Introduction
In eukaryotes, autophagy is a physiological response
for the cell to undergo intracellular protein degradation and
organelle turnover. It is one of the intrinsic cellular properties
for maintaining cellular energy homeostasis under nutrition
deficiency and stress. Recent progress has further extended the
investigation of autophagy to tumorigenesis and cancer therapy.
However, whether it represents a procancer or anticancer mechanism
is far beyond our understanding (1). Autophagy reflects a complicated
cellular process in hepatocellular carcinoma (HCC), a common cancer
in Chinese and Asian populations (2). Deficiency/attenuation of autophagic
function or downregulation of autophagy-related genes (ATGs), key
regulator genes in autophagy, were significantly associated with
the occurrence or poor prognosis of HCC (3–5).
However, autophagy appears to mediate chemotherapy resistance as
noted in in vitro experiments. For example, inhibition of
autophagy was found to facilitate the killing of HCC cells by
chemical drugs (6–14). This is of importance in clinical
implications, as multidrug resistance (MDR) is one of the
detrimental characteristics of HCC (2). An imminent task in cancer research is
to elucidate the role of autophagy in HCC cells under variable
microenvironments before we can address whether the process of
autophagy may become a potential target for HCC therapy.
The high rate of proliferation and other adverse
factors inherent in cancer cells usually result in an overload of
endoplasmic reticulum (ER), leading to accumulation of misfolded
and/or unfolded proteins in ER, a condition referred to as ‘ER
stress’. An unfolded protein response (UPR) is subsequently evoked
to alleviate this stress by activating a group of signal
transduction pathways and the transcription of genes. Tunicamycin
(TM) is an antibiotic, which blocks the formation of
N-acetylglucosamine-lipid intermediates, thereby preventing
glycosylation and maturation of proteins (15). TM is widely accepted as an ER stress
stimulus since it induces the accumulation of unfolded proteins in
the ER lumen. In HCC, a number of environmental factors such as
hypoxia, viral infection, chemicals or radiation stimulation can
trigger ER stress (16–21). Moreover, ER stress is involved in
several signal pathways related to hepatocellular proliferation,
survival and apoptosis (22–24).
ER stress and autophagy in HCC often share the same
stimuli (10,18,21,25,26).
Yet, whether or not ER stress itself triggers autophagy remains
unknown, and the role of ER stress and autophagy in HCC cell
survival and death is still unsolved. The aim of this study was to
investigate autophagic responses in the human HCC cell line HepG2
under ER stress stimulation and its consequent effect on cell
survival and death.
Materials and methods
Reagents
TM from Streptomyces sp. (cat. no. T7765),
3-methyladenine (3-MA; cat. no. M9281), chloroquine diphosphate
salt (CQ; cat. no. C6628) and rapamycin (2.5 mg/ml in DMSO, cat.
no. R8781) were purchased from Sigma-Aldrich (USA). To prepare
stock solutions, 3-MA and CQ were dissolved in sterile ultrapure
water, while TM was dissolved in DMSO with a final concentration of
DMSO in the culture medium no more than 1/1,000 (v/v). Earle’s
balanced salt solution (EBSS) and Dulbecco’s modified Eagle’s
medium (DMEM) were obtained from Invitrogen (USA); fetal bovine
serum (FBS) was obtained from HyClone. The Cell Counting Kit-8
(CCK-8) was purchased from Dojindo Laboratories (Japan). The
primary antibodies used for western blot analysis were anti-LC3
antibody (cat. no. PM036; MBL Co., Ltd.); anti-Beclin 1 antibody
(cat. no. ab16998), anti-GRP78 BiP antibody (cat. no. ab53068; both
from Abcam, Inc., USA) and anti-caspase-3 antibody (cat. no. 9662;
Cell Signaling Technology).
Cell culture
The HCC cell line HepG2 was obtained from the Cell
Bank of Shanghai Institute for Biological Science, Chinese Academy
of Sciences. Cells were routinely cultured in DMEM supplemented
with 10% (v/v) FBS and maintained in a humidified incubator with 5%
CO2 at 37°C.
Cell proliferation and viability
analysis
Cell proliferation assays were performed using
CCK-8. Re-suspended cells were seeded onto 96-well plates at a
concentration of 104 cells/well, and incubated for 24 h
to allow adherence. The cells were then exposed to TM for the
indicated concentrations and time points. For the inhibition assay,
a stock solution of the autophagic inhibitors (3-MA or CQ) or an
equal volume of PBS was added into the culture medium 1 h prior to
TM application. The final concentrations of 3-MA and CQ were 5
mmol/l and 5 μg/ml, respectively. After incubation, cell medium in
each well was substituted with 100 μl pre-prepared
WST®-8 solution (dilution of 1:10 in fresh DMEM); the
plate was then incubated for an additional 1 h at 37°C. Absorbance
at 450 nm for WST-8 formazan was measured using the Elx800
absorbance microplate reader (BioTek Instruments, Inc). Triplicate
measurements were made for each treatment subgroup in one plate,
and the average optical density (OD) of the three wells was
calculated. The average OD450nm of another two cell-free
wells reserved in each plate was calculated as the background
value. The cell viability was calculated according to the formula:
% viability = (OD450nm treated cells -
OD450nm background) / (OD450nm control cells
- OD450nm background) × 100. At least three independent
experiments were performed to generate the mean data for each
intervention.
Transmission electron microscopy
(TEM)
Electron microscopy is a traditional and reliable
method to observe autophagic compartments (27,28).
