Introduction
Noonan syndrome (NS) is an autosomal dominant
disorder, mainly characterized by proportionate short stature,
facial and muscoloskeletal dysmorphisms and congenital heart
defects (most commonly pulmonary valve stenosis and hypertrophic
cardiomiopathy) with an incidence rate between 1:1,000 and 1:2,500
live births (1). Further findings
may include cryptorchidism, bleeding diathesis, lymphatic dysplasia
and mild to moderate developmental delay/intellectual disability
(DD/ID). A myeloproliferative disorder (NS/MPD) can occasionally be
diagnosed in infants with NS. The clinical course of NS/MPD is
usually benign with spontaneous remission. However, various cases
with an aggressive course resembling juvenile myelomonocytic
leukemia (JMML) have been described (2–6). JMML
is a rare clonal myelodysplastic-myeloproliferative disorder
typical of infancy and early childhood, characterized by
spontaneous in vitro proliferation of bone marrow and
peripheral blood hematopoietic progenitors in the absence of
exogenous growth factors, due to selective hypersensitivity to
granulocyte-macrophage colony-stimulating factor (GM-CSF) (7). Hepatosplenomegaly, lymphoadenopathy,
anemia, thrombocytopenia, and fever, variably associated with
symptoms of non-hematopoietic organ infiltration, are common
clinical findings in JMML. The fulfillment of the following
laboratory criteria is required for JMML diagnosis: an absolute
monocyte count >1,000/μl, <20% bone marrow blasts and the
absence of t (9;22) or BCR/ABL rearrangement. Apart from such
mandatory criteria, JMML patients may present with a high white
blood cell count (>10,000/μl), immature myeloid precursors on a
peripheral smear and increased fetal hemoglobin (HbF) for age.
Monosomy 7 is quite frequently noted (8).
Both NS and JMML are characterized by
hyperactivation of the RAS/MAPK signaling pathway. In NS, germline
missense mutations in genes such as PTPN11, KRAS, SOS1, RAF1, BRAF,
SHOC2, NRAS and CBL (9–19) have been documented. In JMML,
mutually exclusive somatic mutations of PTPN11, KRAS, NRAS and NF1
genes can be found in ~75% of cases. NS is associated with germline
PTPN11 mutations in ~50% of the patients, while somatic PTPN11
mutations are found in 35% of children with JMML. This gene encodes
for the ubiquitously expressed non-receptor protein tyrosine
phosphatase (PTP) SHP-2, which is implicated in a variety of
intracellular signaling cascades mediated by growth factors,
cytokines, hormones and cell adhesion molecules (20,21).
SHP-2 is also involved in several developmental processes, in
particular semi-lunar valvulogenesis (22) and hematopoietic cell differentiation
(23,24). PTPN11 mutations favor either the
basal activity or the response to inducing events of the
catalytically active conformation of SHP-2, resulting in gain of
function (11). The PTPN11
mutational spectrum has been shown to be different in JMML, NS/MPD
and NS without any hematological abnormalities (5).
We previously demonstrated that flow cytometric
evaluation of the absolute count of peripheral blood (PB)
CD34+ cells and the apoptotic rate is a simple and
repeatable technique, useful for early detection of clonal
evolution in acquired aplastic anemia. In children with de
novo or secondary refractory anemia with blast excess (RAEB) we
observed a typical pattern of a high PB CD34+ count
associated with a low apoptotic rate, sometimes evident months
before the appearance of the complete clinical image (25).
In the present study, we performed a functional
evaluation of the circulating hematopoietic progenitors in a series
of NS patients. Clonogenic tests in the absence or in the presence
of increasing concentrations of GM-CSF and three-color flow
cytometric analysis for CD45, CD34, and Annexin V were performed
using the PB of 27 patients with NS and 5 patients with JMML. The
different functional patterns were compared to identify a possible
NS subgroup worthy of stringent hematological follow-up for an
increased risk of MPD development.
Materials and methods
Patients
Included in the present study were 27 patients
admitted to Department of Pediatrics with a clinical diagnosis of
NS (independently of their mutational status and the presence of
hematological anomalies). Three patients had a myeloproliferative
disorder (NS/MPD), with monocytosis, atypical monocytoid cells,
myelodysplastic features and granulocyte precursors in PB,
thrombocytopenia and hepato-splenomegaly (2–6). Five
patients with a diagnosis of JMML, fulfilling the EWOG-MDS criteria
(8) were also studied. PB samples
were collected in EDTA at diagnosis. Further blood samples were
collected and analyzed in NS patients when hematological anomalies
(e.g. anemia, thrombocytopenia, leukocytosis, splenomegaly,
lymphadenopathy) and/or alterations of the functional pattern of
circulating hematopoietic progenitors were observed. NS/MPD and
JMML patients were evaluated at various stages during follow-up and
treatment. Ethics committee approval and informed consent of the
parents of the patients were obtained.
