Introduction
Death receptor-3 (DR3) is a member of the tumour
necrosis factor (TNF) receptor (TNFR) superfamily. In the TNFR
superfamily there are 8 death domain (DD) containing receptors,
including TNFR1 (also called DR1); Fas (also called DR2), DR3, DR4,
DR5, DR6, NGFR and EDAR. Upon binding with their ligands, the DD
recruits various proteins that mediate both the death and
proliferation of the cells. Currently, DR3 is known to be the
functional receptor of vascular endothelial growth inhibitor (VEGI)
(1,2). VEGI has been identified as an
anti-angiogenic cytokine that belongs to the TNF superfamily.
Previous investigations have highlighted a potential cancer
inhibitory role for the cytokine. The inhibitory effects of VEGI on
cancer are manifested in three main areas; the direct inhibition on
proliferation of cancer cells, the anti-angiogenic effect on
endothelial cells, and the stimulation of maturation of dendritic
cells (3). Activated DR3 has been
shown to induce rapid apoptosis by activating the caspase cascade
through interaction with TRADD and FADD (4–7). As
with other TNFR family members, DR3 is also able to induce nuclear
factor-κB (NF-κB) and to promote cell survival signals via TRADD
and TRAF2 (2,4,8,9).
Activation of DR3 by VEGI induces caspase activation and apoptosis
in TF-1, human histiocytic lymphoma (U-937), human breast carcinoma
(MCF7), human epithelial carcinoma (HeLa) and human myeloid
lymphoma (ML-1a), but not in T cells (2,10).
However, the activation of DR3 by E-selectin has also been reported
to activate downstream signalling pathways, conferring metastatic
and survival advantages in colon cancer (11).
Breast cancer is one of the leading causes of
cancer-related mortality worldwide; it is by far the commonest form
of cancer in women (12). Despite
improvements in both early detection and treatment, management of
the disease still poses significant challenges. The DR3 receptor is
involved in the modulation of a wide variety of biological
processes. The precise role and mechanism of DR3 in a
physiopathologic context remains unclear and the role of DR3 in
breast cancer remains largely unknown. In the present study, the
expression of DR3 was examined in a cohort of breast cancer
samples. Subsequently, the expression of DR3 was targeted in MCF7
and MDA-MB-231 breast cancer cells before examining the impact on
these cells in vitro.
Materials and methods
Breast tissue sample collection
Breast cancer tissue (n=115) and normal tissue
samples (n=30) were collected immediately after surgery and stored
at −80°C. The clinical follow-up was routinely performed after
surgery, and details were stored in a database. The median
follow-up period was 120 months. The presence of tumour cells in
the collected tissues was verified by a consultant pathologist, who
examined H&E stained frozen sections. Patient clinical data is
shown in Table I.
 | Table IBreast cancer patient cohort clinical
data. |
Table I
Breast cancer patient cohort clinical
data.
| Clinical data | Grouping | Sample no. |
|---|
| Tissue sample | Normal | 30 |
| Tumour | 115 |
| Nottingham prognostic
index (NPI) | NPI-1 (<3.4) | 58 |
| NPI-2 (3.4–5.4) | 38 |
| NPI-3 (>5.4) | 15 |
| Node status | Negative | 62 |
| Node status | Positive | 53 |
| Tumour grade | 1 | 20 |
| 2 | 39 |
| 3 | 54 |
| TNM staging | 1 | 61 |
| 2 | 37 |
| 3 | 7 |
| 4 | 4 |
| Survival
status | Disease free | 81 |
| Metastases | 7 |
| Local
recurrence | 5 |
| Deceased | 14 |
| Prognosis | Good prognosis | 81 |
| Poor prognosis | 26 |
Immunohistochemical staining
Frozen paired tissues from the cohort were cut into
6-μm sections, mounted, air dried and fixed in a mixture of 50%
acetone, 50% methanol for 15 min. These sections were subsequently
rehydrated in Optimax wash buffer (Sigma-Aldrich, Dorset, UK)
before being blocked for 20 min in a wash buffer containing horse
serum. The sections were then incubated for 1 h with an anti-DR3
primary antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA) at a 1:100 concentration. The sections were subjected to
extensive washes before being incubated with a biotinylated
secondary antibody for 30 min. This was subsequently removed
through further washes before adding Avidin Biotin Complex (ABC;
Vector Laboratories, Inc., Nottingham, UK) for 30 min before
further washes. The diaminobenzidine (DAB) chromagen (Vector
Laboratories, Inc.) was added to the sections and incubated in the
dark for 5 min before counterstaining in Mayer's haematoxylin,
dehydrating in ascending grades of methanol, clearing in xylene and
mounting. Sections were subsequently observed under the microscope
before capturing representative images.
Quantitative polymerase chain
reaction
Quantitative polymerase chain reaction, utilising
the Amplifluor technology, was used to detect transcript expression
of DR3 in cDNA prepared from the cohort samples. This methodology
has been previously reported (13).
In brief, primer pairs were designed using the Beacon Designer
programme, with one of the primers containing a Z sequence tag
(Table II). This Z sequence is
complementary to the Universal Z probe (Intergen, Inc., Oxford, UK)
included in the reaction (10 pmol), alongside Hot-start Q-Master
mix (Advanced Biotechnologies, Ltd., Surrey, UK), 10 pmol of
specific forward primer, 1 pmol specific reverse primer containing
Z sequence and cDNA from ~50 ng cohort tissue RNA. The reaction was
prepared and carried out using an iCyclerIQ (Bio-Rad, Hemel
Hempstead, UK) under the following conditions; 94°C for 12 min
followed by 60 cycles of 94°C for 15 sec, 55°C for 40 sec and 72°C
for 40 sec. Transcript expression was determined using an internal
standard, amplified in conjunction with the test samples and
samples were further normalised through calculation and
standardisation in conjunction with cytokeratin-19 sample
levels.
| Table IIPrimer sequences. |
Table II
Primer sequences.