We performed TEM to demonstrate autophagosome formation in HepG2
cells after ER stress stimulation. Briefly, HepG2 cells were grown
in 100-mm-diameter dishes. Following treatment with DMEM (control),
EBSS, 1 μg/ml rapymycin or 2.5 μg/ml TM for the indicated time
periods, respectively, cells were collected and centrifuged at
3,000 rpm. Cell pellets were primarily fixed in a solution with
2.5% glutaraldehyde (v/v) overnight, and then in 1% osmium
tetraoxide (v/v) for 1 h for secondary fixation. After dehydration
in a series of concentrations of ethanol, the cells were finally
embedded in Epon 812. Ultrathin (70 nm) sections were cut on an
NOVA ultramicrotome (LKB, Sweden), stained with uranyl acetate
(saturated aqueous solution) and lead acetate, and then examined
under a transmission electron microscope (JEM-1230; Jeol Ltd.,
Japan).
Western blot analysis
Briefly, cells were washed with pre-cooled PBS twice
and lysed on ice for 30 min in RIPA lysis buffer containing 50
mmol/l Tris-HCl (pH 7.4), 150 mmol/l NaCl, 1 mmol/l
phenylmethylsulfonyl fluoride (PMSF), 1 mmol/l ethylene diamine
tetraacetic acid (EDTA), 0.1% sodium dodecyl sulfate (SDS), 1%
Triton X-100 and 1% sodium deoxycholate. Lysates were centrifuged
at 12,000 rpm for 10 min at 4°C. The supernatant was transferred
into a new tube; the protein concentration was determined using BCA
protein assay. Lysates were incubated with 2X Laemmli sample buffer
(Bio-Rad, Hercules, CA, USA) and heated for 10 min at 95°C. The
proteins from each sample were separated by sodium
dodecyl-sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and
transferred onto polyvinylidene difluoride (PVDF) membranes
(Millipore, Bedford, MA, USA). Membranes were blocked for 2 h at
room temperature in PBS containing 5% defatted milk powder and
washed with Tris-buffered saline/Tween-20 (TBST) three times, then
incubated overnight at 4°C with the diluted primary antibody
(anti-LC3, 1:1,000; anti-GRP78, 1:2,000; anti-Beclin 1, 1:1,000;
anti-β-actin, 1:500) in PBST. After washing in PBST, the membranes
were then incubated for 1 h at room temperature with the secondary
antibody (goat anti-mouse-HRP, 1:10,000). The immunoreactive
proteins were detected using the enhanced chemiluminescence (ECL)
kit (Pierce) and the chemiluminescent signals were captured by
ImageQuant™ LAS-4000 Mini Imager (Fuji, Japan). For quantitative
analysis, the integrated density of each band was obtained using
ImageJ software (US National Institutes of Health).
Data analysis
At least three independent experiments were
performed for the numerical variables unless otherwise stated. Data
are expressed as the means ± standard deviation in each group.
Student’s t-test and one-way ANOVA were used to examine the
differences between two or multiple groups, respectively.
Statistical tests were performed using SPSS 15.0 software. A
probability-value <0.05 was considered to indicate a
statistically significant result.
Results
ER stress triggers accumulation of
autophagic compartments in HepG2 cells
After exposure to TM (2.5 μg/ml) for 24 h, TEM
revealed that there was a greater number of autophagic compartments
accumulated in the TM-treated cells (Fig. 1A) when compared with that in the
non-treated cells (Fig. 1B); the
latter also showed rare autophagic compartments. Autophagosomes
were recognized as double membrane vacuolar structures containing
cytoplasmic contents (Fig. 1A and
C). Other types of autophagic compartments such as
autolysosomes also appeared in the form of membrane vacuolar
structures containing high-density materials (Fig. 1A). Two known autophagy inducers,
starvation medium EBSS (Fig. 1D)
and rapamycin (Fig. 1E), acting via
amino acid deprivation and mTOR inhibition, respectively, were used
as the positive controls, and accumulation of autophagic
compartments in response to these interventions clearly indicated
autophagy.
ER stress induces LC3 conversion and
enhances autophagic flux in HepG2 cells
Western blot analysis was used to detect several key
proteins involved in the process of autophagic flux and ER stress.
GRP78, a resident protein of ER, was used as a molecular marker of
ER stress. The microtubule-associated protein 1-light chain 3
(MAP1-LC3, LC3), also known as Atg8 in yeast, is cleaved by the
Atg4 homologue to form LC3-I (29).
Upon induction of autophagy, LC3-I conjugates to
phosphatidylethanolamine (PE) to become LC3-II which anchors to the
autophagosomal membrane throughout the process of autophagosomal
maturation until degradation by lysosomes (30). Although LC3-II is a reliable marker
of autophagosomes (31), the
process of conjugation of LC3-I to PE to form LC3-II is more
indicative of the autophagic reaction. Thus, the conversion of
LC3-I to LC3-II is considered to be a surrogate of autophagy
induction. Both proteins can be detected by protein electrophoresis
and immunoblotting and the LC3 ratio (calculated by scanned
intensities of LC3-II/LC3-I in each group) is a measure of
autophagic LC3 conversion (32).
