Introduction
Hepatocellular carcinoma (HCC) is one of the most
common types of cancer in the world. HCC is often diagnosed at an
advanced stage as it progresses rapidly due to vascular invasion
and multiple metastases (1). HCC
patients typically relapse after treatment, which leads to a low
5-year survival rate of less than 7% (2). Current treatment options for advanced
HCC include chemotherapy, radiotherapy, proton beam therapy,
chemo-embolization and ablation. However, these treatments remain
unsatisfactory. Gene therapy is a relatively new technique in the
therapeutic battle against cancer (3).
Apoptin, a small protein that is derived from the
chicken anemia virus and consists of 121 amino acids (4,5), has
attracted significant attention as it possesses tumor-specific
cytotoxicity (6,7). The ectopic expression of apoptin
induces apoptosis in various human transformed cells and tumor
cells but has little or no cytotoxic effects in several normal
human cell lines (8,9). The selective toxicity of apoptin is
mainly attributed to its differential subcellular localization in
tumor and normal cells (10,11).
In cancer cells, apoptin predominantly localizes to the nucleus,
whereas in normal cells, its nuclear accumulation is severely
impaired. The present study suggested that apoptin requires
additional interacting molecular partners that involve specific
signaling pathways in cancer cells. Apoptin interacts with various
partners of the human proteome including PI3K/AKT (12,13),
PKCβ (14), APC1 (15), and PML (16), which regulate either apoptin
activation or execution processes. These recent advances
demonstrate that elucidating the mechanism of apoptin-induced
apoptosis can lead to novel tumor-specific pathways that may be
exploitable as anticancer drug targets.
Using kinase software, we found that CDK1 is a
candidate interacting molecule. The HCC cell line HepG2 and the
human normal liver cell line HL-7702 were used to study the role of
CDK1 in apoptin-induced apoptosis in liver cancer cells. These cell
lines showed a marked difference in the expression level of CDK1;
CDK1 expression levels in HepG2 cells were significantly higher
than in HL-7702 cells. Expression vectors bearing genes encoding
GFP-apoptin or Flag-apoptin proteins were developed to evaluate the
tumor-specific toxicity of apoptin in HCC. Apoptin selectively
killed HepG2 cells but had no effect on HL-7702 cell proliferation.
Following shRNA knockdown of CDK1, the tumor-specific killing
effect of apoptin was significantly reduced, and the majority of
apoptin translocated to the cytoplasm from the nucleus. The results
indicated that downregulation of CDK1 leads to the cytoplasmic
relocalization of apoptin.
CDK1 is a highly conserved protein that functions as
a serine/threonine kinase and is a key player in cell cycle
regulation. CDK1 activity during apoptosis was first identified in
lymphoma cells (17). However, the
molecular mechanisms of CDK1 in the processes of apoptosis remain
unknown. Thus, deregulation of cell cycle regulators, such as CDK1,
likely contributes to the tumorigenesis of HCC.
In the present study, we provided strong evidence
for the interaction between CDK1 and apoptin in the process of
tumorigenesis. CDK1 plays a role in the accumulation of apoptin in
the nucleus. This strategy enabled us to identify
apoptin-interacting cellular molecular targets, and the selective
accumulation of apoptin in the nucleus of cancer cells may be
utilized for oncotherapy development.
Materials and methods
Patients and specimens
A total of 48 human primary HCC tissues and matched
control tissues were obtained from patients who underwent
hepatectomy at the the First Affiliated Hospital of Xi’an Jiaotong
University Medical college, between 2009 and 2011. The mean age was
50 years, with 18 females and 30 males. None of these patients had
received preoperative chemotherapy or radiotherapy. Overall
survival, which was defined as the time from the surgery to patient
death or the last follow-up, was used as a measure of prognosis.
Both the tumor and the corresponding non-tumor tissues not less
than 3 cm away from the HCC tissue were sampled, and the diagnosis
was confirmed by pathological examination. After surgical
resection, the fresh cancer tissues and matched normal tissues from
the HCC patients were obtained for western blot analysis.
