was mediated
by PI3K/Akt/caspase and Fas-L/NF-kappaB signaling pathways and
augmented by ubiquitin-proteasome inhibition. Toxicol In Vitro.
24:898–904. 2011. View Article : Google Scholar
|
|
11
|
Chen MC, Yu CH, Wang SW, et al:
Anti-proliferative effects of evodiamine on human thyroid cancer
cell line ARO. J Cell Biochem. 110:1495–1503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang XN, Han X, Xu LN, et al: Enhancement
of apoptosis of human hepatocellular carcinoma SMMC-7721 cells
through synergy of berberine and evodiamine. Phytomedicine.
15:1062–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kan SF, Yu CH, Pu HF, et al:
Anti-proliferative effects of evodiamine on human prostate cancer
cell lines DU145 and PC3. J Cell Biochem. 101:44–56. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ogasawara M, Matsunaga T, Takahashi S, et
al: Anti-invasive and metastatic activities of evodiamine. Biol
Pharm Bull. 25:1491–1493. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Meijer AJ and Codogno P: Regulation and
role of autophagy in mammalian cells. Int J Biochem Cell Biol.
36:2445–2462. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang YP, Liang ZQ, Gao B, et al: Dynamic
effects of autophagy on arsenic trioxide-induced death of human
leukemia cell line HL60 cells. Acta Pharmacol Sin. 29:123–134.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ogier-Denis E and Codogno P: Autophagy: a
barrier or an adaptive response to cancer. Biochim Biophys Acta.
1603:113–128. 2003.PubMed/NCBI
|
|
18
|
Liu D, Yang Y and Liu Q: Inhibition of
autophagy by 3-MA potentiates cisplatin-induced apoptosis in
esophageal squamous cell carcinoma cells. Med Oncol. 28:105–111.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Longo L, Platini F, Scardino A, et al:
Autophagy inhibition enhances anthocyanin-induced apoptosis in
hepatocellular carcinoma. Mol Cancer Ther. 7:2476–2485. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li J, Hou N, Faried A, et al: Inhibition
of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis
in colon cancer cells. Ann Surg Oncol. 16:761–771. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li J, Hou N, Faried A, et al: Inhibition
of autophagy augments 5-fluorouracil chemotherapy in human colon
cancer in vitro and in vivo model. Eur J Cancer. 46:1900–1909.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang J, Wu LJ, Tashino S, et al: Reactive
oxygen species and nitric oxide regulate mitochondria-dependent
apoptosis and autophagy in evodiamine-treated human cervix
carcinoma HeLa cells. Free Radic Res. 42:492–504. 2008. View Article : Google Scholar
|
|
23
|
Lowe SW and Lin AWA: Apoptosis in cancer.
Carcinogenesis. 21:485–495. 2000. View Article : Google Scholar
|
|
24
|
Elmore S: Apoptosis: a review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Van Gurp M, Festjens N, Van Loo G, et al:
Mitochondrial intermembrane proteins in cell death. Biochem Biophys
Res Commun. 304:487–497. 2003.PubMed/NCBI
|
|
26
|
Levine B and Klionsky DJ: Development by
self-digestion: molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kuma A, Mizushima N, Ishihara N, et al:
Formation of the approximately 350-kDa Apg12-Apg5. Apg16 multimeric
complex, mediated by Apg16 oligomerization, is essential for
autophagy in yeast. J Biol Chem. 277:18619–18625. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fujita N, Itoh T, Omori H, et al: The
Atg16L complex specifies the site of LC3 lipidation for membrane
biogenesis in autophagy. Mol Biol Cell. 19:2092–2100. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kabeya Y, Mizushima N, Ueno T, et al: LC3,
a mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hemelaar J, Lelyveld VS, Kessler BM and
Ploegh HL: A single protease, Apg4B, is specific for the
autophagy-related ubiquitin-like proteins GATE-16, MAP1-LC3,
GABARAP, and Apg8L. J Biol Chem. 278:51841–51850. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Suzuki K, Kirisako T, Kamada Y, et al: The
pre-autophagosomal structure organized by concerted functions of
APG genes is essential for autophagosome formation. EMBO J.
20:5971–5981. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mizushima N, Yamamoto A, Hatano M, et al:
Dissection of autophagosome formation using Apg5-deficient mouse
embryonic stem cells. J Cell Biol. 152:657–668. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Seglen PO and Gordon PB: 3-Methyladenine:
specific inhibitor of autophagic/lysosomal protein degradation in
isolated rat hepatocytes. Proc Natl Acad Sci USA. 79:1889–1892.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Petiot A, Ogier-Denis E and Blommaart EF:
Distinct classes of phosphatidylinositol 3′-kinases are involved in
signaling pathways that control macroautophagy in HT-29 cells. J
Biol Chem. 275:992–998. 2000.
|
|
35
|
Maiuri MC, Criollo A and Kroemer G:
Crosstalk between apoptosis and autophagy within the Beclin 1
interactome. EMBO J. 29:515–516. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Laane E, Tamm KP, Buentke E, et al: Cell
death induced by dexamethasone in lymphoid leukemia is mediated
through initiation of autophagy. Cell Death Differ. 16:1018–1029.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Herman-Antosiewicz A, Johnson DE and Singh
SV: Sulforaphane causes autophagy to inhibit release of cytochrome
C and apoptosis in human prostate cancer cells. Cancer Res.
66:5828–5835. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Aoki H, Takada Y, Kondo S, et al: Evidence
that curcumin suppresses the growth of malignant gliomas in vitro
and in vivo through induction of autophagy: role of Akt and
extracellular signal-regulated kinase signaling pathways. Mol
Pharmacol. 72:29–39. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cui Q, Tashiro S, Onodera S, et al:
Autophagy preceded apoptosis in oridonin-treated human breast
cancer MCF-7 cells. Biol Pharm Bull. 30:859–864. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wu YC, Wu WK, Li Y, et al: Inhibition of
macroautophagy by bafilomycin A1 lowers proliferation and induces
apoptosis in colon cancer cells. Biochem Biophys Res Commun.
382:451–456. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hsu KF, Wu CL, Huang SC, et al: Cathepsin
L mediates resveratrol-induced autophagy and apoptotic cell death
in cervical cancer cells. Autophagy. 5:451–460. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Eisenberg-Lerner A, Bialik S, Simon HU, et
al: Life and death partners: apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ito S, Koshikawa N, Mochizuki S, et al:
3-Methyladenine suppresses cell migration and invasion of HT1080
fibrosarcoma cells through inhibiting phosphoinositide 3-kinases
independently of autophagy inhibition. Int J Oncol. 31:261–268.
2007.PubMed/NCBI
|
|
45
|
Goswami A, Ranganathan P and Rangnekar VM:
The phosphoinositide 3-kinase/Akt1/Par-4 axis: a cancer-selective
therapeutic target. Cancer Res. 66:2889–2892. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Opipari AW Jr, Tan L, Boitano AE, et al:
Resveratrol-induced autophagocytosis in ovarian cancer cells.
Cancer Res. 64:696–703. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Trincheri NF, Follo C, Nicotra G, et al:
Resveratrol-induced apoptosis depends on the lipid kinase activity
of Vps34 and on the formation of autophagolysosomes.
Carcinogenesis. 29:381–389. 2008. View Article : Google Scholar : PubMed/NCBI
|