| Primer | Forward | Reverse |
|---|
| GAPDH (PCR) |
5′-AGCTTGTCATCAATGGAAAT |
5′-CTTCACCACCTTCTTGATGT |
| DR3 (PCR) |
5′-CCACAAGAAGATTGGTCTGT |
5′-GCTTCATCTGCAGTAACCAG |
| GAPDH (QPCR) |
5′-CTGAGTACGTCGTGGAGTC |
5′-ACTGAACCTGACCGTACACAGAGATGATGACCCTTTTG |
| DR3 (QPCR) |
5′-CCCTGGTTACTGCAGATG |
5′-ACTGAACCTGACCGTACACTGTCAGGAGGTGCTAGAAG |
| DR3 ribozyme
transgene |
5′-CTGCAGAACATCTGCCTCCAGCCACACTGATGAGTCCGTGAGGA |
5′-ACTAGTCCAGAGCGCTGTGCCGCTGTTTCGTCCTCACGGACT |
Cell lines and culture conditions
The MCF7 and MDA-MB-231 human breast cancer cell
lines were obtained from the American Type Culture Collection
(ATCC; Rockville, MD, USA) and cultured in DMEM/Ham's F12 with
L-Glutamine medium (PAA Laboratories, Somerset, UK), supplemented
with streptomycin, penicillin and 10% fetal calf serum (PAA
Laboratories), and incubated at 37°C, 5% CO2 and 95%
humidity.
Ribozyme transgene targeting of human
DR3
Anti-human DR3 hammerhead ribozyme transgenes were
designed based on the secondary structure of DR3 mRNA generated
using the Zuker RNA mFold programme (14). The ribozymes were subsequently
synthesized and cloned into a pEF6/V5-His-TOPO plasmid vector
(Invitrogen, Paisley, UK). Both DR3 ribozyme transgenes and empty
pEF6 control plasmids were transfected, through electroporation
with an Easyject Plus electroporator (EquiBio, Kent, UK), into MCF7
and MDA-MB-231 breast cancer cells, as previously reported
(15–17). Following transfection, cells were
subjected to a selective period through the addition of 5 μg/ml
blasticidin to the culture medium and, following this period, were
transferred into a maintenance medium (containing 0.5 μg/ml
blasticidin). Cells were routinely tested to confirm stable
transfection and knockdown of DR3 expression. MCF7 cells containing
the ribozyme transgene or control pEF6 plasmid were designated
MCF7DR3KO and MCF7pEF6 respectively.
Similarly, MDA-MB-231 cells containing the ribozyme transgene or
the control pEF6 plasmid were labelled MDA-MB-231DR3KO
and MDA-MB-231pEF6 respectively. The unaltered wild-type
cells were labelled MCF7WT or
MDA-MB-231WT.
RNA extraction and reverse
transcription-polymerase chain reaction (RT-PCR)
Cells were grown to confluence in a 25
cm2 tissue culture flask before being subjected to RNA
extraction using TRI Reagent (Sigma-Aldrich) in accordance with the
supplied protocol. Following extraction, the RNA was quantified
using a spectrophotometer (WPA UV 1101; Biotech Photometer,
Cambridge, UK) and RNA levels throughout the samples were
standardised to 250 ng for use in reverse transcription. An
enhanced avian reverse transcriptase-PCR-100 kit with anchored
oligo(dT) primers was used to carry out reverse transcription in
accordance with the supplied protocol (Sigma-Aldrich). The quality
of the cDNA generated was tested using GAPDH primers before probing
for the expression of DR3 using specific primers. PCR primers were
designed using Beacon Designer (Palo Alto, CA, USA) and synthesized
by Invitrogen. Full primer sequences are outlined in Table II. PCR was performed in a T-Cy
Thermocycler (Creacon Technologies, Ltd., CD Emmen, The
Netherlands) using REDTaq® ReadyMix™ PCR Reaction Mix
(Sigma-Aldrich). PCR was conducted under the following reaction
conditions: initial denaturing at 94°C for 5 min, 34–36 cycles of
denaturing at 94°C for 40 sec, annealing at 55°C for 40 sec and
extension at 72°C for 1 min and final extension at 72°C for 10 min.
Following PCR, products were loaded on a 0.8% agarose gel,
separated electrophoretically, stained in ethidium bromide and
visualised under ultraviolet light.
SDS-PAGE and western blotting
Cancer cells were lysed with an SDS lysis buffer on
a rotor wheel before being spun at 13,000 × g to remove insolubles.
Following lysis, protein levels were quantified using the Bio-Rad
DC Protein Assay kit (Bio-Rad Laboratories, Hercules, CA, USA),
standardised to 2 mg/ml and diluted with Laemmli 2× concentrate
sample buffer (Sigma-Aldrich). Samples were boiled for 5 min before
loading onto a 12% acrylamide gel and being subjected to
electrophoretic separation. Once sufficient separation had occurred
the proteins were blotted onto a Hybond-C extra nitrocellulose
membrane (Amersham Biosciences UK, Ltd., Buckinghamshire, UK),
blocked in 10% milk and subjected to specific antibody probing.
GAPDH was used as an internal control. Proteins were probed using
the respective primary antibodies at a concentration of 1:250 and
specific peroxidase conjugated secondary antibodies at a
concentration of 1:1,000. Monoclonal rabbit anti-DR3 (SC-7909) and
anti-GAPDH (SC-32233) were obtained from Santa Cruz Biotechnology,
Inc. Peroxidase-conjugated anti-mouse and rabbit IgG secondary
antibodies were purchased from Sigma-Aldrich. Protein bands were
visualized using a Supersignal™ West Dura system and documented
using a gel documentation system (UVItec, Cambridge, UK).
Immunocytochemical staining
Twenty thousand cells/well were seeded in glass
chamber slides (Lab-Tek; Fisher Scientific UK, Ltd., Loughborough,
UK). Following overnight incubation, cells were fixed and then
permeabilized with 0.1% Triton X-100 for 5 min in TBS. Following a
blocking with SuperSensitive™ Wash Buffer (BioGenex, CA, USA)
containing horse serum, the primary antibody was added at a
concentration of 1:100 and incubated for 60 min. After extensive
washing, the peroxidase conjugated specific secondary antibody was
added at a concentration of 1:100. This step was followed by
further washes before staining the sections using a DAB kit (Vector
Laboratories), counterstaining in Mayer's haematoxylin for 2 min,
rinsing in tap water and mounting. Slides were subsequently
visualised under the microscope and representative images were
captured.