Histological types were assigned according to the WHO
classification criteria. The protocols used in this study were
approved by the hospital’s Protection of Human Subjects Committee.
The use of human tissues in this study was approved by the
Institutional Review Board of the First Affiliated Hospital of
Xi’an Jiaotong University Medical College and was carried out in
accordance with international guidelines, and written informed
consent was obtained from each patient.
Cell culture
HepG2, Hep3B, SMMC-7721 and HL-7702 cells were
purchased from the Shanghai Institute of Cell Biology (Shanghai,
China). The cells were cultured in Dulbecco’s modified Eagle’s
medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100
U/ml penicillin, and 100 μg/ml streptomycin (Gibco, Grand Island,
NY, USA) in a humidified incubator at 37°C with 5%
CO2.
Expression plasmid construction and
transfection
To construct pcDNA3.1-Flag-apoptin and
pEGFP-apoptin, full-length apoptin (including the full 121 codons)
was excised from plasmid pcDNA3.1-apoptin-V5 after PCR with
EcoRI and BamHI and then cloned into the
pcDNA3.1-Flag and pEGFP vectors at the EcoRI and
BamHI sites. The 4 complementary primers used were
5′-GCGGATCCGCCACCATGG ATTACAAGGATGACGACGATAAGAACGCTCTCCAAG
AAGATA-3′ and 5′-GCGAATTCTTACAGTCTTATACA CCTTCTTGC-3′,
5′-CGGAATTCGCCACCATGAACGC TCTCCAAGAAGATAC-3′ and 5′-CGGGATCCTTACAGT
CTTATACACCTTCTTGC-3′. The fidelity of the plasmid constructs was
confirmed by DNA sequencing.
Cells were plated 24 h before transfection in a
6-well plate at a density of 2×105. Transfection with
4.0 μg pcDNA3.1-Flag-apoptin/pEGFP-apoptin vector or 4.0 μg
pcDNA3.1/pEGFP empty vector (as a positive control) was performed
using Lipofectamine 2000 (Invitrogen, Paisley, UK) according to the
manufacturer’s protocol.
MTT proliferation
Cell viability was determined by the colorimetric
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. HepG2 and/or HL-7702 cells were transfected with
pcDNA3.1-Flag-apoptin/pEGFP-apoptin vector, pcDNA3.1/pEGFP empty
vector or siRNA-CDK1. Cells were seeded at 5×104,
104, and 103 per well in 96-well plates for
MTT assays. On days 0, 1, 2, 3, 4 and 5 post-infection, the cells
were washed twice with PBS and incubated with 0.5 mg/ml MTT (Sigma)
for 4 h. After the incubation period, cells were washed with PBS,
solubilized with dimethyl sulfoxide (DMSO), and quantified using a
microplate reader at the absorbance of 550 nm. The average values
were obtained from triplicates.
Flow cytometric analysis
The apoptosis was evaluated by Annexin-V binding and
7-AAD uptake using an Annexin V-PE/7-AAD kit (Keygen, China).
Briefly, cells were plated at a density of 1×105
cells/well into 6-well plates for 24 h. The cells were transfected
with pEGFP-apoptin vector and shRNA-CDK1 for 24 and 48 h,
respectively. The cells were washed three times with cold PBS and
resuspended in Annexin V binding buffer. The cells were stained
with Annexin V-PE for 15 min, washed, and then stained with 7-AAD.
The cells were analyzed on a BD FACSCanto II (Becton-Dickinson,
Oxford, UK) and 10,000 events were acquired per sample.
Subsequently, the cells were analyzed by flow cytometry using
CellQuest software (BD Biosciences).
