Introduction
Recently, a growing number of studies have shown
that cancer is a stem cell disease (1). Cancer stem cells possess unlimited
potential for self-renewal and differentiation and exhibit high
tumourigenicity and drug resistance. Compared with other cell types
in tumours, cancer stem cells exhibit a greater capacity for
proliferation, differentiation and invasion. They have also been
shown to tolerate radiation and chemotherapy. These cells are not
only the origin of the tumour cell population but are also the
cause of tumour metastasis and recurrence (1). Under the guidance of the stem cell
theory, researchers have used various methods to obtain and
identify cancer stem cells in leukaemia, breast cancer, brain
glioblastoma, prostate cancer, pancreatic cancer, colon cancer,
ovarian cancer, liver cancer, lung cancer and other tumours
(2–11). However, no systematic testing has
been developed to explore the ideal surface markers for stem cells
in nasopharyngeal carcinoma (NPC), which is one of the most common
malignancies in Southeast Asia. Because of the scarcity of
available human NPC tissues and the difficulties associated with
collecting them, studies investigating stem cells within primary
tumour cell populations have not previously been reported.
The CD133 antigen is a glycoprotein with a molecular
weight of 117 kDa exhibiting 5 transmembrane domains (12). CD133 was originally reported as a
specific marker for stem cells (13); however, subsequent studies have
found it to be expressed in various tumour cell lines. Tumour cells
expressing CD133 have been confirmed to be stem cells in human
brain tumours, colon cancer, prostate cancer, liver cancer,
melanoma, pancreatic cancer and other types of solid tumours
(8,14–19),
suggesting that CD133 may be a broad-spectrum marker of cancer stem
cells (20).
In the present study, a cell population with a
CD133+ phenotype from the NPC cell line and xenograft
tumours was isolated using magnetic activated cell sorting (MACS)
technology. The proliferation, differentiation of CD133+
cells and the tumourigenicity of CD133+ cells in nude
mice were further investigated.
Materials and methods
The present animal study was approved by the Ethics
Committee of the First Hospital Affiliated of Sun Yat-sen
University (no. 2009–01, 2009–02). The study conformed to the
provisions of the Declaration of Helsinki.
Cell culture
The CNE2 cell line was obtained from the Cancer
Centre, Sun Yat-sen University (21). The cell culture medium used was
RPMI-1640 (Guangzhou Land Bio, China) supplemented with 5% fetal
calf serum (HyClone, USA). The cells were cultured in a
water-saturated atmosphere under 5% CO2 at 37°C.
Conversely, we separated tumour tissues from 20 NPC patients and
performed a primary culture in vitro. Keratinocyte-SFM
medium (Gibco, USA) was used to culture these primary human NPC
cells. Features of the growth pattern of primary human NPC cells
were observed. Flow cytometry was performed to quantify the
expression of CD133 (Miltenyi, Germany) on the surface of NPC
cells. The test was repeated 3 times.
CD133 cell sorting using immunomagnetic
beads
A single cell suspension of ~1×108 CNE2
cells was used for cell sorting. Cells were incubated with CD133/l
immunomagnetic beads (Miltenyi) for 30 min at 4°C. For magnetic
separation, a MACS cell separation column was used to retain the
positive cells linked with the beads. The CD133+ cells
obtained from the column were centrifuged and resuspended in
RPMl-1640 (with 5% inactivated foetal calf serum) or serum-free
medium. The purity of CD133+ and CD133− cells
was evaluated by standard flow cytometric analysis.
CD133+ and CD133− cells were harvested, and
their characteristics were determined for activities of
proliferation, sphere formation and differentiation.
Immunofluorescence
Slides with CD133+ and CD133−
cells were immersed in PBS for 5 min, and then were permeabilised
with 0.1% Triton X-100 for 10 min. After washing with PBS 3 times
for 5 min, the CD133+ and CD133− cells were
blocked with 5% BSA (HyClone, USA) at 37°C for 30 min. The cells
were then incubated with primary antibodies, including Oct3/4
(Santa Cruz Biotechnology, Inc. Santa Cruz, CA, USA), Nanog
(Abgent, USA) and Sox2 (Abcam, UK), in 5% BSA at 4°C for 16 h. The
cells were then incubated with fluorescent secondary antibodies
diluted with 1% BSA at 37°C for 60 min. Finally, the cell nuclei
were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Qiyun
Biotechnology, Co., Ltd., China) (1:200), and the slides were
visualised using fluorescence microscopy (IX71; Olympus,
Japan).
Proliferation assay
The proliferation of the cells was detected using an
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay on days 1, 3, 5 and 7. Three groups (including
CD133+, CD133− cells and unsorted cells) were
plated onto 96-well plates (2,000 cells in 0.2 ml of cell culture
medium/well). A blank well containing medium alone was used as the
control. The cells were then incubated with MTT (5 mg/ml; Sigma,
USA) in 20 μl at 4 h before the collection. The culture medium was
finally removed, and 150 μl DMSO was added into the well. After
shaking thoroughly for 10 min, the plates were read for the
absorbance in an enzyme immunoassay instrument at 570 nm. Six wells
were analysed for each group.
Sphere formation
For subculturing of suspended cell spheres,
CD133+ cells and CD133− cells
(1×103/ml) were cultured in 6-well plates in serum-free
DMEM-F12 medium with bFGF (10 ng/ml), EGF (20 ng/ml), B27 (10
μl/ml) and insulin (5 μg/ml). The cells were cultured under
conditions of 5% CO2 at 37°C for 3 days, and the culture
medium was replaced every other day.
Differentiation assay
CD133+ cells obtained by immunomagnetic
bead sorting were cultured in 6-well plates. On days 0, 3, 10, 15
and 21, the percentages of CD133+ cells were further
determined by flow cytometry. The experiment was repeated 3 times,
and the average values were calculated.