In vitro growth assay
An in vitro growth assay was used to examine
the growth rates of MCF7 and MDA-MB-231 control cells and those
transfected with the ribozyme transgenes. Cells were seeded, at a
density of 3,000 cells/well, into 96-well plates before being
incubated for 4 h, 1, 3 or 5 days. Plates were then fixed in 4%
formaldehyde (v/v) and stained with 0.5% (w/v) crystal violet.
Crystal violet stain was subsequently extracted using 10% acetic
acid (v/v) and cell density was determined using an ELx800
spectrophotometer (Bio-Tek Instruments, Inc., Winooski, VT, USA) to
measure the absorbance at 540 nm. Percentage increase in cell
density, compared to the 4 h reference plate, was used as a measure
of cell growth.
In vitro Matrigel invasion assay
An in vitro Matrigel invasion assay was used
to assess the invasiveness of control and transfected breast cancer
cells. This assay has previously been described by our group
(18). Briefly, 15,000 control or
transfected cells were seeded into transwell inserts
(Becton-Dickinson Labware, Franklin Lakes, NJ, USA), containing 8.0
μm pores, which had previously been coated with 50 μg/insert of
Matrigel Matrix Basement Membrane (BD Biosciences, Oxford, UK) and
incubated for 3 days. Following this incubation period, cells that
had invaded through the Matrigel membrane and migrated to the
underside of the transwell insert were fixed in 4% formaldehyde
(v/v) and stained with 0.5% (w/v) crystal violet. The number of
invaded cells in several representative fields was quantified by
counting cells/field under ×20 objective magnification.
In vitro Matrigel adhesion assay
The cell-matrix adhesive potential of control and
transfected cells was examined using an in vitro Matrigel
adhesion assay. This assay was adapted from a previously reported
method (19). In brief, 45,000
control or transfected cells/well were seeded into a 96-well plate
that had been pre-coated with 5 μg of Matrigel artificial basement
membrane. This plate was subsequently incubated for 45 min before
being subjected to vigorous washing in BSS. Following washings, any
remaining adherent cells were then fixed in 4% formaldehyde (v/v)
and stained with 0.5% (w/v) crystal violet. Adherent cells were
counted in several random fields under ×20 objective magnification
and quantified to determine adhesive capacity.
In vitro migration/wounding assay
Control and transfected cell migration rates were
examined using an in vitro migration/wounding assay. This
technique was modified from a previously described method reported
by our group (20). Cell migration
rates were calculated based on the capacity to close an artificial
wound in the monolayer. In brief, cells were cultured in a 24-well
plate to form a monolayer; this monolayer was subsequently wounded
through scratching with a blunted needle. The closure of this
wound, through the migration of cells at the wound fronts, was
subsequently tracked and recorded over 90 min using a time-lapsed
video system (Panasonic UK, Ltd., London, UK). Cellular migration
was then calculated at set time points, in reference to the initial
time point, using the ImageJ analysis software.
Statistical analysis
Experimental procedures were repeated independently
a minimum of 3 times. Statistical comparisons were made between
cells transfected with the DR3 ribozyme transgenes
(MCF7DR3KO and MDA-MB-231DR3KO) and the
respective control cell line containing the empty pEF6 plasmid
(MCF7pEF6 and MDA-MB-231pEF6). Data was
statistically analysed using a two-sample, two-tailed t-test or two
way ANOVA and the Minitab 14 software package. A p-value <0.05
was taken to indicate statistically significant differences.
Patient survival was analysed using the Kaplan-Meier method
(SPAW18).
Results
Expression profile of DR3 in clinical
breast cohort and association with clinical, pathological and
long-term survival of the patients
DR3 transcript levels were examined in the breast
cohort using quantitative PCR. A decreased level of DR3 expression
was revealed in breast tumours (n=115) (854±473 copies/50 ng RNA),
compared to normal tissues (n=30) (24975±12176 copies/50 ng RNA,
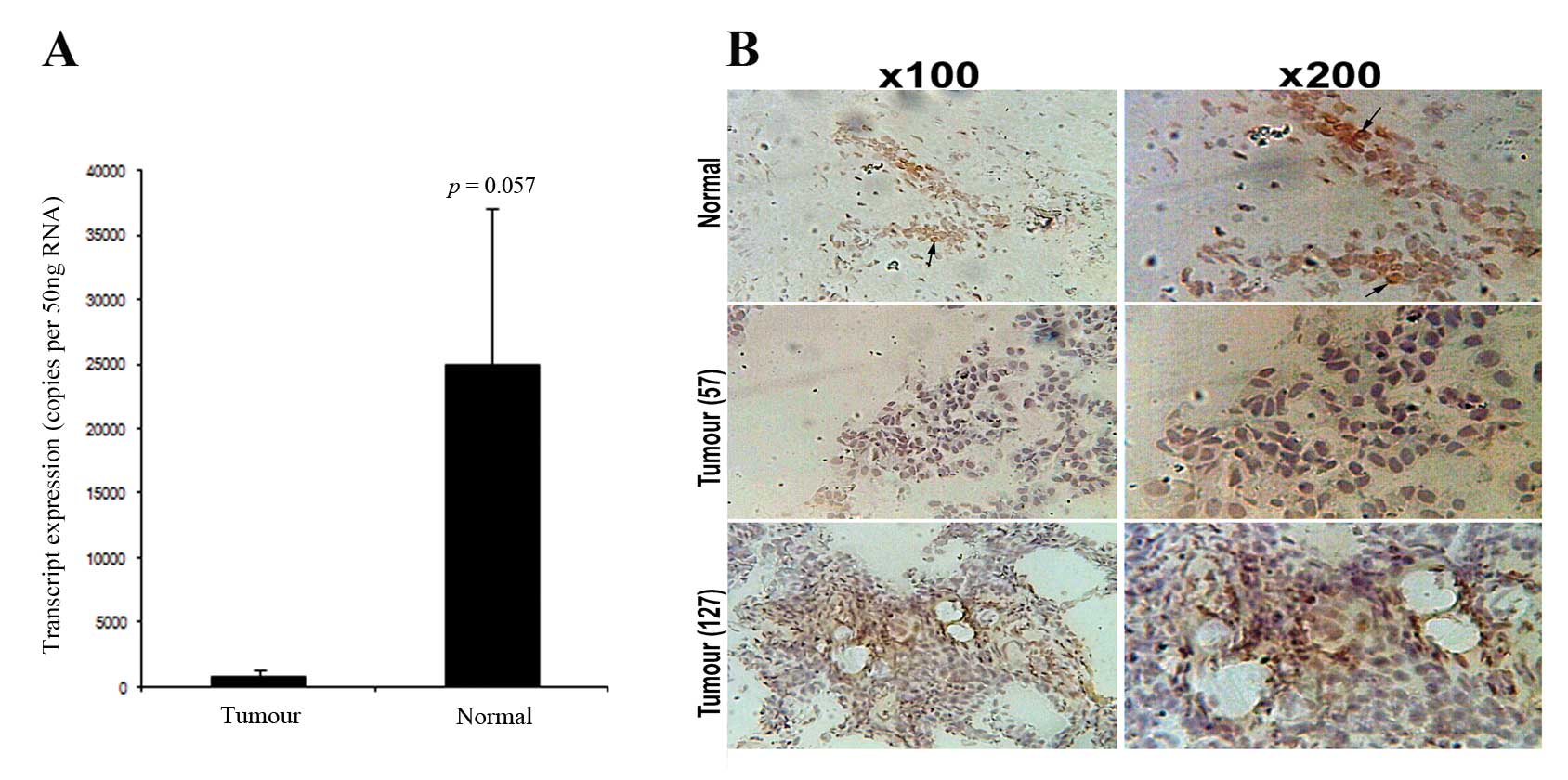
p=0.057) (Fig. 1A).