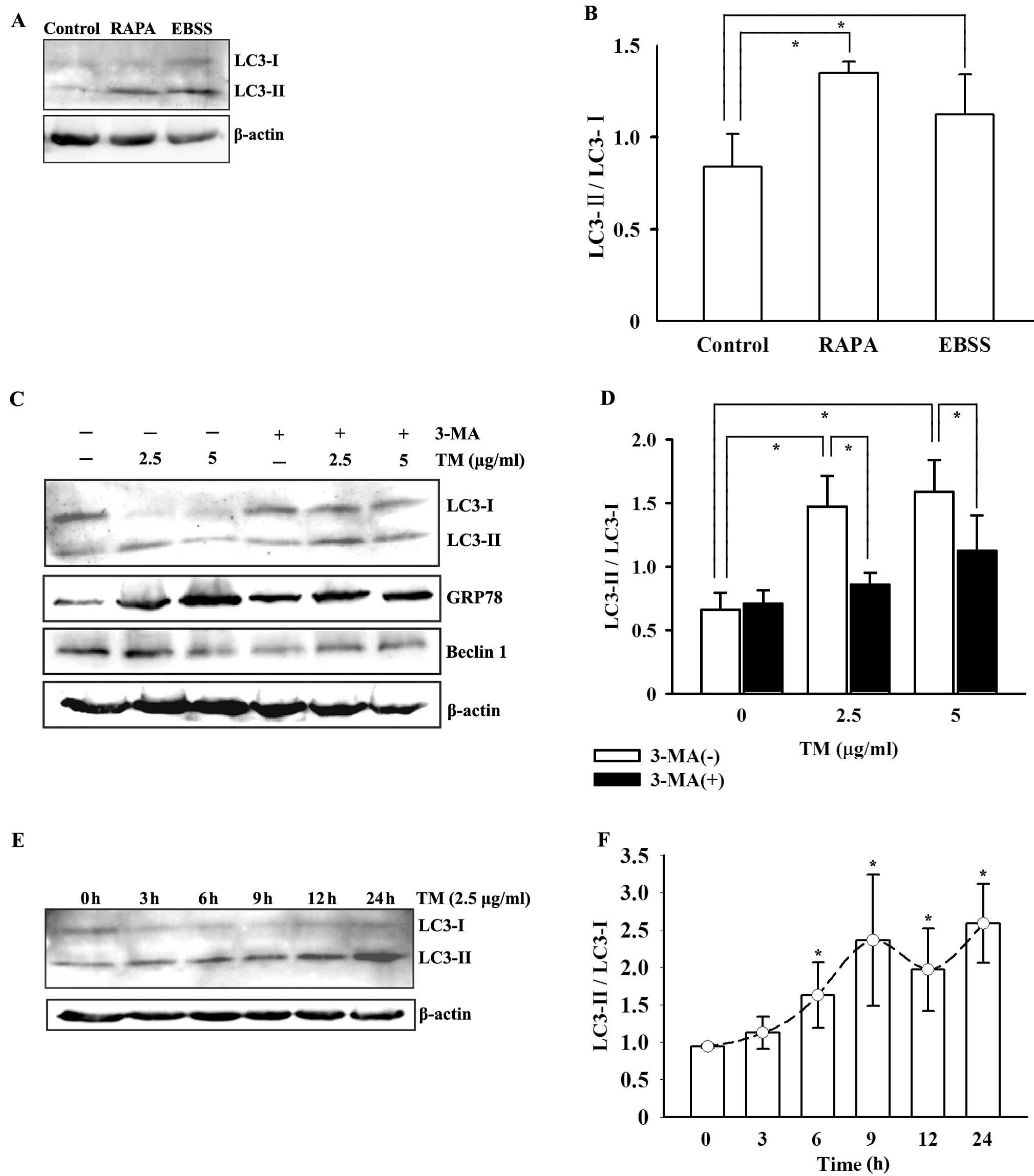
Initially, we performed two positive controls for
autophagy detection: the two well-known autophagy inducers EBSS
(incubated for 6 h) and rapamycin (1 μg/ml for 24 h). Both induced
overexpression of LC3-II and an increase in the LC3 ratio (Fig. 2A and B). Consequently, decreased
expression of LC3-I and increased expression of LC3-II were
observed in HepG2 cells after ER stress stimulation (Fig. 2C). Accordingly, the LC3 ratio
(LC3-II/I) was elevated in the cells treated with 2.5 and 5 μg/ml
TM when compared to the ratio in the unstressed cells (P<0.05).
This indicates an increase in conversion of LC3-I to LC3-II after
ER stress (Fig. 2D). 3-MA, a PI3K
class III inhibitor (also named Vps34) suppressed the LC3
conversion which suggests the involvement of PI3K ClassIII/Vps34 in
the observed autophagy by ER stress. In parallel, we ascertained
the occurrence of ER stress in HepG2 cells through evidence of
GRP78 overexpression after TM stimulation (Fig. 2C). Furthermore, we also demonstrated
a time-dependent increase in autophagy induction in the ER-stressed
HepG2 cells by treating the cells with TM for 3, 6, 9, 12 and 24 h
(Fig. 2E and F). Another
autophagy-related protein, Beclin 1, appeared to be unaffected
either by the ER stressor or co-existing 3-MA (Fig. 2C).