Cell cycle phase distribution
A total of 1×106 CD133+ and
CD133− cells were centrifuged at 1,000 rpm for 5 min,
resuspended in 0.2 ml PBS and then fixed in l ml of 70% ethanol at
4°C for 16 h. After washing with PBS, the cells were then incubated
with 300 μl of DNA dye at room temperature for 30 min. Cell cycle
status was assessed by flow cytometry (Elite; Beckman-Coulter,
USA). The relative proportions of cells in the G0/G1, S and G2/M
phases were analysed, and the percentages of cells in each phase
were calculated. The cell proliferation index (Pla) of each subset
was calculated according to the following equation: Pla = 100 × (S
+ G2/M)/(G0/Gl + S + G2/M).
Tumourigenicity in animals
Twelve male 4- to 6-week-old nude BALB/c mice from
the Animal Centre at Sun Yat-sen University were used. Six mice
received a subcutaneous inoculum of 1×104
CD133+ cells in the upper right dorsum, and the other
six mice received a transplantation of 1×105
CD133+ cells. CD133− cells with a similar
cell density (either 1×104 or 1×105) were
subcutaneously inoculated into the upper left dorsum of the mice.
At 4 weeks after the transplantation, the xenograft tumours were
collected and fixed in 4% paraformaldehyde for H&E staining or
were incubated in explant tissue cultures with 3 ml of
keratinocyte-SFM medium (Gibco) for cell culture. Finally, the
expression of CD133 in the xenograft tumour cells was assessed by
flow cytometry.
CD133+ in primary human NPC
cells
For the CD133+ cells isolated from
primary human NPC cells, the expression, proliferation,
differentiation and tumourigenicity of the CD133 cells were
assessed as described above. Immunohistochemical analyses using a
rat cytokeratin (CK) primary antibody against human cells and
keratin monoclonal antibodies (Wuhan Boster Biological Technology,
Ltd., Wuhan, China) were performed to identify the source of the
cells.
Statistical analysis
The statistical software SPSS11.0 was applied for
data processing. Measured data are expressed as the mean ± SD.
Independent sample t-test was used for performing comparisons
between two groups. One-way ANOVA with Tamhane was used for
comparisons among more than two groups. A P-value of 0.05 was
considered to indicate a statistically significant result. The
Chi-square test was used to compare the relative tumourigenicity of
the 2 tumour cell subsets in the nude mice.
Results
CD133+ cells in CNE2
cells
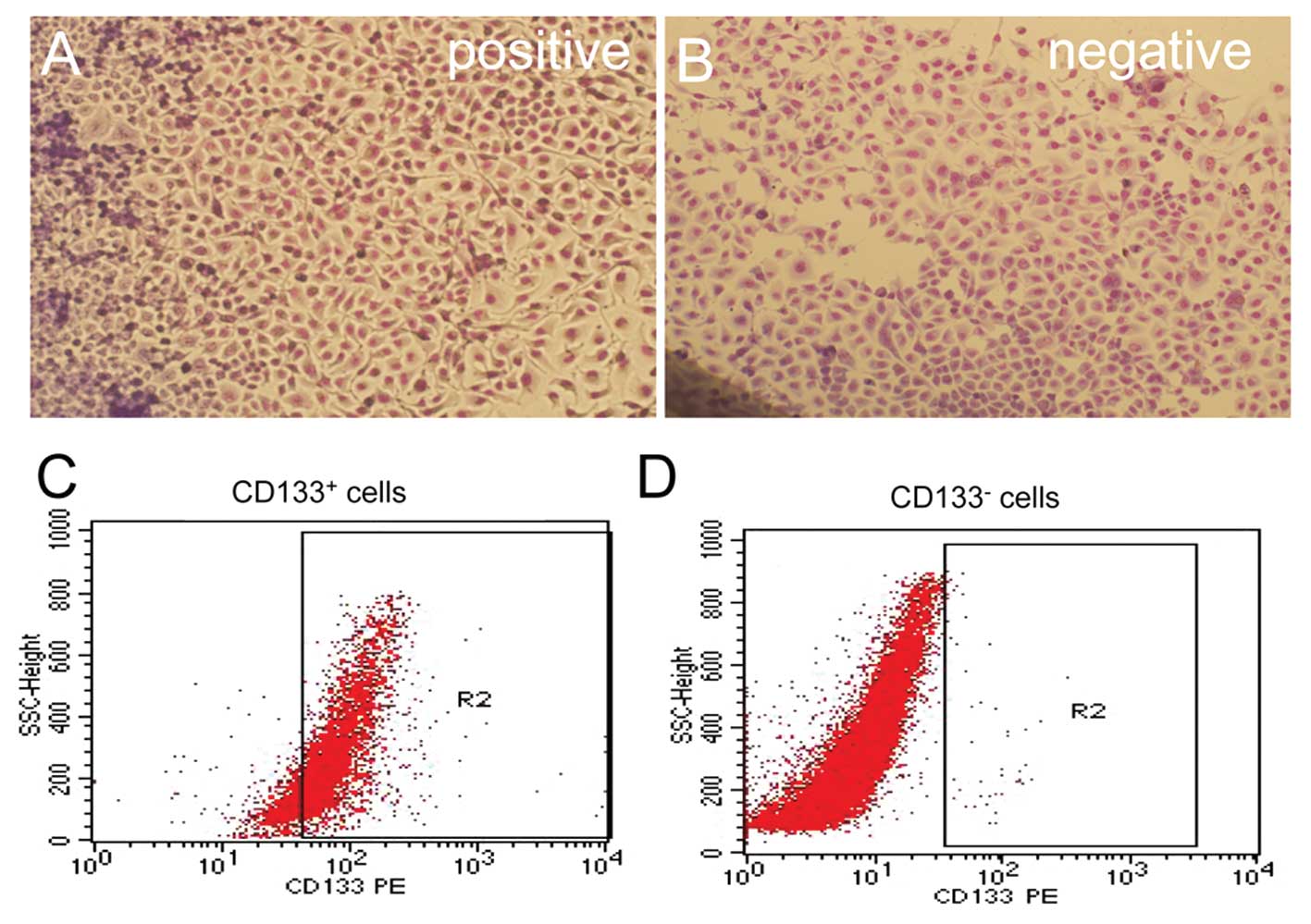
Three independent measurements for the flow
cytometric analysis detected CD133 expression rates of 3.23, 3.75
and 3.09%, with an average of 3.36±0.35%. After sorting,
CD133+ and CD133− tumour cells were cultured
in RPMI-1640 medium containing 5% fetal calf serum. The
CD133+ cells grew faster than the CD133−
cells. There were no significant differences for Giemsa staining
between the 2 subsets of cells. After immunomagnetic sorting with
CD133 beads, the cells were analysed by flow cytometry. Three
measurements for CD133 expression in the CD133+-sorted
population indicated a high CD133+ expression rate of
89.15±7.80%. However, the CD133+ expression rate in the
CD133−-sorted population was only 0.23±0.04% (Fig. 1).