Genomic mutational analysis
Genomic DNA was isolated from 200 μl of PB by the
QIAamp DNA Blood Mini kit (Qiagen, Germantown, MD, USA). A
molecular analysis of PTPN11, KRAS, SOS1, RAF1, BRAF, SHOC2, NRAS
and CBL was performed as previously described (11–20).
Absolute count of CD34+ cells
and the apoptotic index
Flow cytometric analysis was performed within 2 h
after venipuncture. The absolute count of CD34+ cells
and the apoptotic rate were evaluated by a three-color fluorescence
for CD45, CD34 and Annexin V as follows. A total of
5×105 nucleated cells were incubated for 20 min at 4°C
with anti-CD34 PE (8G12; Becton-Dickinson, San José, CA, USA) and
anti-CD45 PerCP (2D1; BD Biosciences, Franklin Lakes, NJ, USA).
After incubation and red cell lysis by ammonium chloride, the
samples were washed in cold phosphate-buffered saline (PBS) and
incubated with Annexin V-fluorescein isothiocyanate (FITC)
(Apoptosis Detection kit; R&D Systems, Minneapolis, MN, USA),
according to the manufacturer’s instructions. The cells were then
analyzed in a Coulter Epics XL2 (IL, Bedford, MA, USA) cytometer
equipped with an argon laser. CD34+ cells were
identified by a sequential gating strategy according to the ISHAGE
protocol (26). Absolute CD34
counts were assessed by a two-platform method with a Sysmex K4500
counter (Sysmex Corporation, Kobe, Japan). At least 100
CD34+ cells were evaluated in each experiment.
Cell cultures
Low-density mononuclear cells (2×105)
obtained from the patient PB by density centrifugation over
Ficoll-Hypaque gradient were plated in multi well plates in 250 μl
Iscove’s modified Dulbecco’s medium (IMDM) containing 30% fetal
calf serum (FCS) (both from Sigma-Aldrich, St. Louis, MO, USA),
0.3% noble agar and 100 U/ml rhIL-3 and decreasing concentrations
(20, 10, 5, 1, 0.1 ng/ml) of rhGM-CSF (both from Invitrogen Life
Technologies, Carlsbad, CA, USA). After 14 days, single aggregates
of >40 cells were scored as CFU-GMs. GM-CFU assay was also
performed without GM-CSF stimulation. GM-CFU assay in the same
culture conditions was also performed in 21 pediatric controls
(median age, 9.0; range, 1–18 years).
Statistical analysis
The patients were divided into 3 groups: NS, NS/MPD
and JMML. Historical controls (n=68) from our laboratory were
utilized for the absolute count of CD34+ in PB and the
apoptotic rate (25).
For the absolute count of CD34+ cells and
apoptotic rate, the values for each patient were compared with the
mean control value adjusted for age, as previously published
(25).
The cell culture data and the absolute count of
CD34+ cells and the apoptotic rate results were analyzed
using the non-parametric Kruskal-Wallis test. Pairwise comparisons
for the disease groups were performed for each of the GM-CSF
concentration utilized in the cell cultures, as well as for the 4
variables analyzed in the test (absolute CD34+,
percentage of CD34+/Annexin V+, absolute
CD34+ to the mean age group value ratio, percentage of
CD34+/Annexin V+ to the mean age group value
ratio). Fisher’s exact probability test was utilized for
correlations between the groups.
Results
Mutational spectrum of the patients
The mutational spectrum of our series of NS, NS/MPD
and JMML patients is shown in Table
I.
 | Table IMutational spectrum, WBC, PLT and
monocyte counts, CD34+ absolute count and apoptotic
rate, CFU-GM from peripheral blood in a series of NS, NS/MPD and
JMML patients. |
Table I
Mutational spectrum, WBC, PLT and
monocyte counts, CD34+ absolute count and apoptotic
rate, CFU-GM from peripheral blood in a series of NS, NS/MPD and
JMML patients.