Western blot analysis
Whole-cell extracts were prepared from
pcDNA3.1-Flag-apoptin-treated, pEGFP-apoptin-treated or
control-treated cells cultured in 6-well plates. Following
transfection, cells were harvested and resuspended in lysis buffer,
washed three times with ice-cold PBS and lysed in extraction buffer
(40 mmol/l Tris-HCl, pH 7.5, 150 mmol/l KCl, 1 mmol/l EDTA, 1%
Triton X-100, 100 mmol/l NaVO3, 1 mmol/l PMSF)
supplemented with the protease inhibitor cocktail. Proteins (60 μg)
were separated by 10% SDS-PAGE and transferred to PVDF membranes.
The membranes were blocked with 10% non-fat milk in Tris-buffered
saline (TBS) at 37°C for 3 h and then incubated with mouse
anti-Flag antibody (1:1,000), mouse anti-GFP antibody (1:1,000) or
mouse anti-β-actin antibody (1:2,000) in TBS containing 10% non-fat
milk for 12 h at 4°C. Horseradish peroxidase-linked anti-mouse IgG
(1:5,000) was used as a secondary antibody (in TBS containing 10%
non-fat milk for 3 h at room temperature), and antigen-antibody
complexes were detected using an enhanced chemiluminescence kit
(ECL Plus, Amersham, Freiburg, Germany). Densitometry values for
western blotting and antibody array experiments were estimated by
the ImageQuant TL software (GE Healthcare, Buckinghamshire, UK) and
expressed as arbitrary units (a.u.). Multiple film exposures were
used to verify the linearity of the samples analyzed and to avoid
saturation of the film. All antibodies used in the study were
purchased from Cell Signaling Technology.
Immunoprecipitation
Cells were seeded in 10-cm dishes and transfected
with pcDNA3.1-Flag-apoptin/pEGFP-apoptin vector or pcDNA3.1/pEGFP
empty vector. At 24 h after transfection, cells were washed three
times with ice-cold PBS, lysed in 1 ml of ice-cold cell lysis
buffer (40 mmol/l HEPES, 120 mmol/l NaCl, 1% Triton X-100, 10
mmol/l pyrophosphate, 10 mmol/l glycerophosphate, 50 mmol/l NaF,
1.5 mmol/l Na3VO4, 1 mmol/l EDTA, and
complete protease inhibitor cocktail, pH 7.5) on ice for 30 min.
The cell debris was removed, and anti-GFP (CST, UK) or anti-CDK1
antibody (CST, UK) was added to the cell lysate for 30 min at room
temperature under constant agitation. A total of 20 μl of washed
Bio-Adembeads PAG (Invitrogen) was added to the cell lysate with an
antibody for 2–3 h at 4°C and complexes were collected using a
magnet rack. Then, the immunocomplex was analyzed by a western blot
assay.
Immunofluorescence
Cells were plated on coverslips in a 12-well plate
the day before transfection. Transfection was performed using
Lipofectamine 2000 (Invitrogen). At 24 h after transfection, cells
were fixed with 4% paraformaldehyde, permeabilized in 0.1% Triton
X-100, blocked in 3% bovine serum albumin (BSA), and incubated with
mouse anti-Flag monoclonal antibody or anti-CDK1 antibody (CST,
USA) for 1 h and Cy3-red or FITC-green IgG secondary antibody (KPL,
Gaithersburg, MD, USA) for 1 h. The cells were washed and covered
with DAPI (4,6-diamidino-2-phenylindole) mounting medium (Vector
Laboratories, Cambridgeshire, UK) and visualized by using a
fluorescence microscope (Leica spII).