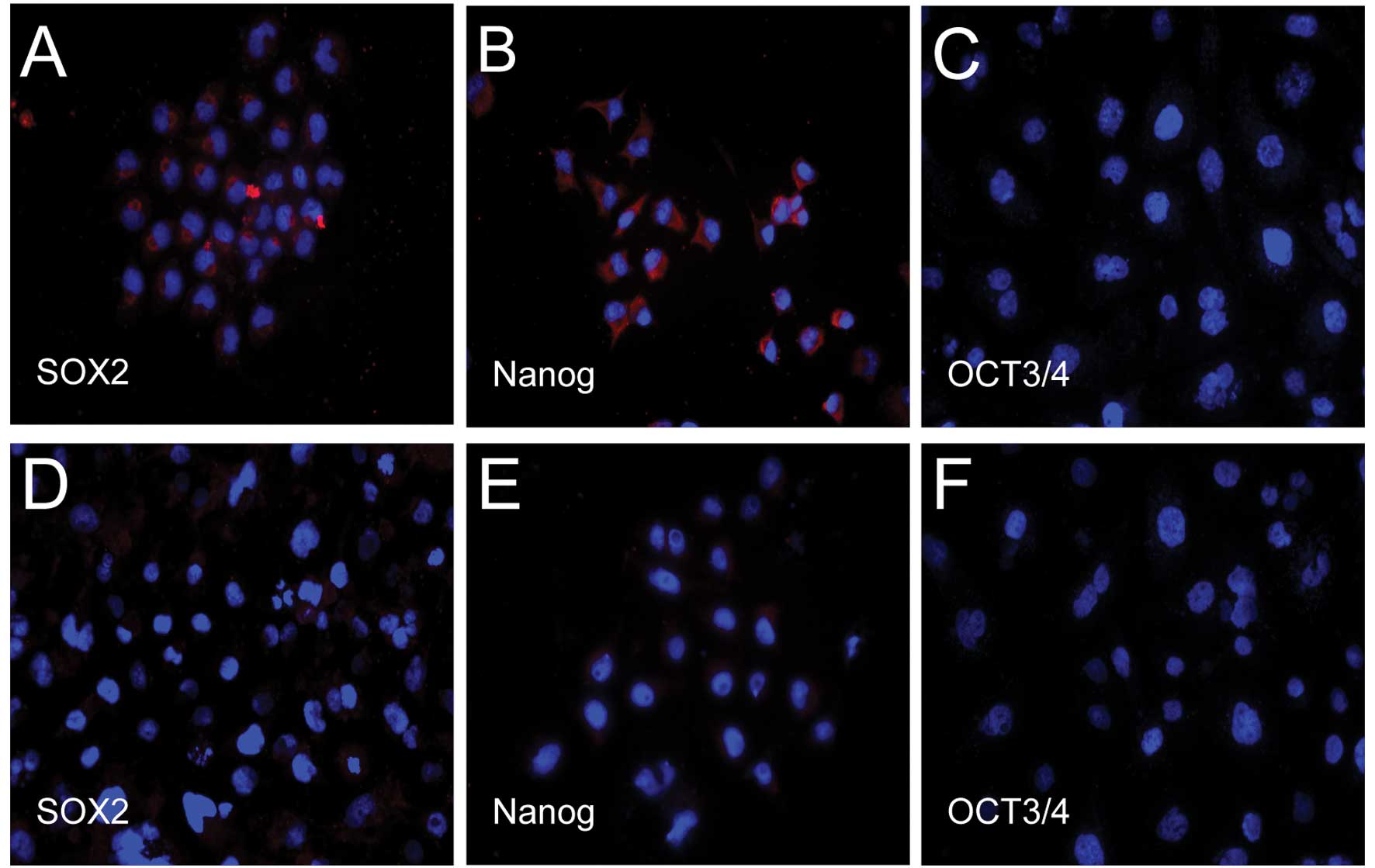
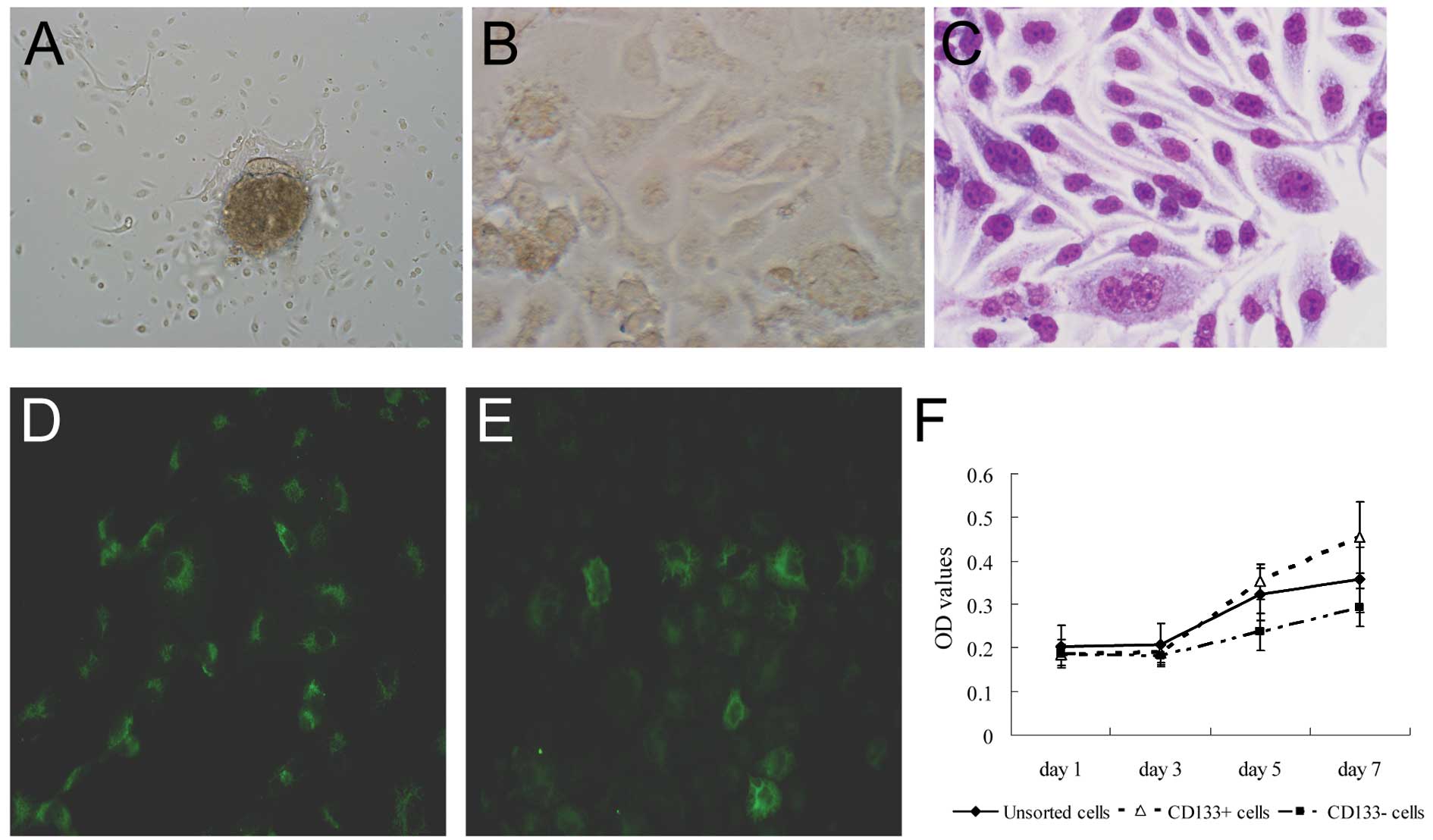
Expression of cellular markers as
detected by immunofluorescence
We found that both transcription factors, Nanog and
Sox2, the main regulators of human embryonic stem cell pluripotency
and self-renewal capacities, but not Oct3/4, were expressed in
CD133+ cells. However, only weak expression of Nanog and
Sox2, but no expression of Oct3/4, was observed in the
CD133− cells (Fig.
2).
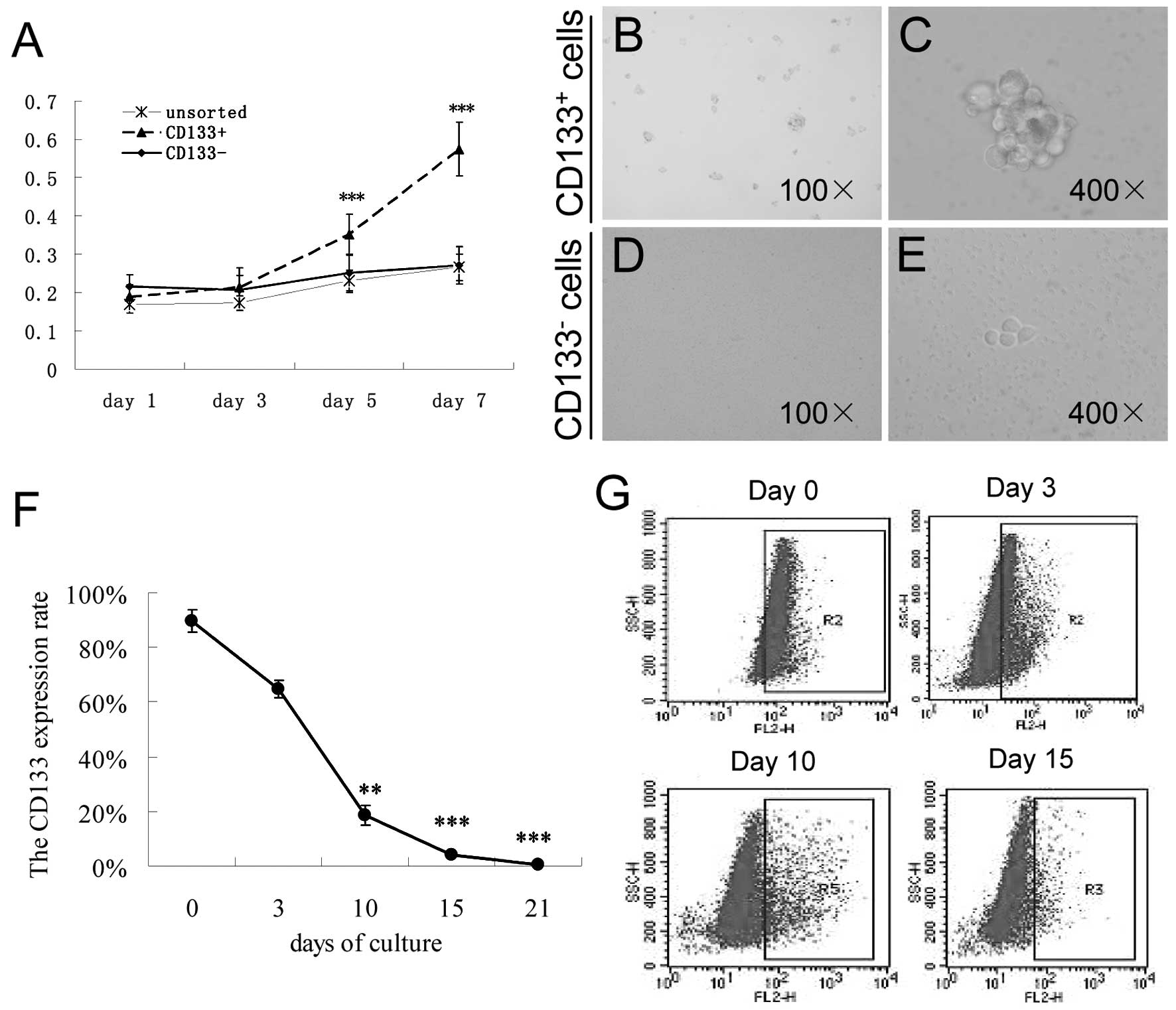
Proliferation assay
CD133+ cells exhibited a high
proliferation rate when compared with the rate in the
CD133− and unsorted cells on days 5 (0.351±0.012) and 7
(0.573±0.0165) (both P<0.001), but not on days 1 and 3 (Fig. 3A).