| Patient | Mutated gene | Mutation | Hypersensitivity to
GM-CSF | Unstimulated colony
growth | Annexin
V+/CD34+ (%) | CD34 (/μl) | WBC
(103/μl) | Monocytes
(103/μl) | PLT
(103/μl) |
|---|
| NS1 | PTPN11 | Gln79Arg | No | No | 8.5 | L | 3.6 | N | 11.9 | 0.740 | 284 |
| NS2 | PTPN11 | Asn58His | No | No | 26.1 | H | 7 | N | 11.6 | 0.400 | 262 |
| NS3 | PTPN11 | Tyr63Cys | No | No | 16.1 | N | 2.5 | L | 8.3 | 0.300 | 428 |
| NS4 | PTPN11 | Gly503Arg | No | No | 2.56 | L | 3.42 | L | 17.1 | 1.720 | 331 |
| NS5 | PTPN11 | Gly503Glu | No | No | 7.6 | L | 3.6 | N | 7.3 | 0.900 | 249 |
| NS6 | PTPN11 | Asn308Ser | No | No | 12 | L | 6.2 | H | 10.3 | 0.690 | 297 |
| NS7 | PTPN11 | Leu261His | No | No | 10 | L | 2.9 | N | 5.8 | 0.470 | 416 |
| NS8 | PTPN11 | Leu261His | No | No | 0 | L | 4.8 | N | 12.0 | 0.770 | 376 |
| NS9 | PTPN11 | Leu261His | No | No | 10 | L | 17.52 | H | 7.3 | 0.600 | 277 |
| NS10 | PTPN11 | Phe285Ile | No | No | 7.9 | L | 3.8 | N | 7.7 | 0.410 | 218 |
| NS11 | PTPN11 | Glu76Asp | Yes | Yes | 1.0 | L | 12 | H | 12.0 | 1.140 | 300 |
| NS12 | PTPN11 | Glu139Asp | No | No | 17.8 | L | 2.2 | L | 7.3 | 0.510 | 198 |
| NS13 | PTPN11 | Asp61Asn | Yes | No | 4.1 | L | 13.8 | H | 15.3 | 0.840 | 212 |
| NS14 | SOS | Ile252Thr | No | No | 27.7 | H | 2.6 | L | 6.4 | 0.450 | 259 |
| NS15 | SOS | Thr266Lys | No | No | 8.8 | L | 16.2 | H | 12.5 | 0.620 | 291 |
| NS16 | SOS | Arg552Gly | No | No | 18.2 | L | 1.3 | L | 4.3 | 0.340 | 259 |
| NS17 | SOS | Glu433Lys | No | No | 0 | L | 7.75 | N | 15.5 | 1.420 | 422 |
| NS18 | RAF1 | Ser257Leu | No | No | 8 | L | 2.5 | L | 6.4 | 0.540 | 253 |
| NS19 | SOS | Met269Thr | Yes | No | 4.4 | L | 5.2 | N | 8.7 | 1.100 | 182 |
| NS20 | n.d. | n.d. | No | No | 11.2 | N | 4.5 | L | 12.2 | 1.110 | 184 |
| NS21 | KRAS | Gln22Arg | No | No | 15.7 | L | 5.6 | N | 9.44 | 0.630 | 551 |
| NS22 | BRAF | Leu597Val | No | No | 0.09 | L | 7.1 | H | 7.9 | 1.600 | 397 |
| NS23 | RAF1 | Pro261Ser | No | No | 25 | H | 5 | N | 12.6 | 1.450 | 384 |
| NS24 | SHOC2 | Ser2Gly | No | No | 6.8 | L | 8.5 | N | 17.0 | 0.700 | 482 |
| NS/MPD1 | PTPN11 | Phe285Ser | Yes | Yes | 2.4 | L | 58.0 | H | 14.5 | 2.500 | 399 |
| NS/MPD2 | PTPN11 | Asp61Asn | Yes | Yes | 0.2 | L | 1,374 | H | 43.5 | 5.520 | 54 |
| NS/MPD3 | PTPN11 | Asp61Asn | Yes | No | 1.6 | L | 205.7 | H | 20.2 | 2.550 | 167 |
| JMML1 | n.d. | n.d. | Yes | Yes | 2.1 | L | 193.6 | H | 9.3 | 1.900 | 55 |
| JMML2 | PTPN11 | Glu76Gly | Yes | Yes | 12.1 | N | 44 | H | 8.8 | 1.380 | 31 |
| JMML3 | PTPN11 | Gly503Val | Yes | No | 7.2 | L | 49.6 | H | 3.1 | 1.070 | 45 |
| JMML4 | NF1 | n.d. | Yes | Yes | 0.4 | L | 109.8 | H | 11.2 | 1.430 | 15 |
| JMML5 | n.m. | n.d. | Yes | Yes | 0.4 | L | 232 | H | 36.2 | 7.610 | 71 |
Monocyte counts
All JMML patients showed monocytosis >1,000/μl.