Results
Apoptin-induced specific killing of HepG2
cells
HepG2 and HL-7702 cells were transfected with
pcDNA3.1-Flag-apoptin/pEGFP-apoptin or pcDNA3.1/pEGFP to
investigate whether apoptin had differential cytotoxicity toward
HCC and normal liver cells. First, western blot analysis was used
to detect the expression of apoptin (Fig. 1B). Using the MTT assay, the efficacy
of tumor-specific killing activity was evaluated on days 1, 2, 3, 4
and 5 after transfection. As shown in Fig. 1C, the cell viability of HepG2 cells
was significantly decreased after transfection with
pcDNA3.1-Flag-apoptin or pEGFP-apoptin as compared to the control,
whereas the normal liver cell line HL-7702 was not affected by
apoptin. Expression of apoptin-induced apoptosis in HepG2 cells had
little or no cytotoxic effect on HL-7702 cells, which indicates
that apoptin can be applied as a potential therapeutic target in
HCC treatment. To further examine the subcellular localization of
apoptin in HepG2 and HL-7702 cells, the cells were transfected with
pcDNA3.1-Flag-apoptin, and the intracellular distribution of the
expressed Flag-apoptin was observed by immunofluorescent microscopy
24 h later. Flag-apoptin exhibited the same intracellular
localization as GFP-apoptin and native apoptin in HepG2 or HL-7702
cells; i.e., apoptin rapidly translocated to the nucleus in HepG2
cells, whereas it remained dispersed predominantly in the cytoplasm
in HL-7702 cells (Fig. 1D).
CDK1 expression in human hepatic cancer
tissues and cell lines
Western blot analysis and real-time PCR analyses
were performed on 48 pairs of human hepatic cancer tissues and
their corresponding adjacent non-tumor tissues to determine the
expression levels of CDK1. All 48 pairs of cancer tissues showed a
significantly higher CDK1 expression level than their matched
non-tumor tissues (P<0.001; Fig. 2A
and B). In addition, the extent of their activation varied
significantly, suggesting that the pathways leading to the
activation of CDK1 can be regulated independently. Similar results
were also observed in the HCC cell lines HepG2, Hep3B, SMMC-7721
and the normal liver cell line HL-7702 (Fig. 2C). These results indicated that the
abnormal activation of CDK1 was associated with the cancerous or
precancerous state. Therefore, the dysregulation of this cellular
pathway may be involved in the activation process of
apoptin-induced tumor-specific apoptosis.
Knockdown of CDK1 effectively inhibits
apoptin-induced apoptosis
To determine whether a causal link exists between
CDK1 and apoptin-induced tumor-specific apoptosis, HepG2 cells were
transfected with both pcDNA3.1-Flag-apoptin/pEGFP-apoptin or
pcDNA3.1/pEGFP and a lentiviral vector expressing CDK1 shRNA.
Transfection with a construct expressing a scrambled shRNA was
performed as a negative control. Western blotting demonstrated
successful knockdown of endogenous CDK1 protein by shRNA (Fig. 3A). An MTT cell survival assay showed
a strong reduction in the pro-apoptotic activity of apoptin after
transfection with shRNA (Fig. 3B).
Similar results were also observed by FACS analysis (Fig. 3C). These results collectively
indicated that CDK1 was an important apoptin-interacting protein
and played an essential role in the regulation of apoptin-induced
apoptosis. However, other intricate pathways may also be involved,
and further research is required to elucidate these pathways.