An important consideration in autophagy detection
was whether or not the observed accumulation of autophagosomes (or
its surrogate, LC3-II) was indicative of an authentic autophagy
induction or simply the blockage of autophagosomal degradation in
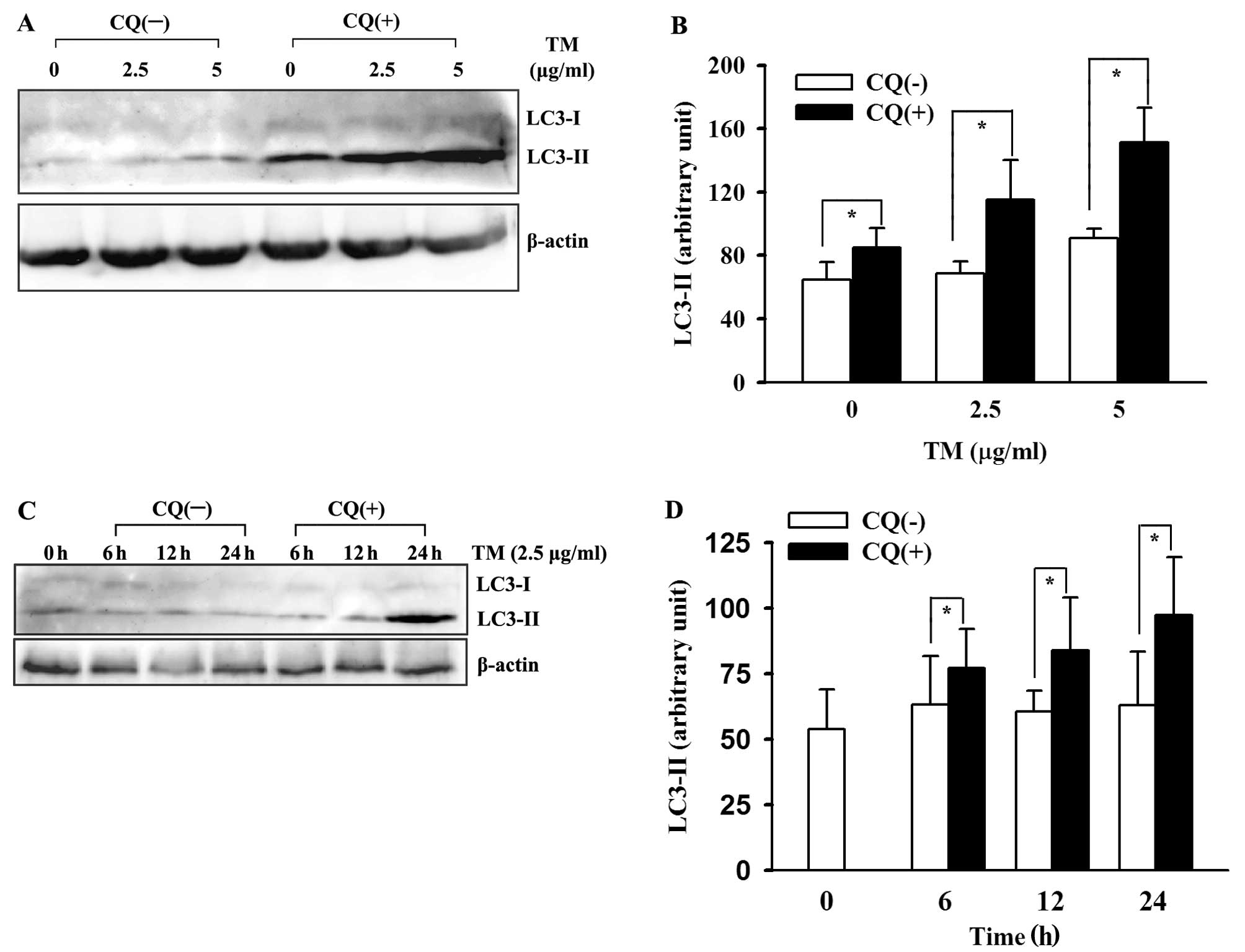
the lysosomes. Thus, performing an ‘autophagic flux’ assay by using
a lysosomal inhibitor was necessary to distinguish between the
aforementioned two possibilities (31). We performed ‘LC3 turnover assay’ to
demonstrate autophagic flux enhancement in the ER-stressed HepG2
cells. Briefly, we pretreated the cultured cells with lysosomal
inhibitor CQ for 1 h before TM, then compared changes in LC3
expression with those in the absence of CQ. The results showed a
marked increase in LC3-II accumulation after lysosomal inhibition.
As shown in Fig. 3, the upregulated
LC3-II was lysosomal-dependent. This indicated an authentic
induction of autophagy in the HepG2 cells by TM, not a defect in
autophagosomal degradation.