Immunohistochemical staining (IHC) showed that DR3 staining in
normal mammary epithelial cells (Fig.
1B) is largely nuclear staining. Fig. 1B confirmed reduced levels of DR3
staining in tumour tissues (shown are grade 1 and 3 tumours)
compared to normal tissues (Fig.
1B).
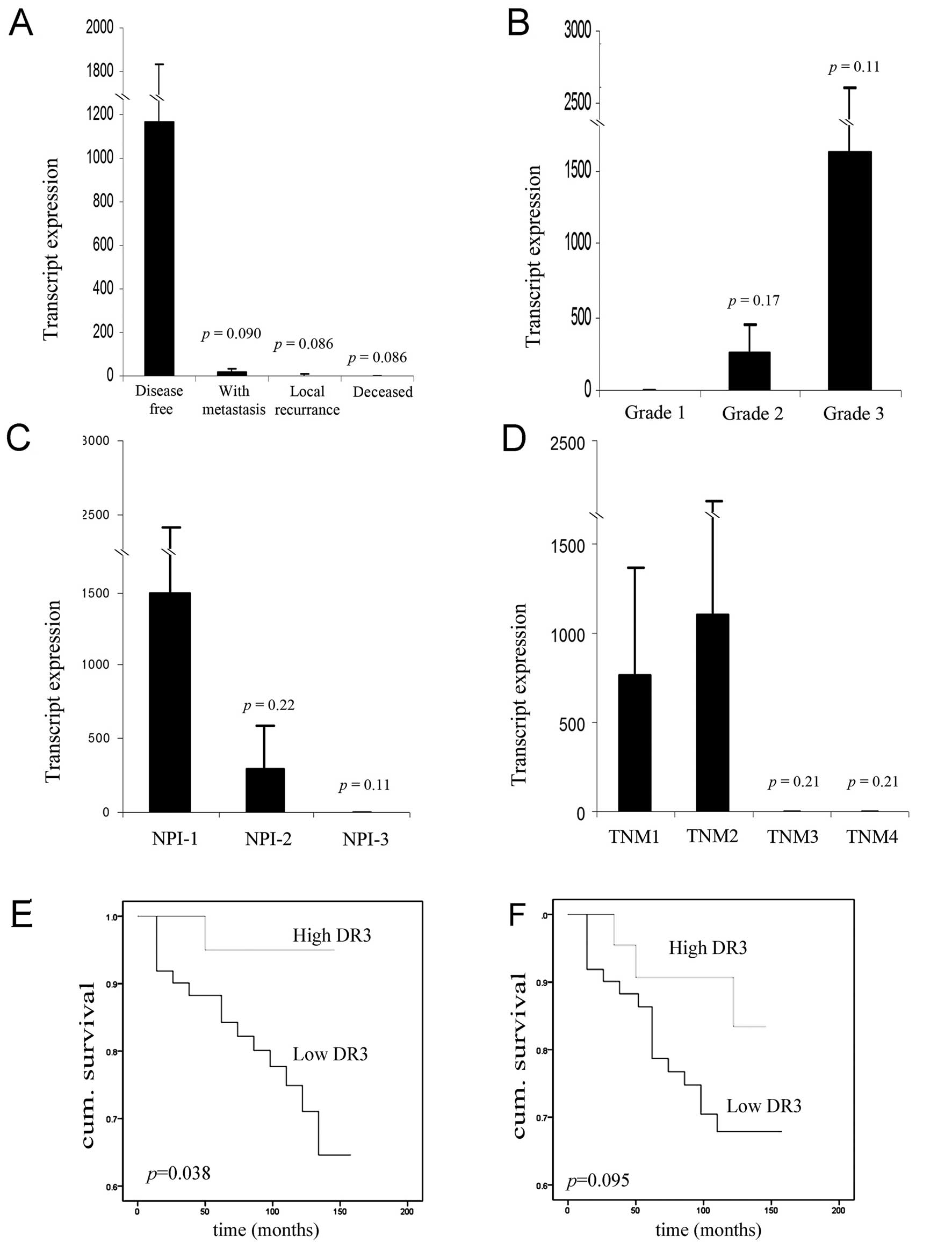
Further analysis of DR3 transcript levels against
the clinical aspect demonstrated that decreased DR3 expression was
correlated with poor disease prognosis over the 10 year follow-up
period (Fig. 2A). DR3 levels were
found to be elevated in patients who remained disease free
(1165±669 copies/50 ng RNA), when compared to those with metastasis
(16.1±16 copies/50 ng RNA, p=0.090), with local recurrence
(2.69±2.1 copies/50 ng RNA, p=0.086) and those who succumbed to
breast cancer (2.27±1.2 copies/50 ng RNA, p=0.086) with close to
significant levels being reached. A similar trend was observed in
respect to Nottingham prognostic index (NPI) (Fig. 2C) with DR3 expression levels being
higher in NPI-1 (1497±915 copies/50 ng RNA) compared to NPI-2
(300±292 copies/50 ng RNA, p=0.22) and NPI-3 (0.110±0.072 copies/50
ng RNA, p=0.11). By contrast, DR3 expression levels were found to
be elevated in the poorly differentiated, higher grade 2 (264±190
copies/50 ng RNA, n=39, p=0.17) and grade 3 (1629±993 copies/50 ng
RNA, n=54, p=0.11) tumours when compared to grade 1 tumours
(0.026±0.026 copies/50 ng RNA, n=20) (Fig. 2B). No significant correlations were
observed between DR3 expression and TNM classification, although
again the expression levels of DR3 tended to be lower in the higher
TNM classification tumours (Fig.