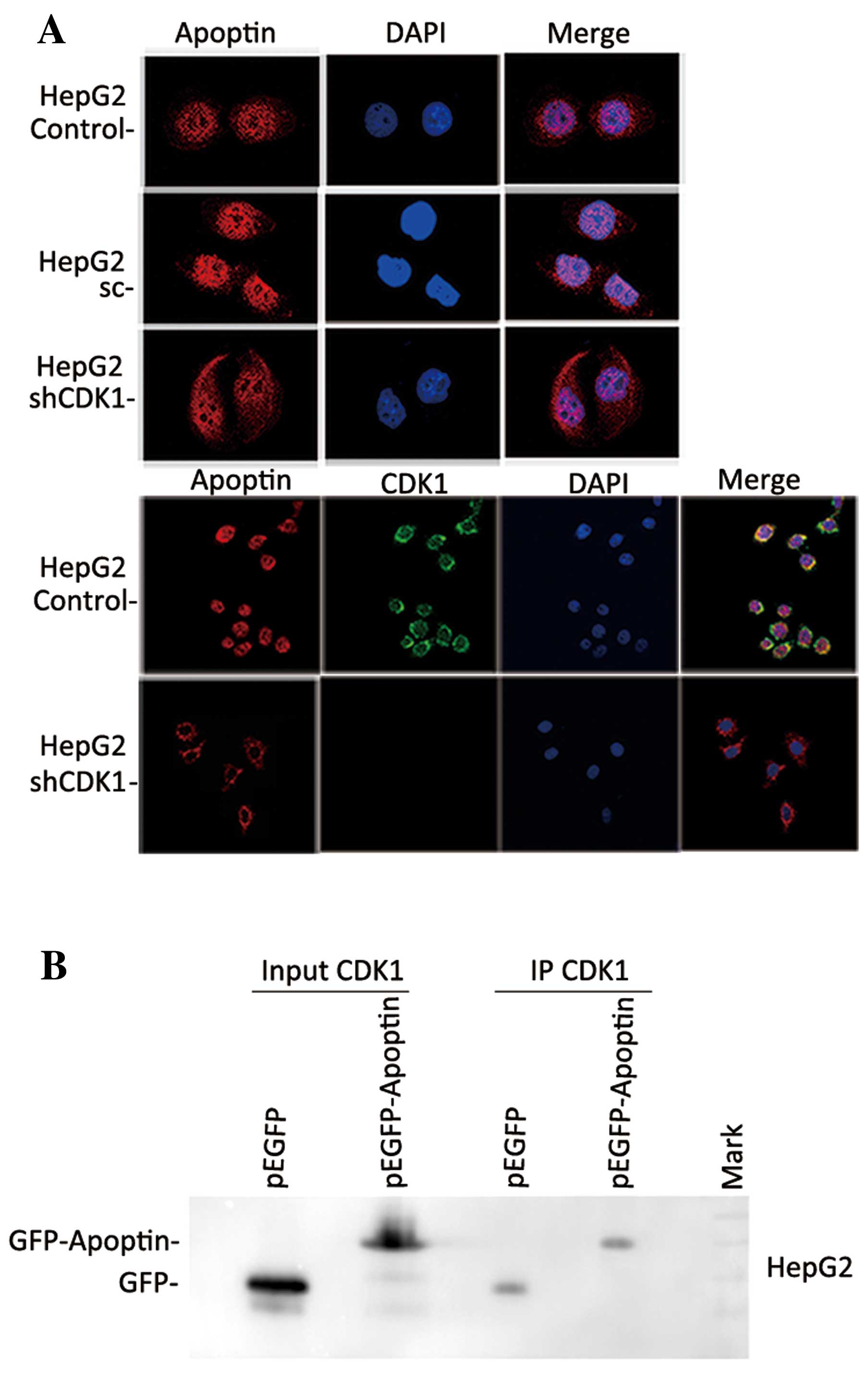
shRNA-mediated knockdown of CDK1 activity
affects the subcellular localization of apoptin
The tumor-specific activity of apoptin is dependent
on its ability to localize in the nucleus of tumor cells but not on
its ability to localize in the nucleus of primary normal cells
(10,18). To confirm the association between
CDK1 activity and the subcellular localization of apoptin, HepG2
cells were co-transfected with pcDNA3.1-Flag-apoptin and a
lentiviral vector expressing CDK1 shRNA. At 24 h post-transfection,
the nuclear translocation of apoptin was evaluated by
immunofluorescent microscopy. Flag-apoptin exhibited the same
intracellular distribution pattern in untransfected or scrambled
shRNA-transfected parental cells as the native apoptin (Fig. 4A). However, in cells transfected
with shRNA, Flag-apoptin relocalized predominantly to the cytoplasm
(Fig. 4A). The above data confirmed
that the activation of CDK1 may regulate the subcellular
localization of apoptin and that the suppression of CDK1 activation
had a significant effect on apoptin nuclear aggregation. To
determine whether apoptin interacts with CDK1 in HepG2 cells,
immunoprecipitation (IP) was performed. As shown in Fig. 4B, western blot analysis of the IP
complexes confirmed co-immunoprecipitation of CDK1 with the
GFP-apoptin fusion protein but not with GFP alone (Fig. 4B). These results suggest that there
was definitive interaction between CDK1 and apoptin in HepG2
cells.