Ten out of the 27 NS patients showed monocytosis >1,000/μl,
which included the 3 NS/MPD patients (Tables I and II).
| Table IICirculating monocytes, peripheral
blood CD34+ cells, their apoptotic rate and CFU-GMs in a
series of NS, NS/MPD and JMML patients. |
Table II
Circulating monocytes, peripheral
blood CD34+ cells, their apoptotic rate and CFU-GMs in a
series of NS, NS/MPD and JMML patients.
| Clinical
parameters | Controls | NS | NS/MPD | JMML |
|---|
| Monocytes
(/μl) | 600 (200–900) | 695
(300–1,720) | 2,550
(2,500–5,520) | 1,600
(1,070–7,600) |
| CD34+
(/μl) | 5.2 (1.8–23.1) | 4.9 (1.3–17.5) | 205.7
(58–1,374) | 109.8 (44–232) |
| Annexin
V+/CD34+ (%) | 17.6
(2.8–49.6) | 8.6 (0.0–27.7) | 1.4 (0.2–2.4) | 2.1 (0.4–12.1) |
| GM-CSF (20
ng/ml) | 9 (0–26) | 4 (0–34) | 36 (0–84) | 46 (18–258) |
| GM-CSF (10
ng/ml) | 5 (0–18) | 2 (0–30) | 38 (0–76) | 40 (10–244) |
| GM-CSF (5
ng/ml) | 3 (0–18) | 1 (0–24) | 38 (0–50) | 42 (6–262) |
| GM-CSF (1
ng/ml) | 1 (0–16) | 1 (0–23) | 34 (0–62) | 32 (6–264) |
| GM-CSF (0.1
ng/ml) | 0 (0–14) | 0 (0–8) | 26 (0–36) | 30 (2–240) |
| Unstimulated | 0 (0–0) | 0 (0–4) | 4 (0–32) | 18 (0–84) |
Platelet counts
All JMML patients showed thrombocytopenia as well as
1 out of the 3 NS/MPD patients. In the other NS patients, the
platelet counts were in the range of normality, without a
correlation with monocyte counts (R=0.016) (Table I).
Absolute CD34+ cell count and
apoptotic rate
The PB absolute CD34+ cell counts and
apoptotic rates in the different groups of patients are shown in
Tables I and II. In JMML and NS/MPD patients, we
observed high levels of circulating CD34+ cells with a
low apoptotic rate. In NS patients, CD34+ cell counts
were normal, whereas their apoptotic rate was significantly lower
than that in the controls (p<0.01). Concerning the absolute
CD34+cell count, statistically significant differences
were noted among the NS and JMML (p=0.001), NS and NS/MPD
(p<0.05), controls and JMML (p<0.01), and controls and NS/MPD
patients (p<0.05). In contrast, no differences in the absolute
CD34+ cell count were observed between the controls and
NS or between the NS/MPD and JMML patients. Normalizing the
absolute CD34+cell count for age (absolute
CD34+ to mean age group value ratio), the results from
the pairwise comparison did not change.
A pairwise comparison of the Annexin V+
percentage showed a statistically significant decrease in the
apoptotic rate in each disease group when compared with the
controls: NS (p<0.01), NS/MPD (p<0.05), JMML(p<0.01);
whereas no significant difference was observed between NS/MPD and
JMML, JMML and NS, and NS/MPD and NS patients. When the percentage
of Annexin V+ cells was normalized for age (percentage
of Annexin V+/CD34+ cells to mean age group
value ratio), the results for the pairwise comparison did not
change.
Cell cultures
In the JMML patients, the clonogenic assays from PB
showed hypersensitivity to GM-CSF and spontaneous CFU-GM growth
(except in one patient previously treated with chemotherapy at
another center who did not show spontaneous CFU-GM growth)
(Table I). In 3 out of 3 NS/MPD
patients we observed hypersensitivity to GM-CSF (in patient NS/MPD3
during the follow-up) and in 2 out of 3 spontaneous CFU-GM growth
was noted. In 2 NS patients (NS13 and NS19) we observed
hypersensitivity to GM-CSF, and no spontaneous CFU-GM growth. One
NS patient (NS11) showed hypersensitivity to GM-CSF and spontaneous
CFU-GM growth. This patient had a favorable clinical outcome, and
the clonogenic assays performed 6 months later showed normal
results.