2D).
Low levels of DR3 transcripts were associated with a
significantly shorter overall survival compared with those who had
tumours with high levels [127.8 (115.1–140.5) months vs. 139.2
(130–148.4) months, p=0.038] (Fig.
2E). There was a trend of decreased disease free survival for
patients with low DR3 transcript, although this failed to reach
statistical significance (p=0.095) (Fig. 2F).
Generation of breast cancer cell lines
with suppressed DR3 expression
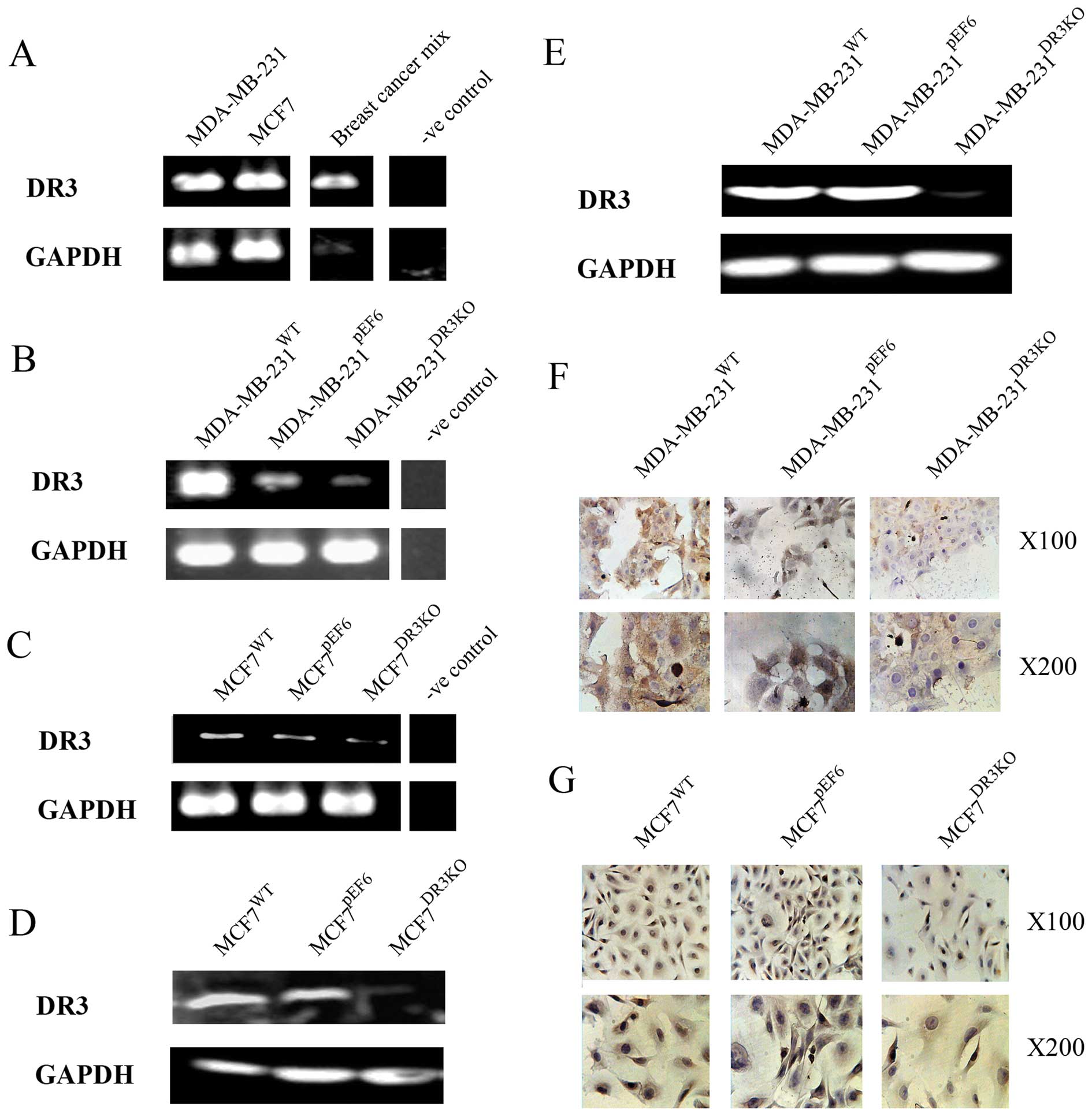
The expression of DR3 was examined in MCF7 and
MDA-MB-231 cells using RT-PCR (Fig.
3A). MCF7 and MDA-MB-231 cells were found to express modest
levels of the DR3 transcript and were subsequently transfected with
anti-DR3 ribozyme transgenes. RT-PCR demonstrated that DR3 mRNA
expression was successfully knocked down in MCF7DR3KO
and MDA-MB-231DR3KO cells by the ribozyme transgene in
comparison to the level of expression in wild-type cells
(MCF7WT and MDA-MB-231WT) and in empty
plasmid control cells (MCF7pEF6 and
MDA-MB-231pEF6) (Fig. 3B and
C).
Additionally, western blotting was used to probe for
DR3 protein levels in both the control and transfected cell lines.
Similar to the trends seen at the mRNA level, DR3 protein was found
to be expressed in all of the control cell lines
(MCF7WT, MDA-MB-231WT, MCF7pEF6
and MDA-MB-231pEF6), and the expression of DR3 protein
exhibited a dramatic reduction in the ribozyme transgene
transfected cell lines (MCF7DR3KO and
MDA-MB-231DR3KO) (Fig. 3D
and E). Immunocytochemical (ICC) staining was similarly used to
detect protein levels of DR3 in these cells. ICC staining indicated
a decreased protein level of DR3 in cells transfected with the
ribozyme transgene (Fig. 3F and
G).