Discussion
Several viral proteins that kill tumor cells have
been identified. Apoptin is one of the family members of
proline-rich proteins derived from the chicken anemia virus.
Apoptin has an efficient apoptotic effect on cancer cells (9). An additional characteristic
demonstrated by this chicken anemia virus-derived protein is its
tumor-specific cytotoxicity that differs from other chemo- or
bio-therapeutic strategies (6,7,9). These
findings indicated that apoptin and its cellular interacting and
regulatory targets may be important candidates for anticancer
therapeutics. Apoptin-induced, tumor-specific apoptosis presumably
requires additional interacting partners that may activate specific
signaling pathways in cancer cells. Although a number of
apoptin-interacting molecules have been identified, the molecular
mechanism underlying the action of apoptin remains poorly
understood. These interactions may be important for the nuclear
localization of apoptin or its tumor-selective cytotoxicity
(12–16). Based on our hypothesis, due to the
tumor-specific cytotoxicity of apoptin, the mediator that interacts
with apoptin in cancer cells should demonstrate a significantly
different activity or expression in cancer cells as compared to
normal cells. In this study, as well as in previous investigations,
HCC tissues showed a significantly higher CDK1 expression than
their matched non-tumor tissues. Similar results were also observed
in HCC cell lines. These results suggested that CDK1 acts as a
tumor-specific mediator, affecting apoptin-induced cytotoxicity in
HCC cells. CDK1 plays an important role in cell division (19), and several CDK1 substrates, such as
histone H1 and PI3K/AKT, play crucial roles in cell cycle
modulation (20,21). CDKs form a family of Ser/Thr kinases
that phosphorylate hundreds of protein substrates. [S/T*]PX[K/R] is
the classic recognition site of CDK1 (22–24)
and, coincidentally, the amino acid sequences surrounding the
phosphorylation site of apoptin is TTTPSRPR (25), which indicates that CDKs are
candidate kinases for apoptin. Consistent with this finding, the
results of our previous calculation using the KinasePhos software
(http://kinasephos.mbc.nctu.edu.tw)
suggested that CDKs, including CDK1, CDK2 and CDK4/6, are
noteworthy candidates for interaction with apoptin. Several studies
have shown an interaction between CDK2 and apoptin. Maddika et
al(26) found that CDK2 is an
important kinase in apoptin-induced apoptosis. In this study, to
clarify the mechanism of the tumor-specific killing effect induced
by apoptin, we investigated the interaction of apoptin with CDK1.
Although our co-IP result confirmed that CDK1 interacted with
apoptin, the detailed mechanism of the CDK1-apoptin interaction
remains unclear, and further experiments are required to reveal
this. CDK1 participates in a subset of apoptotic programs aside
from cell cycle regulation (17,27).
However, there was no evidence to indicate the relevance between
CDK1 and exogenous cell-killing proteins. Our study is the first to
establish the relationship between CDK1 and apoptin and highlights
a new mechanism of cell death induced by CDK1.
Apoptin is predominantly localized in the nucleus of
cancer cells, whereas in normal cells, its nuclear accumulation is
severely impaired (10,11,18).