Role of autophagy in the maintenance of
viability of ER-stressed HepG2 cells
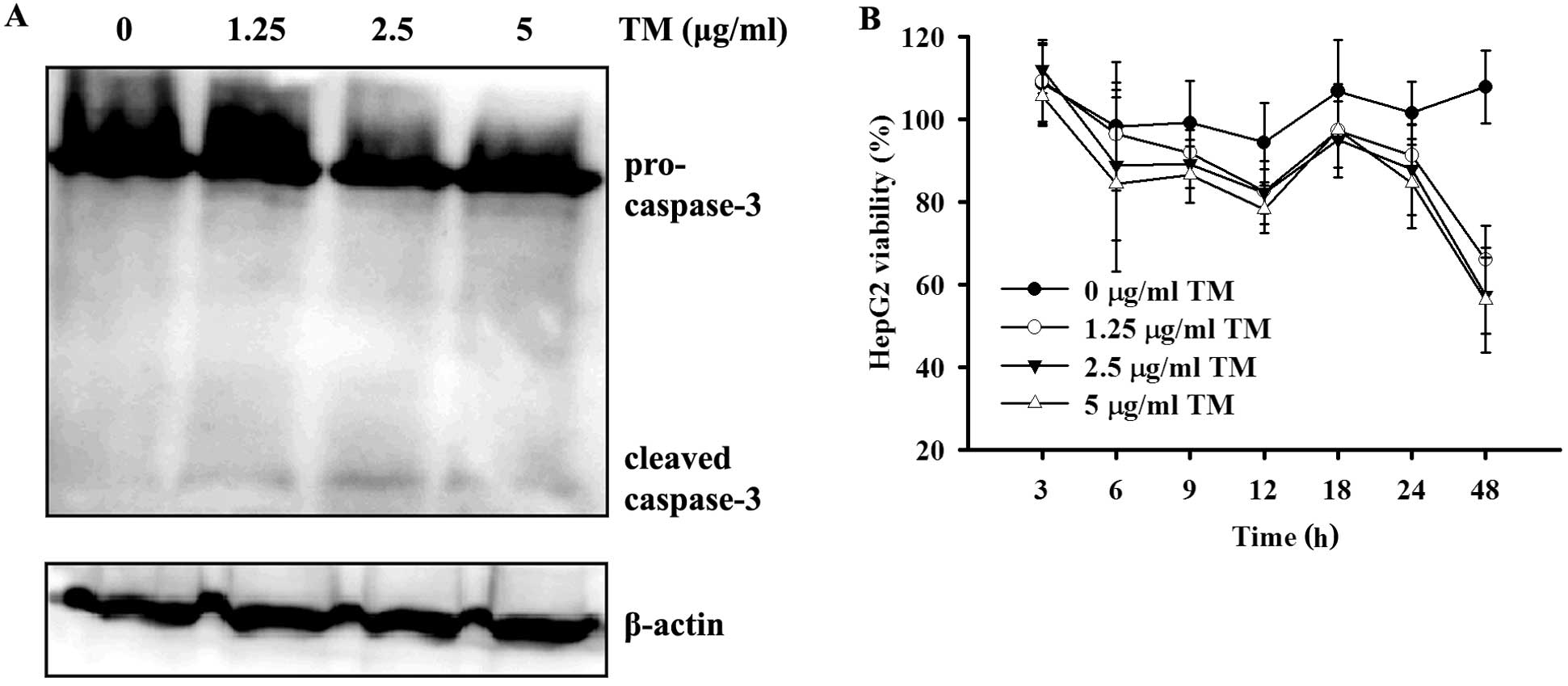
ER-stress associated cell death was demonstrated in
HepG2 cells by the cleavage and activation of caspase-3 after
treatment with 1.25, 2.5 or 5 μg/ml TM for 48 h (Fig. 4A). Furthermore, results of the CCK-8
test also showed a decrease in HepG2 cell viability after ER stress
stimulation, and the cell viability was well correlated with the
dose and duration of TM incubation (Fig. 4B). We confirmed induction of
autophagy in the ER-stressed HepG2 cells. To further ascertain the
role of the observed autophagy in ER-stress related cell death and
survival, we introduced two autophagy inhibitors which blocked the
autophagic pathway at different sites; 3-MA, an inhibitor of
phosphatidyl inositol 3-kinase, blocks initiation of autophagic
vesicles and CQ, a lysosomal inhibitor, affects the downstream
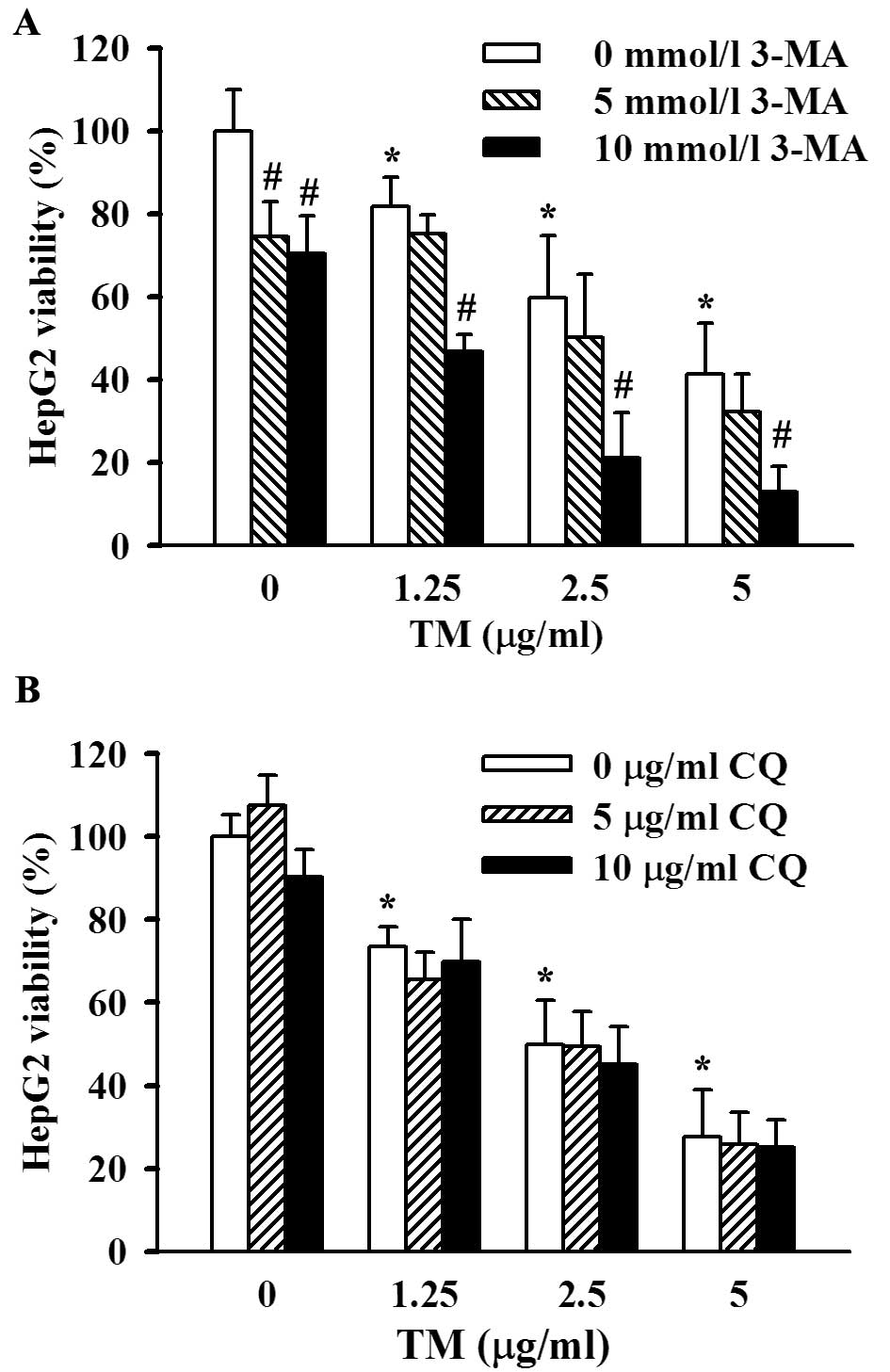
process of autophagy. In the presence of 3-MA (5 or 10 mmol/l at
final concentrations), 48 h of TM stimulation induced a more
significant decrease in cell viability than that without 3-MA
co-incubation, and 10 mmol/l 3-MA exerted a greater degree of cell
viability suppression than 5 mmol/l 3-MA (Fig. 5A). The data described above showed
that inhibition of autophagy facilitated ER stress-associated cell
death, i.e., enhancement of autophagy induced by ER stress
contributed to cell viability maintenance. However, CQ did not
appear to exert an effect on the viability of ER-stressed HepG2
cells (Fig. 5B), although lysosome
inhibition was considered to a certain degree as a dysfunction of
autophagy.
Discussion
Induction of autophagy in HepG2 cells by
TM
Although both UPR and autophagy have been
interpreted as stress responses in eukaryotes, direct evidence
linking ER stress to autophagy was first published in yeast
(33), following by evidence in
normal and transformed human cell lines (34–39).
However, the so-called ‘increase in autophagy’ in most published
studies (in fact, accumulation of autophagosomes) can be attributed
not only to induction of autophagy but also blockage of the
degradation in lysosomes (31). Few
studies reportedly have performed lysosomal inhibition assay to
verify the fluency of the whole autophagic flux. Thus, the purpose
of the present study was to demonstrate the induction of autophagy
and enhancement of autophagic flux in HepG2 cells following ER
stress, by observing ultramicroscopic accumulation of
autophagosomes, alterations in biochemical markers and functional