Suspension cell culture
The CD133+ and CD133− cells
obtained after immunomagnetic cell sorting were harvested and
cultured in serum-free culture medium containing various growth
factors. After 3 days, many individual cells in the
CD133+ cell culture were observed to survive and
proliferate in suspension. These cells gradually formed sphere
colonies with different sizes and irregular shapes. However, most
of the cells finally died in the same serum-free medium in the
CD133− cell culture. Only a few CD133− cells
adhered to the wall and grew slowly; additionally, no clear sphere
colony was found (Fig. 3B-E).
Differentiation of CD133+
cells
Using flow cytometry, we found that CD133 expression
decreased from 89.15±7.80% on day 0 to 18.4±3.7% on day 10
(P<0.01), 4±0.45% on day 15 (P<0.001) and 0.37±0.10% on day
21 (P<0.001) in the culture system (Fig. 3F and G). This finding indicates that
CD133+ cells have the potential to differentiate into
other types of cells.
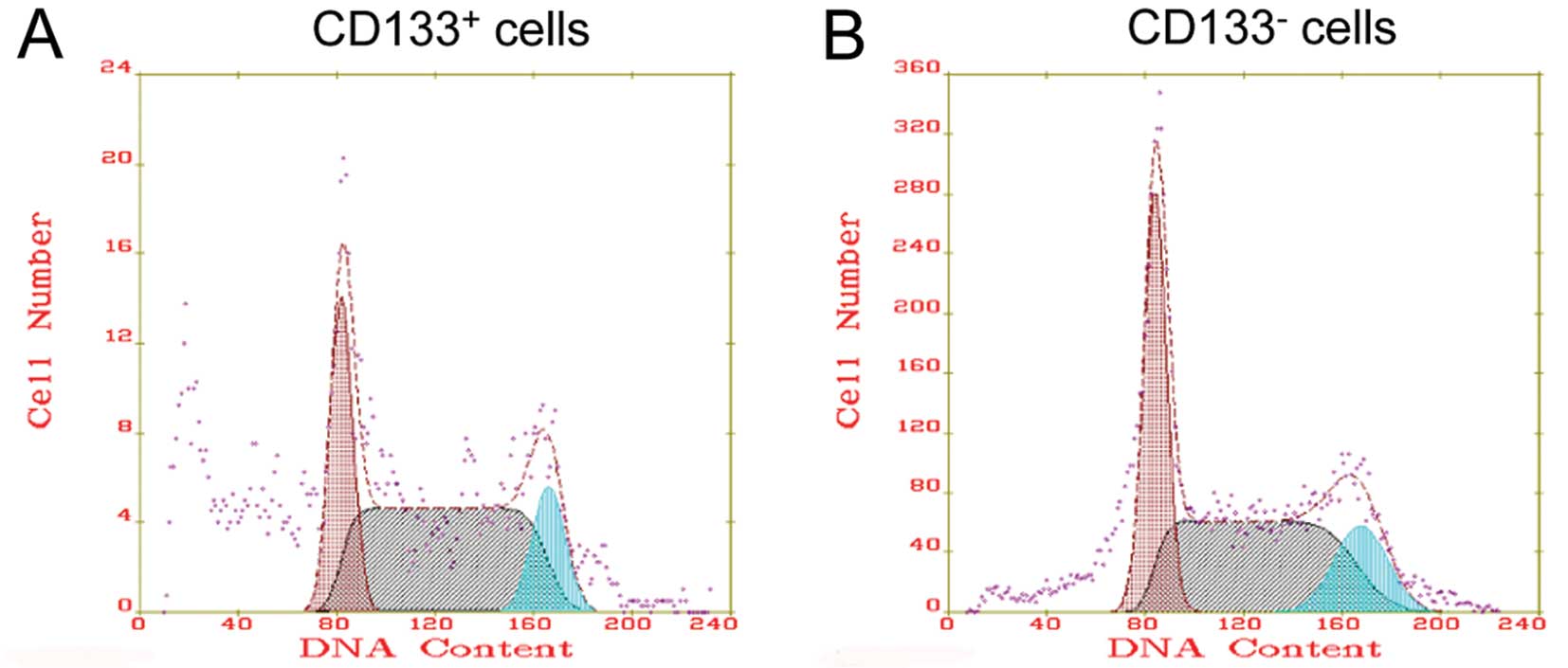
Cell cycle distribution
We examined the cell cycle distribution and cell
proliferation index for the CD133+ and CD133−
cells. There were 38.97±11.76% cells in the G0/G1 phase and
42.2±17.46% cells in the S phase for the CD133+ cells
(Fig. 4A). There was no significant
difference in the cell cycle distribution between the
CD133+ and CD133− cells (Fig. 4B).