Here, we provided evidence that the nuclear accumulation of apoptin
in tumor cells is mediated by CDK1. We also investigated whether
there is a causal link between CDK1 and apoptin-induced,
tumor-specific apoptosis. A strong reduction in apoptin-induced
apoptosis was observed after successful knockdown of endogenous
CDK1 protein by shRNA. These results suggested that CDK1 was an
important protein interacting with apoptin and played a significant
role in the regulation of apoptin-induced apoptosis. Previous
studies have shown that the selective toxicity of apoptin is mainly
attributed to its differential subcellular localization in tumor
and normal cells (10). In cancer
cells, apoptin is predominantly localized in the nucleus, whereas
in normal cells, apoptin is detected in the cytoplasm (10,11,18).
Hence, the subcellular localization of apoptin could indirectly
reflect its apoptotic activity. shRNA knockdown of CDK1
significantly reduced nuclear accumulation of apoptin in HepG2
cells, which further confirmed the association between the CDK1
activation and the cytotoxic activity of apoptin in HepG2 cells.
Moreover, Lee et al(28)
asserted that GFP-apoptin did not exhibit the same intracellular
distribution pattern as wild-type apoptin. By contrast, our results
showed the same intracellular localization of Flag-apoptin and
GFP-apoptin as wild-type apoptin in HCC and normal liver cells. Our
results are consistent with previous findings (29). A different cell line would be a
possible factor causing this divergence. However, the present study
identified a previously unrecognized mechanism of interaction
between CDK1 and apoptin.
In conclusion, the abnormal activation of CDK1 was
generally associated with the cancerous or precancerous state. A
definitive interaction between CDK1 and apoptin was also detected;
CDK1 may regulate the subcellular localization of apoptin. Our
study provides a novel mechanism for apoptin regulation involving
the CDK1 pathway.
Acknowledgements
This study was supported by the China National
Natural Scientific Foundation (no. 81071692 and 81272488), the
Shaanxi Province Natural Scientific Foundation (no. 2010JM4021) and
the First Affiliated Hospital of Xi’an Jiaotong University Medical
College Foundation (no. 2009–13).
References
|
1
|
Zhang Y, Wang S, Li D, et al: A systems
biology-based classifier for hepatocellular carcinoma diagnosis.
PloS One. 6:e224262011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Almogy G, Lieberman S, Gips M, et al:
Clinical outcomes of surgical resections for primary liver sarcoma
in adults: results from a single centre. Eur J Surg Oncol.
30:421–427. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tanaka S and Arii S: Molecularly targeted
therapy for hepatocellular carcinoma. Cancer Sci. 100:1–8. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Noteborn MH, Todd D, Verschueren CA, et
al: A single chicken anemia virus protein induces apoptosis. J
Virol. 68:346–351. 1994.PubMed/NCBI
|
|
5
|
Noteborn MH, de Boer GF, van Roozelaar DJ,
et al: Characterization of cloned chicken anemia virus DNA that
contains all elements for the infectious replication cycle. J
Virol. 65:3131–3139. 1991.PubMed/NCBI
|
|
6
|
Tavassoli M, Guelen L, Luxon BA and Gaken
J: Apoptin: specific killer of tumor cells? Apoptosis. 10:717–724.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Noteborn MH: Proteins selectively killing
tumor cells. Eur J Pharmacol. 625:165–173. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guelen L, Paterson H, Gaken J, Meyers M,
Farzaneh F and Tavassoli M: TAT-apoptin is efficiently delivered
and induces apoptosis in cancer cells. Oncogene. 23:1153–1165.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Danen-Van Oorschot AA, Fischer DF,
Grimbergen JM, et al: Apoptin induces apoptosis in human
transformed and malignant cells but not in normal cells. Proc Natl
Acad Sci USA. 94:5843–5847. 1997.PubMed/NCBI
|
|
10
|
Danen-Van Oorschot AA, Zhang YH, Leliveld
SR, et al: Importance of nuclear localization of apoptin for
tumor-specific induction of apoptosis. J Biol Chem.
278:27729–27736. 2003.PubMed/NCBI
|
|
11
|
Heilman DW, Teodoro JG and Green MR:
Apoptin nucleocytoplasmic shuttling is required for cell
type-specific localization, apoptosis, and recruitment of the
anaphase-promoting complex/cyclosome to PML bodies. J Virol.