enhancement of autophagic degradation in lysosomes.
We confirmed an authentic induction in autophagy and
enhancement of autophagic flux in the ER-stressed HepG2 cells.
However, in another study, ER stress-induced autophagic degradation
of apoB in the HepG2 cell line was unable to be demonstrated
(39). Yet, we did not refute our
theory as the authors of that study also demonstrated enhancement
of autophagic apoB degradation in two other hepatoma cell lines,
and alternative degradation pathways for apoB may coexist in HepG2
cells; the propensity of apoB degradation varies in different cell
lines.
ER stress signals to autophagy are still unresolved.
PI3K class III/Vps34 is clearly involved in ER stress-induced
autophagy in HepG2 cells, as 3-MA can suppress autophagy induction
by TM in HepG2 cells. We also detected Beclin 1 expression in HepG2
cells. Decreased expression of autophagy protein Beclin 1 is
associated with poor prognosis of HCC (5), and induction of autophagy in prostate
cancer cells involves interaction between Bcl-2 and Beclin 1 at the
ER (40). Yet, we failed to find a
difference in its expression between ER-stressed and unstressed
cells; possibly a distinct signaling pathway exists in ER stress
triggered autophagic flux in HCC.
Role of ER stress-induced autophagy in
the survival of HepG2 cells
ER stress leads to accumulation of unfolded protein,
and autophagy is associated with the degradation of protein and
organelles (41). Based on this
finding, ER stress and related autophagy are cancer cell survival
mechanisms in response to hypoxia, nutrition deficiency and stress.
However, autophagic cell death (ACD, type II cell death) was
proposed recently by the Nomenclature Committee on Cell Death
(NCCD) as a new sub-routine of cell death (42), yet the role of autophagy in cell
death has not yet been clarified (43–47).
One study reported that ER stress-induced autophagy exerts
differential effects on cell survival: pro-survival effects in
cancer cells and pro-death effects in normal cells (34). Our results demonstrated a decrease
in cell survival in the TM-treated HepG2 cells in the presence of
the autophagic inhibitor 3-MA, indicating that a pro-survival
effect of autophagy played a dominant role in ER-stressed HepG2
cells, similar to that in neuroblastoma cells (38). Autophagy inhibition induced by
chemical drugs in HepG2 cells and other hepatocarcinoma cell lines
also exhibited enhancement of cell death (48,49),
but whether ER stress was involved in these mechanisms is
unknown.