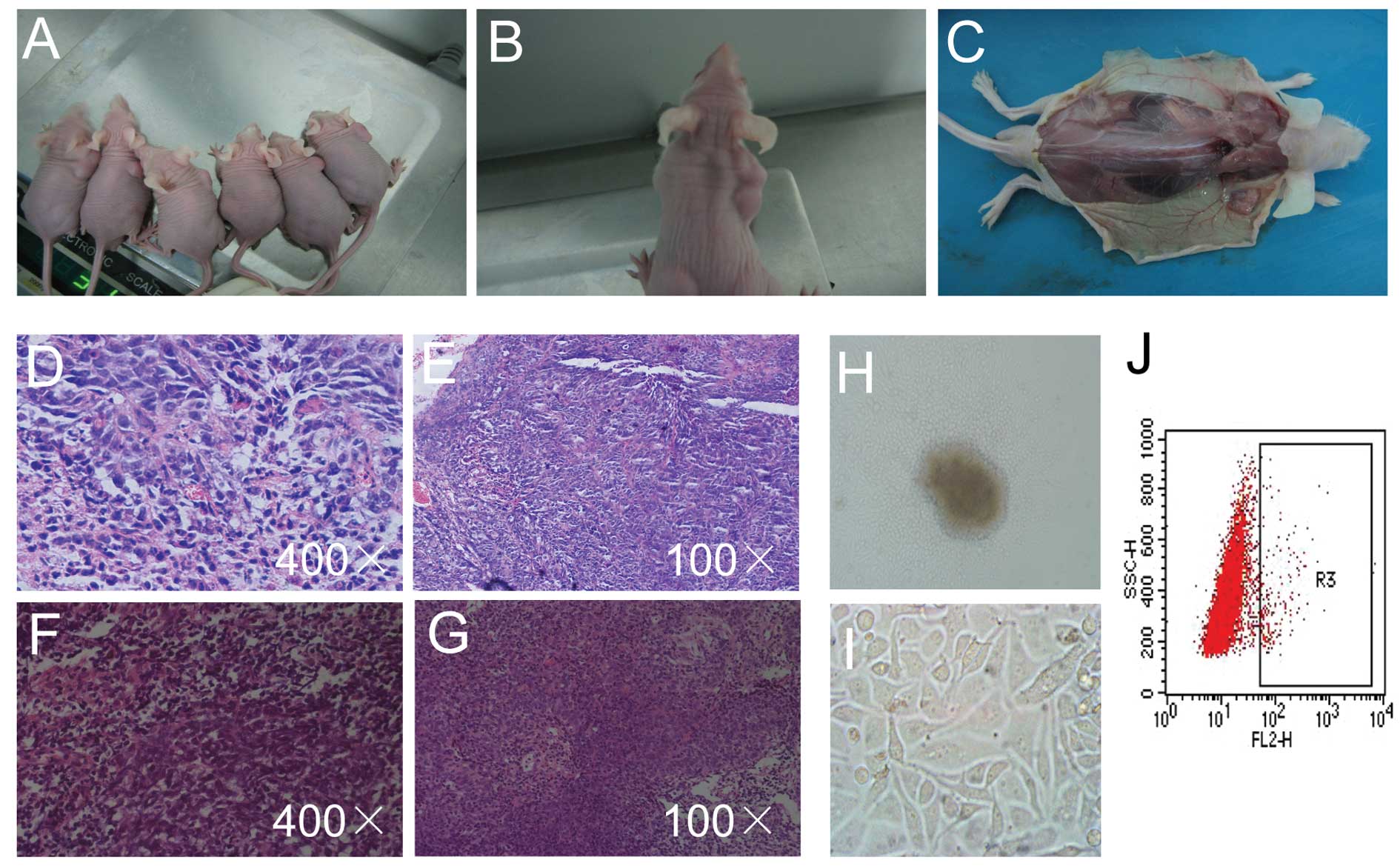
Tumourigenic potential in mice
No tumour formation was observed in mice that
received a xenograft of 1×104 cells regardless of CD133
expression status. Tumour masses were found in 5 of 6 nude mice at
6–9 days after transplantation with 1×105
CD133+ cells. The tumours grew to >1 cm in diameter
in 4 of 6 nude mice at 30 days, and necrosis was found in the
centre of the tumour masses. However, no tumour mass was found
after the transplantation of 1×105 CD133−
cells. The difference was statistically significant (P<0.05)
(Fig. 5A-C).