80:7535–7545. 2006. View Article : Google Scholar
|
|
12
|
Maddika S, Wiechec E, Ande SR, et al:
Interaction with PI3-kinase contributes to the cytotoxic activity
of apoptin. Oncogene. 27:3060–3065. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maddika S, Bay GH, Kroczak TJ, et al: Akt
is transferred to the nucleus of cells treated with apoptin, and it
participates in apoptin-induced cell death. Cell Prolif.
40:835–848. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang J, Cole D, Westwood N, et al:
Crucial roles for protein kinase C isoforms in tumor-specific
killing by apoptin. Cancer Res. 70:7242–7252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Teodoro JG, Heilman DW, Parker AE and
Green MR: The viral protein Apoptin associates with the
anaphase-promoting complex to induce G2/M arrest and apoptosis in
the absence of p53. Genes Dev. 18:1952–1957. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Janssen K, Hofmann TG, Jans DA, Hay RT,
Schulze-Osthoff K and Fischer U: Apoptin is modified by SUMO
conjugation and targeted to promyelocytic leukemia protein nuclear
bodies. Oncogene. 26:1557–1566. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shi L, Nishioka WK, Th’ng J, Bradbury EM,
Litchfield DW and Greenberg AH: Premature p34cdc2 activation
required for apoptosis. Science. 263:1143–1145. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kuusisto HV, Wagstaff KM, Alvisi G and
Jans DA: The C-terminus of apoptin represents a unique tumor
cell-enhanced nuclear targeting module. Int J Cancer.
123:2965–2969. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Murray AW: Recycling the cell cycle:
cyclins revisited. Cell. 116:221–234. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brown NR, Noble ME, Endicott JA and
Johnson LN: The structural basis for specificity of substrate and
recruitment peptides for cyclin-dependent kinases. Nat Cell Biol.
1:438–443. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Loog M and Morgan DO: Cyclin specificity
in the phosphorylation of cyclin-dependent kinase substrates.
Nature. 434:104–108. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brotherton DH, Dhanaraj V, Wick S, et al:
Crystal structure of the complex of the cyclin D-dependent kinase
Cdk6 bound to the cell-cycle inhibitor p19INK4d. Nature.
395:244–250. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jeffrey PD, Russo AA, Polyak K, et al:
Mechanism of CDK activation revealed by the structure of a
cyclinA-CDK2 complex. Nature. 376:313–320. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Honda R, Lowe ED, Dubinina E, et al: The
structure of cyclin E1/CDK2: implications for CDK2 activation and
CDK2-independent roles. EMBO J. 24:452–463. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Los M, Panigrahi S, Rashedi I, et al:
Apoptin, a tumor-selective killer. Biochim Biophys Acta.
1793:1335–1342. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Maddika S, Panigrahi S, Wiechec E, et al:
Unscheduled Akt-triggered activation of cyclin-dependent kinase 2
as a key effector mechanism of apoptin’s anticancer toxicity. Mol
Cell Biol. 29:1235–1248. 2009.PubMed/NCBI
|
|
27
|
Golsteyn RM: Cdk1 and Cdk2 complexes
(cyclin dependent kinases) in apoptosis: a role beyond the cell
cycle. Cancer Lett. 217:129–138. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee YH, Cheng CM, Chang YF, Wang TY and
Yuo CY: Apoptin T108 phosphorylation is not required for its
tumor-specific nuclear localization but partially affects its
apoptotic activity. Biochem Biophys Res Commun. 354:391–395. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Poon IK, Oro C, Dias MM, Zhang J and Jans
DA: Apoptin nuclear accumulation is modulated by a CRM1-recognized
nuclear export signal that is active in normal but not in tumor
cells. Cancer Res. 65:7059–7064. 2005. View Article : Google Scholar : PubMed/NCBI
|