We did not observe an effect of the lysosomal
acidification inhibitor, CQ, which blocks the downstream process of
autophagy. This finding suggests that the linkage between autophagy
signaling and death signals may be located upstream in the process
of autophagy, as the targets of 3-MA were located at the initiation
of autophagy. In fact, ER itself can function as a switching point
in autophagy and death by interaction between Bcl-2 family proteins
occurring on the ER membrane (50,51).
In conclusion, ER stress in HepG2 cells elicits
enhancement of autophagic flux. This contributes to the maintenance
of cell viability. Our results may help to eluciate the
understanding of carcinogenesis and drug resistance of HCC.
Acknowledgements
We thank Professor Qi-Xing Zhu of Anhui Medical
University for providing the experimental platform and
encouragement to complete the study. This study was supported by
grants from National Natural Science Foundation of China (no.
81071986 and no. 81272739), and by a key project of the Anhui
Provincial Office of Education, China (no. KJ2011A154). C.W.
gratefully acknowledges support from the Biotechnology and
Biological Sciences Research Council (BBSRC) (BB/G015554/1;
BB/I025379/1).
References
|
1
|
Levine B: Cell biology: autophagy and
cancer. Nature. 446:745–747. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar
|
|
3
|
Kisen GO, Tessitore L, Costelli P, et al:
Reduced autophagic activity in primary rat hepatocellular carcinoma
and ascites hepatoma cells. Carcinogenesis. 14:2501–2505. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Qu X, Yu J, Bhagat G, et al: Promotion of
tumorigenesis by heterozygous disruption of the beclin 1 autophagy
gene. J Clin Invest. 112:1809–1820. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ding ZB, Shi YH, Zhou J, et al:
Association of autophagy defect with a malignant phenotype and poor
prognosis of hepatocellular carcinoma. Cancer Res. 68:9167–9175.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Song J, Qu Z, Guo X, et al:
Hypoxia-induced autophagy contributes to the chemoresistance of
hepatocellular carcinoma cells. Autophagy. 5:1131–1144. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Du H, Yang W, Chen L, et al: Role of
autophagy in resistance to oxaliplatin in hepatocellular carcinoma
cells. Oncol Rep. 27:143–150. 2012.PubMed/NCBI
|
|
8
|
Ding ZB, Hui B, Shi YH, et al: Autophagy
activation in hepatocellular carcinoma contributes to the tolerance
of oxaliplatin via reactive oxygen species modulation. Clin Cancer
Res. 17:6229–6238. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xie BS, Zhao HC, Yao SK, et al: Autophagy
inhibition enhances etoposide-induced cell death in human hepatoma
G2 cells. Int J Mol Med. 27:599–606. 2011.PubMed/NCBI
|
|
10
|
Guo XL, Li D, Hu F, et al: Targeting
autophagy potentiates chemotherapy-induced apoptosis and
proliferation inhibition in hepatocarcinoma cells. Cancer Lett.
320:171–179. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi YH, Ding ZB, Zhou J, et al: Targeting
autophagy enhances sorafenib lethality for hepatocellular carcinoma
via ER stress-related apoptosis. Autophagy. 7:1159–1172. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shimizu S, Takehara T, Hikita H, et al:
Inhibition of autophagy potentiates the antitumor effect of the
multikinase inhibitor sorafenib in hepatocellular carcinoma. Int J
Cancer. 131:548–557. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu YL, Yang PM, Shun CT, Wu MS, Weng JR
and Chen CC: Autophagy potentiates the anti-cancer effects of the
histone deacetylase inhibitors in hepatocellular carcinoma.
Autophagy. 6:1057–1065. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hui B, Shi YH, Ding ZB, et al: Proteasome
inhibitor interacts synergistically with autophagy inhibitor to
suppress proliferation and induce apoptosis in hepatocellular
carcinoma. Cancer. 118:5560–5571. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tkacz JS and Lampen O: Tunicamycin
inhibition of polyisoprenyl N-acetylglucosaminyl pyrophosphate
formation in calf-liver microsomes. Biochem Biophys Res Commun.
65:248–257. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Koumenis C: ER stress, hypoxia tolerance
and tumor progression. Curr Mol Med. 6:55–69. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shuda M, Kondoh N, Imazeki N, et al:
Activation of the ATF6, XBP1 and grp78 genes in human
hepatocellular carcinoma: a possible involvement of the ER stress
pathway in hepatocarcinogenesis. J Hepatol. 38:605–614. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Al-Rawashdeh FY, Scriven P, Cameron IC,
Vergani PV and Wyld L: Unfolded protein response activation
contributes to chemoresistance in hepatocellular carcinoma. Eur J
Gastroenterol Hepatol. 22:1099–1105. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu Z, Jensen G and Yen TS: Activation of
hepatitis B virus S promoter by the viral large surface protein via
induction of stress in the endoplasmic reticulum. J Virol.
71:7387–7392. 1997.PubMed/NCBI
|
|
20
|
Li B, Gao B, Ye L, et al: Hepatitis B
virus X protein (HBx) activates ATF6 and IRE1-XBP1 pathways of
unfolded protein response. Virus Res. 124:44–49. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sir D, Chen WL, Choi J, Wakita T, Yen TS
and Ou JH: Induction of incomplete autophagic response by hepatitis
C virus via the unfolded protein response. Hepatology.