We observed a significant difference in the
distribution and in the median values of CFU-GM among the 3 groups
(JMML, NS, NS/MPD) (Table II).
Table III (pairwise comparisons
of GM-CSF-stimulated clonogenic assays) showed that at different
GM-CSF concentrations, JMML patients were significantly more
responsive to GM-CSF than both the controls and NS patients.
Concerning unstimulated cultures, a significant growth advantage
was observed also in NS/MPD when compared with the controls and NS
patients (Table III).
Specifically, clonogenic assays without GM-CSF were able to
distinguish between NS and NS/MPD patients. Fisher’s exact
probability test showed a significant correlation between the
groups in the unstimulated colony growth tests (0% in controls,
4.2% in NS, 80% in JMML, 66.7% in NS/MPD, p<0.001).
| Table IIIPairwise comparisons of the
GM-CSF-stimulated clonogenic assay in the disease groups. |
Table III
Pairwise comparisons of the
GM-CSF-stimulated clonogenic assay in the disease groups.
| Pair | GM-CSF (20
ng/ml) | GM-CSF (10
ng/ml0 | GM-CSF (5
ng/ml) | GM-CSF (1
ng/ml) | GM-CSF (0.1
ng/ml) | Unstimulated |
|---|
| Controls-NS | 0.248 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
|
Controls-NS/MPD | 1.000 | 1.000 | 1.000 | 1.000 | 0.460 | 0.010 |
| Controls-JMML | 0.079 | 0.038 | 0.022 | 0.010 | 0.007 | 0.000 |
| NS-JMML | 0.001 | 0.002 | 0.004 | 0.004 | 0.003 | 0.000 |
| NS-NS/MPD | 0.671 | 0.808 | 0.903 | 0.786 | 0.674 | 0.016 |
| NS/MPD-JMML | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
Follow-up of NS and NS/MPD patients
Six NS patients showed isolated monocytosis
>1,000/μl, 2 NS patients showed isolated hyper-responses to
GM-CSF, 5 NS patients showed an isolated increase in circulating
CD34+ cells. In all of these patients a 12-month
follow-up showed no alterations in clinical findings. Patients NS11
and NS/MPD1 showed monocytosis >1,000/μl, a hyper-response to
GM-CSF, CFU-GM growth without GM-CSF stimulation, high circulating
CD34+ counts with a low apoptotic rate. In these 2 NS
patients a clinical and laboratory follow-up was performed. Patient
NS11 carrying the Glu76Asp PTPN11 mutation, was phenotypically
characterized by polyhydramnios in the prenatal history, typical