DR3 expression does not alter cell
growth
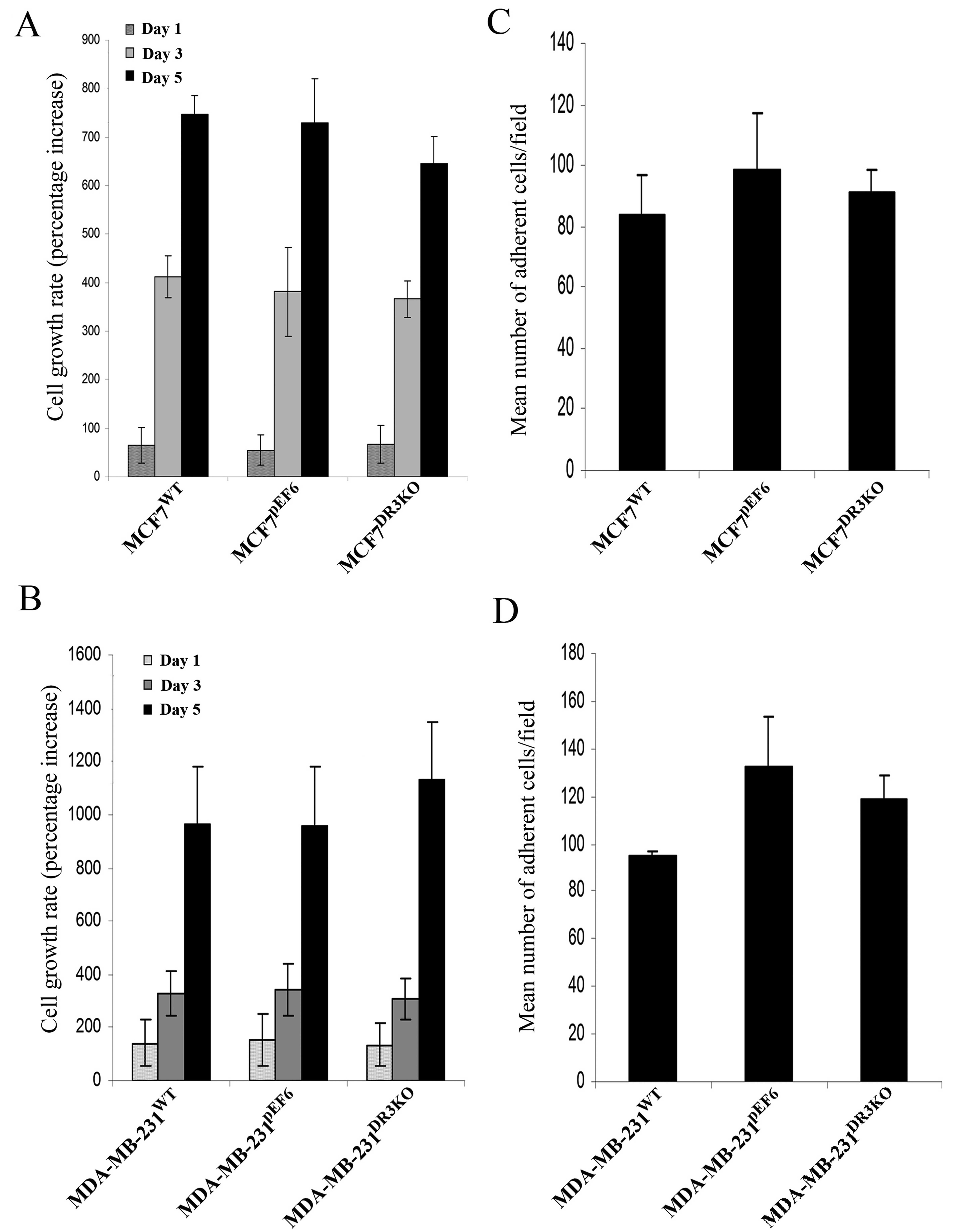
The effect of DR3 on the growth of MCF7 and
MDA-MB-231 cells was examined using an in vitro tumour cell
growth assay. Transfection with the DR3 ribozyme transgene did not
appear to impact the cell growth rate of either MCF7 or MDA-MB-231
cells. Knockdown of DR3 resulted in no significant difference in
growth rate between the MCF7DR3KO and
MCF7pEF6 cell lines over the 1-, 3- and 5-day incubation
periods (Day 1, 67.3±38.9 vs. 55.99±31.75, p=0.536; Day 3,
366.0±37.6 vs. 381.2±91.6, p=0.86; Day 5, 645.0±57.5 vs.
728.4±92.6, p=0.486) (Fig. 4A). A
similar trend was seen between the MDA-MB-231DR3KO and
MDA-MB-231pEF6 cell lines over the 1-, 3- and 5-day
incubation period (Day 1, 136.0±81.8 vs. 153.8±97.4, p=0.980; Day
3, 306.8±78.9 vs. 339.8±96.9, p=0.798; Day 5, 1131.4±220.2 vs.
958.5±223.5, p=0.596) (Fig.
4B).
Knockdown of DR3 does not affect cell
adhesion
The capacity of MCF7 and MDA-MB-231 breast cancer
cells to adhere to an artificial Matrigel basement membrane was
examined using an in vitro Matrigel adhesion assay.
Transfection with the DR3 ribozyme transgene did not affect the
adhesive properties of either MCF7 or MDA-MB-231 cells to an
artificial Matrigel basement membrane over a 40-min incubation
period (Fig. 4C and D). No
significant difference in adhesive capacity was seen between cells
containing the ribozyme transgene and their respective pEF6 control
cells (MCF7DR3KO vs. MCF7pEF6, 91.0±7.7 vs.
98.8±18.3, p=0.714; MDA-MB-231DR3KO vs.
MDA-MB-231pEF6, 118.6±10.1 vs. 132.5±21.4, p=0.637).