48:1054–1061. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang HC, Huang W, Lai MD and Su IJ:
Hepatitis B virus pre-S mutants, endoplasmic reticulum stress and
hepatocarcinogenesis. Cancer Sci. 97:683–688. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cho HK, Cheong KJ, Kim HY and Cheong J:
Endoplasmic reticulum stress induced by hepatitis B virus X protein
enhances cyclo-oxygenase 2 expression via activating transcription
factor 4. Biochem J. 435:431–439. 2011. View Article : Google Scholar
|
|
24
|
Joyce MA, Walters KA, Lamb SE, et al: HCV
induces oxidative and ER stress, and sensitizes infected cells to
apoptosis in SCID/Alb-uPA mice. PLoS Pathog. 5:e10002912009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sir D, Tian Y, Chen WL, Ann DK, Yen TS and
Ou JH: The early autophagic pathway is activated by hepatitis B
virus and required for viral DNA replication. Proc Natl Acad Sci
USA. 107:4383–4388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mizui T, Yamashina S, Tanida I, et al:
Inhibition of hepatitis C virus replication by chloroquine
targeting virus-associated autophagy. J Gastroenterol. 45:195–203.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ylä-Anttila P, Vihinen H, Jokitalo E and
Eskelinen EL: Monitoring autophagy by electron microscopy in
Mammalian cells. Methods Enzymol. 452:143–164. 2009.PubMed/NCBI
|
|
28
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
He H, Dang Y, Dai F, et al:
Post-translational modifications of three members of the human
MAP1LC3 family and detection of a novel type of modification for
MAP1LC3B. J Biol Chem. 278:29278–29287. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kabeya Y, Mizushima N, Ueno T, et al: LC3,
a mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Klionsky DJ, Abeliovich H, Agostinis P, et
al: Guidelines for the use and interpretation of assays for
monitoring autophagy in higher eukaryotes. Autophagy. 4:151–175.
2008. View Article : Google Scholar
|
|
32
|
Karim MR, Kanazawa T, Daigaku Y, Fujimura
S, Miotto G and Kadowaki M: Cytosolic LC3 ratio as a sensitive
index of macroautophagy in isolated rat hepatocytes and H4-II-E
cells. Autophagy. 3:553–560. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yorimitsu T, Nair U, Yang Z and Klionsky
DJ: Endoplasmic reticulum stress triggers autophagy. J Biol Chem.
281:30299–30304. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ding WX, Ni HM, Gao W, et al: Differential
effects of endoplasmic reticulum stress-induced autophagy on cell
survival. J Biol Chem. 282:4702–4710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Qin L, Wang Z, Tao L and Wang Y: ER stress
negatively regulates AKT/TSC/mTOR pathway to enhance autophagy.
Autophagy. 6:239–247. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kawakami T, Inagi R, Takano H, et al:
Endoplasmic reticulum stress induces autophagy in renal proximal
tubular cells. Nephrol Dial Transplant. 24:2665–2672. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sakaki K, Wu J and Kaufman RJ: Protein
kinase Ctheta is required for autophagy in response to stress in
the endoplasmic reticulum. J Biol Chem. 283:15370–15380. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ogata M, Hino S, Saito A, et al: Autophagy
is activated for cell survival after endoplasmic reticulum stress.
Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Qiu W, Zhang J, Dekker MJ, et al: Hepatic
autophagy mediates endoplasmic reticulum stress-induced degradation
of misfolded apolipoprotein B. Hepatology. 53:1515–1525. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lian J, Karnak D and Xu L: The
Bcl-2-Beclin 1 interaction in (−)-gossypol-induced autophagy versus
apoptosis in prostate cancer cells. Autophagy. 6:1201–1203.
2010.
|
|
41
|
Lum JJ, Bauer DE, Kong M, et al: Growth
factor regulation of autophagy and cell survival in the absence of
apoptosis. Cell. 120:237–248. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Galluzzi L, Vitale I, Abrams JM, et al:
Molecular definitions of cell death subroutines: recommendations of
the Nomenclature Committee on Cell Death 2012. Cell Death Differ.
19:107–120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Levine B and Yuan J: Autophagy in cell
death: an innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kroemer G and Levine B: Autophagic cell
death: the story of a misnomer. Nat Rev Mol Cell Biol. 9:1004–1010.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wirawan E, Vanden Berghe T, Lippens S,
Agostinis P and Vandenabeele P: Autophagy: for better or for worse.
Cell Res. 22:43–61. 2012. View Article : Google Scholar
|
|
46
|
Denton D, Nicolson S and Kumar S: Cell
death by autophagy: facts and apparent artefacts. Cell Death
Differ. 19:87–95. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cheng Y and Yang JM: Survival and death of
endoplasmic-reticulum-stressed cells: role of autophagy. World J
Biol Chem. 2:226–231. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Daniel F, Legrand A, Pessayre D, Vadrot N,
Descatoire V and Bernuau D: Partial Beclin 1 silencing aggravates
doxorubicin- and Fas-induced apoptosis in HepG2 cells. World J
Gastroenterol. 12:2895–2900. 2006.PubMed/NCBI
|
|
49
|
Longo L, Platini F, Scardino A, Alabiso O,
Vasapollo G and Tessitore L: Autophagy inhibition enhances
anthocyanin-induced apoptosis in hepatocellular carcinoma. Mol
Cancer Ther. 7:2476–2485. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Rovetta F, Stacchiotti A, Consiglio A, et
al: ER signaling regulation drives the switch between autophagy and
apoptosis in NRK-52E cells exposed to cisplatin. Exp Cell Res.
318:238–250. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Szegezdi E, Macdonald DC, Ni Chonghaile T,
Gupta S and Samali A: Bcl-2 family on guard at the ER. Am J Physiol
Cell Physiol. 296:C941–C953. 2009. View Article : Google Scholar : PubMed/NCBI
|