We further identified that the pathologic morphology
of the xenograft tumours in the nude mice was consistent with that
of undifferentiated and non-keratinised human nasopharyngeal
tissue. Similar to human NPC tissues, the xenografts exhibited a
specific morphology with large nuclei and dark staining. They
exhibited similar nuclear atypia and mitotic frequency. We found
low differentiation, abundant blood vessels and some necrosis in
the centres of the tumours (Fig.
5D-G). In the 5 primary cell cultures, 3 samples successfully
survived and proliferated to passage. The tumour cells slowly grew
out from the centre of the tissues with large nuclei and little
cytoplasm at day 3. The cells grew as irregular polygons to fused
large cells similar to that of CNE2 cells from an initial spindle
shape. After passaging, the xenograft tumour cells took ~4 days to
reach confluence in a 25-cm2 flask, a period that was
approximately twice as long as that for the CNE2 cell line
(Fig. 5H and I). There were
2.17±0.46% CD133+ cells in the xenograft tumour cells
(Fig. 5J).
Characterisation of the primary human NPC
cells
Nine of 20 primary human NPC cell samples
successfully survived and proliferated to passage. Under culture in
keratinocyte-SFM medium, the tumour cells grew out from the centre
of the primary tissue and adhered to the dish exhibiting an
irregular polygon shape on day 3. Four samples had no passage, and
4 samples had 2–3 cell propagations. One clone with 6 propagations
was used for subsequent experiments (Fig. 6A-C). The cells demonstrated
expression of epithelial markers of CK (Fig. 6D-F). The expression rate of membrane
CD133 was 2.8±0.17% for 3 independent experiments. Additionally, we
found that the CD133+ cells displayed a higher
proliferative capacity when compared with that of the
CD133− cells on days 5 and 7 (P<0.05) and compared
with that of the unsorted cells on day 7 (P<0.05). Furthermore,
the unsorted cells displayed a higher proliferative capacity when
compared with the CD133− cells on days 5 and 7
(P<0.05) (Table I and Fig. 6F).
 | Table IUltraviolet absorption of
CD133+, CD133− and unsorted tumour cells in
primary NPC cells. |
Table I
Ultraviolet absorption of
CD133+, CD133− and unsorted tumour cells in
primary NPC cells.
|
CD133+ |
CD133− | Unsorted | F-value | P-value |
|---|
| Day 1 | 0.184±0.025 | 0.187±0.032 | 0.202±0.048 | 1.388 | 0.259 |
| Day 3 | 0.190±0.023 | 0.183±0.022 | 0.207±0.049 | 2.342 | 0.106 |
| Day 5 | 0.352±0.041 | 0.237±0.043 | 0.323±0.059 | 27.650 | 0.000 |
| Day 17 | 0.454±0.083 | 0.293±0.044 | 0.357±0.075 | 24.606 | 0.000 |
Discussion
The cancer stem cell theory has provided a novel and
reasonable explanation for the mechanism of the occurrence and
recurrence of nasopharyngeal carcinoma. In recent years, the
presence of nasopharyngeal cancer stem cells has been reported in
the literature. Approximately 0.3% of the tumour cells in nude mice
with human NPC xenograft tumours were label-retaining stem cells
(22). Wang et al(21) reported side population cells (SP
cells) in NPC cell lines with high tumourigenic ability and some
chemotherapy tolerance. However, ideal surface markers for stem
cells in nasopharyngeal carcinoma remain unidentified. Our study
identified that CNE2 cells contain a small population of
CD133+ cells with a strong potential for self-renewal,
proliferation and differentiation, suggesting that CD133 represents
a marker of NPC stem cells.
In the present study, CNE2 cells stably expressed
the CD133 antigen with an expression rate of 3.36%. In particular,
CD133+ cells, but not CD133− cells, expressed
high levels of both Nanog and Sox2, marker genes of normal human
stem cells. Previous research has shown that various cancer stem
cells share consistent differentiation markers with normal stem
cells. The CD34+CD38− surface antigen
phenotype is not only specific to tumour stem cells in acute
myeloid leukaemia but also to human hematopoietic stem cells
(2). CD133 and nestin have been
demonstrated to exhibit identical expression levels in both glioma
cancer stem cells and normal neural stem cells (23). Our results suggest that CD133 may be
an ideal candidate surface marker for cancer stem cells in
nasopharyngeal carcinoma. CD133+ cells presented a
significantly greater proliferative capacity than CD133−
cells. More importantly, injection of 1×105
CD133+ cells, but not CD133− cells, resulted
in a tumourigenicity rate of 83.3%. Pathological staining of the
xenograft tumours and primary tumour cultures demonstrated that the
tissues of the xenograft tumours were morphologically similar to
the tissues of the pathological tumours of human nasopharyngeal
carcinoma. Although culturing primary human NPC tissues was very
difficult, we successfully cultured one clone of NPC stem cells in
6 NPC clones. Similarly to the results from the CNE2 cell line, the
CD133+ cells from primary human tissues displayed a
higher proliferative capacity compared with CD133− and
unsorted cells. Our findings suggest that CD133+ cells
present a higher proliferative capacity. When assessing the
differentiation capacity of purified CD133+ NPC cells,
we found that purified CD133+ NPC cells rapidly lost the
expression of CD133 after differentiation. This finding indicates
that most of the CD133+ NPC cells differentiate to
tumour cells except for a few that maintain the activity of cancer
stem cells.