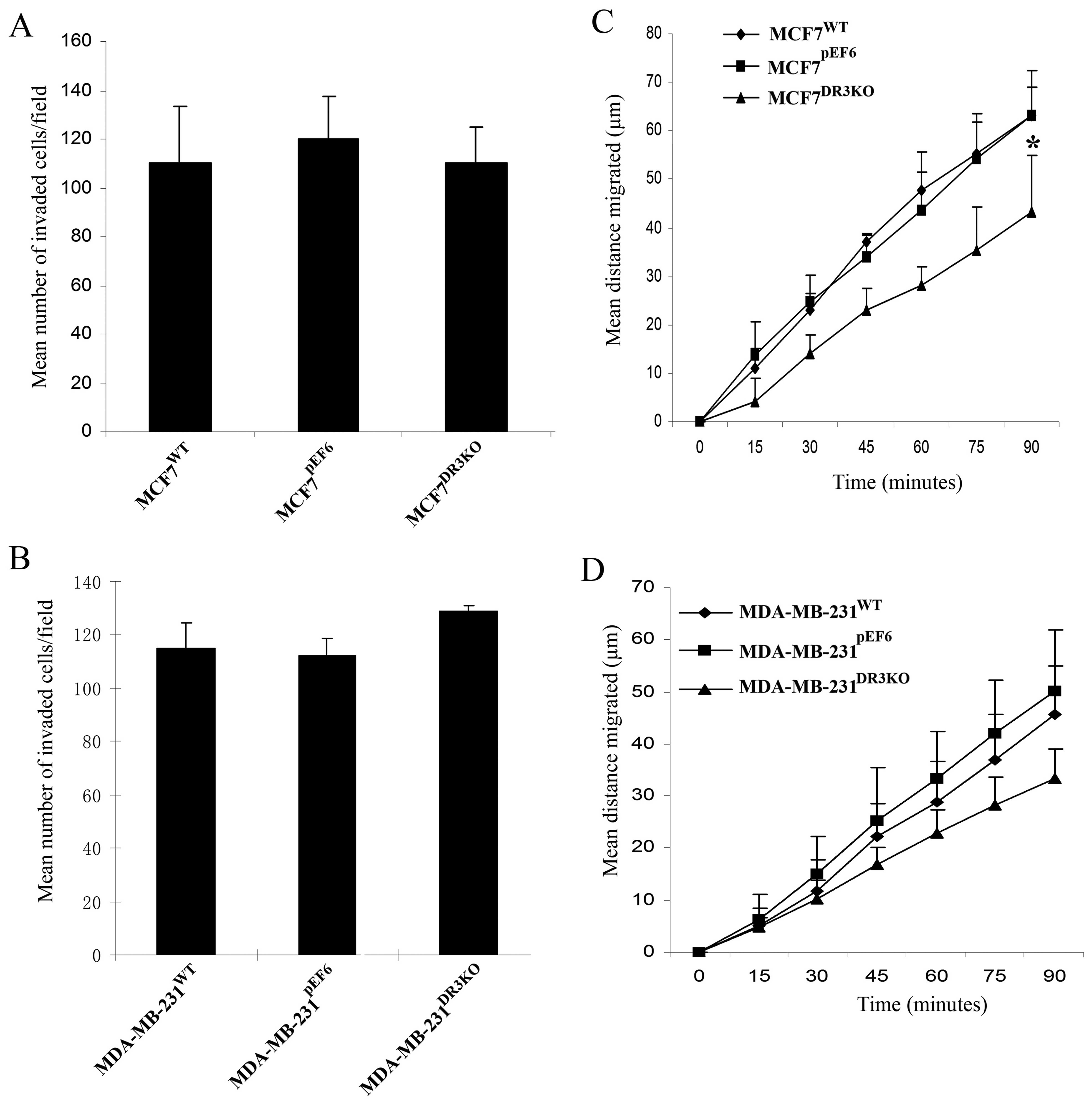
Loss of endogenous DR3 does not affect
cell invasion
No significant difference in cellular invasion was
found between the MDA-MB-231DR3KO and
MDA-MB-231pEF6 cell lines over the 3-day incubation
period (128.85±1.98 vs. 112.15±6.43, respectively, p=0.111)
(Fig. 5B). Similarly, no
significant differences were seen between the invasive capacity of
MCF7DR3KO and the control MCF7pEF6 cell lines
(110.1±15.0 vs. 120.1±2.93, respectively, p=0.688) (Fig. 5A).
Loss of DR3 can impact on cell
migration
The effect of DR3 expression on cell migration was
assessed using a migration/wounding assay. Knockdown of DR3
negatively impacted the migration of MCF7 cells (Fig. 5C). The rate of cell migration in
MCF7DR3KO was significantly lower than that in MCF7
cells containing the empty pEF6 control plasmid after 90 min
(p=0.011). Similarly, migratory rates of MDA-MB-231DR3KO
tended to be reduced compared to the pEF6 control cell line after
90 min, although this did not reach statistical significance
(p=0.065) (Fig. 5D).
Discussion
DR3 is a member of the TNF receptor superfamily. At
present, DR3 is known as the functional receptor of VEGI. Studies
have demonstrated that VEGI can inhibit angiogenesis and breast
cancer tumour growth and display prognostic relevance as breast
cancer patients with an overall poor prognosis express
significantly lower levels of VEGI compared to those with a
favourable prognosis (21,22). In the current study, we reported
that DR3 expression was decreased in higher NPI stage breast cancer
and lower levels correlated with a poorer outcome and a shorter
overall survival.
In the current study, we also examined the
expression of DR3 at mRNA and protein levels in breast cancer
tissues. Decreased transcript expression of DR3 was also observed
in the breast cancer tissue sections compared to normal tissues
(p=0.057). In line with this, decreased DR3 protein expression in
cancerous sections compared to normal sections was apparent in the
IHC staining, which also showed that DR3 proteins in normal
epithelial cells are largely seen in the nuclei. Taken together,
this indicates that a loss of DR3 may occur as normal tissues and
cells progress to a cancerous state.
However, further analysis of the breast cancer
cohort indicated an increase in DR3 transcript expression levels in
higher histological grade tumours. We deduced that DR3 may have
several ligands and play diverse roles in breast cancer, however,
further experimental study is needed in regards to this and to
fully identify the exact mechanisms of action. Of note, a recent
study showed that DR3 may be a new receptor for E-selectin, which
has been linked with cancer metastasis. The findings of that study
indicated that the activation of DR3 in response to E-selectin
triggers the transendothelial migration of cancer cells and
protects them against apoptosis, suggesting that DR3 has evolved to
provide metastatic advantages to colon cancer cells (11) and this molecule may thus play a
number of complex roles in cancer.
In addition to exploring DR3 in a clinical cohort,
we also examined this molecule in vitro in a number of
breast cancer cell lines. Knockdown of DR3 in MDA-MB-231 cells
(MDA-MB-231DR3KO) and MCF7 cells (MCF7DR3KO)
resulted in a reduction of cell migration in vitro. The rate
of migration in MCF7DR3KO was significantly reduced in
comparison to MCF7pEF6 (p=0.011). However,
MDA-MB-231DR3KO, whilst not quite significantly reduced
(p=0.065), did also demonstrate a reduction in migration rates in
comparison to MDA-MB-231pEF6 control cells. This
capacity is essential for tumour cells to disseminate to secondary
sites. In this respect, the data are somewhat conflicting, as lower
levels of DR3 were apparent in patients with metastatic spread and
poorer prognosis whilst in vitro suppression of DR3 levels
resulted in reduced migration rates. This again suggests a complex
role for DR3 in the development and progression of breast tumours
which requires further study to fully elucidate and to better
understand the mechanism of action and role played by this
molecule.
Acknowledgements
Dr Z. Ge is a recipient of the Cardiff University
China Medical Scholarship. The authors thank the Albert Hung
Foundation and Cancer Research Wales for supporting this study.
References
|
1
|
Zhai Y, Ni J, Jiang GW, et al: VEGI, a
novel cytokine of the tumor necrosis factor family, is an
angiogenesis inhibitor that suppresses the growth of colon
carcinomas in vivo. FASEB J. 13:181–189. 1999.PubMed/NCBI
|
|
2
|
Migone TS, Zhang J, Luo X, et al: TL1A is
a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell
costimulator. Immunity. 16:479–492. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang N, Sanders AJ, Ye L and Jiang WG:
Vascular endothelial growth inhibitor in human cancer (Review). Int
J Mol Med. 24:3–8. 2009.PubMed/NCBI
|
|
4
|
Muppidi JR, Tschopp J and Siegel RM: Life
and death decisions: secondary complexes and lipid rafts in TNF
receptor family signal transduction. Immunity. 21:461–465. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Locksley RM, Killeen N and Lenardo MJ: The
TNF and TNF receptor superfamilies: integrating mammalian biology.
Cell. 104:487–501. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kitson J, Raven T, Jiang YP, et al: A
death-domain-containing receptor that mediates apoptosis. Nature.
384:372–375. 1996. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chinnaiyan AM, O'Rourke K, Yu GL, et al:
Signal transduction by DR3, a death domain-containing receptor
related to TNFR-1 and CD95. Science. 274:990–992. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wen L, Zhuang L, Luo X and Wei P:
TL1A-induced NF-kappaB activation and c-IAP2 production prevent
DR3-mediated apoptosis in TF-1 cells. J Biol Chem. 278:39251–39258.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marsters SA, Sheridan JP, Donahue CJ, et
al: Apo-3, a new member of the tumor necrosis factor receptor
family, contains a death domain and activates apoptosis and
NF-kappa B. Curr Biol. 6:1669–1676. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Haridas V, Shrivastava A, Su J, et al:
VEGI, a new member of the TNF family activates nuclear factor-kappa
B and c-Jun N-terminal kinase and modulates cell growth. Oncogene.
18:6496–6504. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gout S, Morin C, Houle F and Huot J: Death
receptor-3, a new E-Selectin counter-receptor that confers
migration and survival advantages to colon carcinoma cells by
triggering p38 and ERK MAPK activation. Cancer Res. 66:9117–9124.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Porter PL: Global trends in breast cancer
incidence and mortality. Salud Publica Mex. 51(Suppl 2): 141–146.
2009. View Article : Google Scholar
|
|
13
|
Jiang WG, Watkins G, Fodstad O,
Douglas-Jones A, Mokbel K and Mansel RE: Differential expression of
the CCN family members Cyr61, CTGF and Nov in human breast cancer.
Endocr Relat Cancer. 11:781–791. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zuker M: Mfold web server for nucleic acid
folding and hybridization prediction. Nucleic Acids Res.
31:3406–3415. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang WG, Davies G and Fodstad O: Com-1/P8
in oestrogen regulated growth of breast cancer cells, the ER-beta
connection. Biochem Biophys Res Commun. 330:253–262. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sanders AJ, Parr C, Mason MD and Jiang WG:
Suppression of hepatocyte growth factor activator inhibitor-1 leads
to a more aggressive phenotype of prostate cancer cells in
vitro. Int J Mol Med. 20:613–619. 2007.PubMed/NCBI
|
|
17
|
Jiang WG, Martin TA, Lewis-Russell JM,
Douglas-Jones A, Ye L and Mansel RE: Eplin-alpha expression in
human breast cancer, the impact on cellular migration and clinical
outcome. Mol Cancer. 7:712008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jiang WG, Hiscox S, Hallett MB, Horrobin
DF, Mansel RE and Puntis MC: Regulation of the expression of
E-cadherin on human cancer cells by gamma-linolenic acid (GLA).
Cancer Res. 55:5043–5048. 1995.PubMed/NCBI
|
|
19
|
Jiang WG, Hiscox S, Hallett MB, Scott C,
Horrobin DF and Puntis MC: Inhibition of hepatocyte growth
factor-induced motility and in vitro invasion of human colon cancer
cells by gamma-linolenic acid. Br J Cancer. 71:744–752. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang WG, Hiscox SE, Parr C, et al:
Antagonistic effect of NK4, a novel hepatocyte growth factor
variant, on in vitro angiogenesis of human vascular endothelial
cells. Clin Cancer Res. 5:3695–3703. 1999.PubMed/NCBI
|
|
21
|
Parr C, Gan CH, Watkins G and Jiang WG:
Reduced vascular endothelial growth inhibitor (VEGI) expression is
associated with poor prognosis in breast cancer patients.
Angiogenesis. 9:73–81. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhai Y, Yu J, Iruela-Arispe L, et al:
Inhibition of angiogenesis and breast cancer xenograft tumor growth
by VEGI, a novel cytokine of the TNF superfamily. Int J Cancer.
82:131–136. 1999. View Article : Google Scholar : PubMed/NCBI
|