It has been reported that most in vivo cancer
stem cells are in a quiescent state (G0 phase) (24). However, we found no difference in
the distribution of the G0/G1, S and G2/M phases between the
CD133+ and CD133− cells, perhaps because of
changes in the micro-environment (25). From the moment of separation,
significant changes were noted in the micro-environment (niche) of
the cells that caused the cancer stem cells to rapidly enter the
cell cycle to restore the normal proportion of non-cancer stem
cells (26). A previous report
concerning the HEP-2 tumour cell line reported that
CD133+ cells exhibiting a high capacity for self-renewal
and strong in vivo tumourigenicity have a cell cycle
distribution similar to that of unsorted cells (27). These data suggest that some types of
cancer stem cells may be in a quiescent state.
In summary, using cell lines and primary cancer
cells, we found that CD133 may be a useful surface marker for
cancer stem cells of nasopharyngeal carcinoma. CD133+
cells possess a strong potential for self-renewal, proliferation
and differentiation and exhibit high tumourigenicity.
Acknowledgements
This study was supported by the Natural Science
Foundation of Guangdong Province (grant no. 10251008901000023).
References
|
1
|
Marotta LL and Polyak K: Cancer stem
cells: a model in the making. Curr Opin Genet Dev. 19:44–50. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bonnet D and Dick JE: Human acute myeloid
leukemia is organized as a hierarchy that originates from a
primitive hematopoietic cell. Nat Med. 3:730–737. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Al-Hajj M, Wicha MS, Benito-Hernandez A,
Morrison SJ and Clarke MF: Prospective identification of
tumorigenic breast cancer cells. Proc Natl Acad Sci USA.
100:3983–3988. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Singh SK, Clarke ID, Terasaki M, et al:
Identification of a cancer stem cell in human brain tumors. Cancer
Res. 63:5821–5828. 2003.PubMed/NCBI
|
|
5
|
Fang D, Nguyen TK, Leishear K, et al: A
tumorigenic subpopulation with stem cell properties in melanomas.
Cancer Res. 65:9328–9337. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Collins AT, Berry PA, Hyde C, Stower MJ
and Maitland NJ: Prospective identification of tumorigenic prostate
cancer stem cells. Cancer Res. 65:10946–10951. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li C, Heidt DG, Dalerba P, et al:
Identification of pancreatic cancer stem cells. Cancer Res.
67:1030–1037. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
O'Brien CA, Pollett A, Gallinger S and
Dick JE: A human colon cancer cell capable of initiating tumour
growth in immunodeficient mice. Nature. 445:106–110. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Baba T, Convery PA, Matsumura N, et al:
Epigenetic regulation of CD133 and tumorigenicity of
CD133+ovarian cancer cells. Oncogene. 28:209–218. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma S, Chan KW, Hu L, et al: Identification
and characterization of tumorigenic liver cancer stem/progenitor
cells. Gastroenterology. 132:2542–2556. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim CF, Jackson EL, Woolfenden AE, et al:
Identification of bronchioalveolar stem cells in normal lung and
lung cancer. Cell. 121:823–835. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shmelkov SV, St Clair R, Lyden D and Rafii
S: AC133/CD133/Prominin-1. Int J Biochem Cell Biol. 37:715–719.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Klonisch T, Wiechec E, Hombach-Klonisch S,
et al: Cancer stem cell markers in common cancers - therapeutic
implications. Trends Mol Med. 14:450–460. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Galli R, Binda E, Orfanelli U, et al:
Isolation and characterization of tumorigenic, stem-like neural
precursors from human glioblastoma. Cancer Res. 64:7011–7021. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bao S, Wu Q, McLendon RE, et al: Glioma
stem cells promote radioresistance by preferential activation of
the DNA damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Richardson GD, Robson CN, Lang SH, Neal
DE, Maitland NJ and Collins AT: CD133, a novel marker for human
prostatic epithelial stem cells. J Cell Sci. 117:3539–3545. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yin S, Li J, Hu C, Chen X, et al: CD133
positive hepatocellular carcinoma cells possess high capacity for
tumorigenicity. Int J Cancer. 120:1444–1450. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Frank NY, Margaryan A, Huang Y, et al:
ABCG5-mediated doxorubicin transport and chemoresistance in human
malignant melanoma. Cancer Res. 65:4320–4333. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hermann PC, Huber SL, Herrler T, et al:
Distinct populations of cancer stem cells determine tumor growth
and metastatic activity in human pancreatic cancer. Cell Stem Cell.
1:313–323. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tirino V, Desiderio V, d'Aquino R, et al:
Detection and characterization of CD133+cancer stem
cells in human solid tumours. PLoS One. 3:e34692008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang J, Guo LP, Chen LZ, Zeng YX and Lu
SH: Identification of cancer stem cell-like side population cells
in human nasopharyngeal carcinoma cell line. Cancer Res.
67:3716–3724. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang HB, Ren CP, Yang XY, et al:
Identification of label-retaining cells in nasopharyngeal epithelia
and nasopharyngeal carcinoma tissues. Histochem Cell Biol.
127:347–354. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang EH, Heidt DG, Li CW and Simeone DM:
Cancer stem cells: a new paradigm for understanding tumor
progression and therapeutic resistance. Surgery. 141:415–419. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Soltysova A, Altanerova V and Altaner C:
Cancer stem cells. Neoplasma. 52:435–440. 2005.
|
|
25
|
Li L and Neaves WB: Normal stem cells and
cancer stem cells: the niche matters. Cancer Res. 66:4553–4557.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Postovit LM, Margaryan NV, Seftor EA, et
al: Human embryonic stem cell microenvironment suppresses the
tumorigenic phenotype of aggressive cancer cells. Proc Natl Acad
Sci USA. 105:4329–4334. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wei XD, Zhou L, Cheng L, Tian J, Jiang JJ
and Maccallum J: In vivo investigation of CD133 as a putative
marker of cancer stem cells in Hep-2 cell line. Head Neck.
31:94–101. 2009. View Article : Google Scholar : PubMed/NCBI
|