facial dysmorphisms, pulmonic stenosis, bilateral cryptorchidism
and normal neuropsychomotor development. His clinical follow-up in
the following months was normal, and a laboratory hematologic
evaluation performed 12 months later showed normal response to
GM-CSF, no spontaneous colony growth and normal CD34+
cell and monocyte counts.
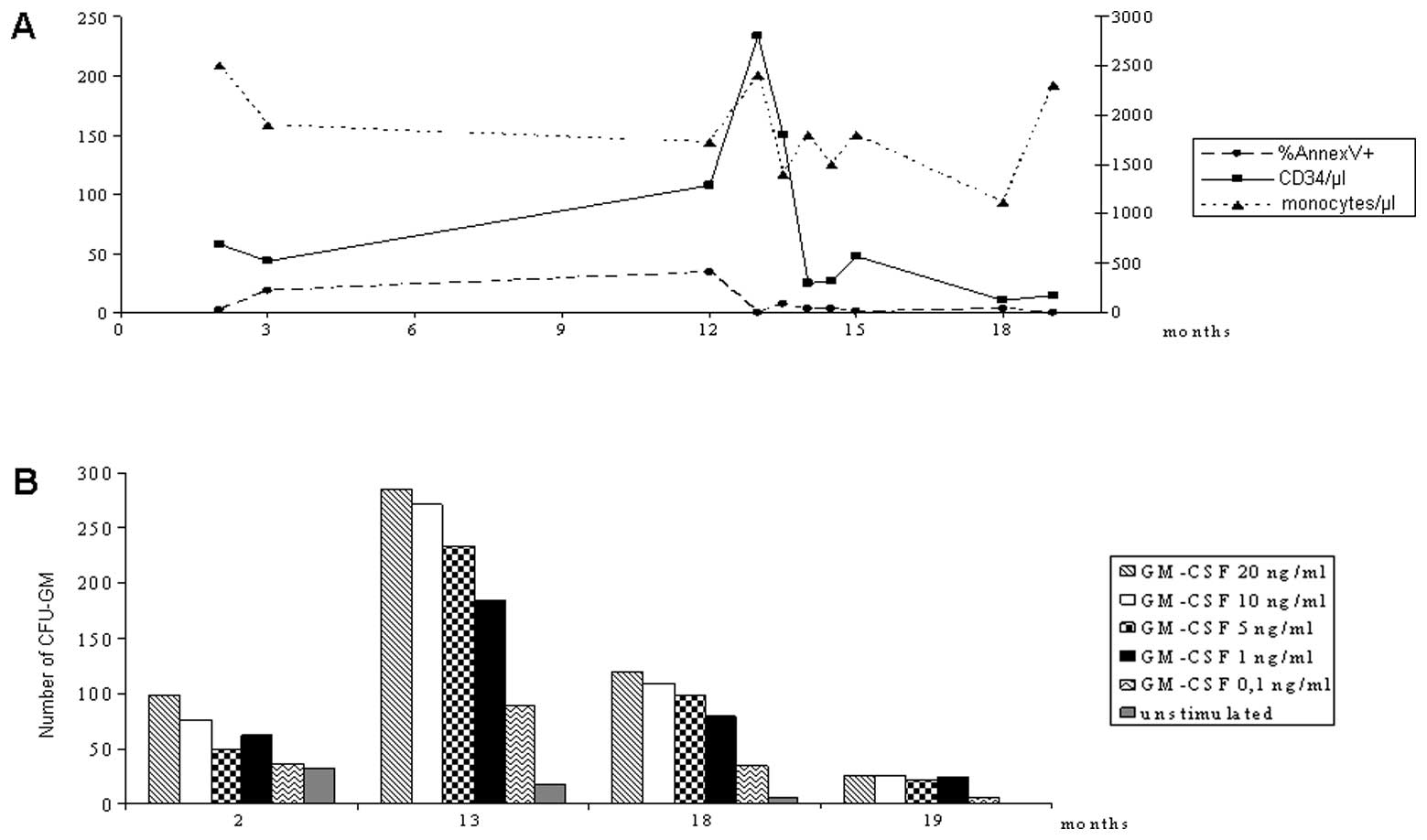
Patient NS/MPD1 was first evaluated at the age of 2
months while she was in the cardiac surgery unit due to obstructive
hypertrophic cardiomyopathy. A clinical diagnosis of NS was
confirmed by the molecular analysis of the PTPN11 gene, that
revealed the Phe285Ser mutation. A preliminary hematological
evaluation evidenced only mild hepatomegaly and monocytosis.
Clonogenic assays and flow cytometry showed hypersensitivity to
GM-CSF, spontaneous CFU-GM growth, increased circulating
CD34+ cells with a low apoptotic rate. At the age of 12
months, the patient showed splenomegaly, thrombocytopenia and
monocytosis, with the presence in the peripheral blood smear of 10%
of atypical monocytoid cells, moderate myelodysplastic features and
granulocyte precursors. Bone marrow aspirate showed mild
myelodysplasia and the presence of 15% of atypical monocytoid
elements. A polymerase chain reaction (PCR) for bcr/abl
rearrangement was negative. The HUMARA assay performed on
peripheral blood populations was normal, excluding a frank JMML
evolution and suggesting a polyclonal myeloproliferative disorder,
described in NS (2). Hence, a
diagnosis of NS/MPD was made. Thrombocytopenia and splenomegaly
persisted over the following months, and the clinical course
worsened, with a progressive development of lymphatic dysplasia
with thoracic duct ectasia, progressive severe respiratory
insufficiency, pleural effusion and exitus at the age of 20 months
for acute cardiopulmonary failure.
Patients NS/MPD2 and NS/MPD3 were diagnosed in the
first month of life presenting a myeloproliferative disorder.
Table IV shows the clinical and
laboratory follow-up of the NS/MPD patients, and Fig. 1 shows a detailed follow-up of
patient NS/MPD1.
| Table IVFunctional follow-up of 3 NS/MPD
patients with myeloproliferative evolution. |
Table IV
Functional follow-up of 3 NS/MPD
patients with myeloproliferative evolution.
| NS/MPD1 | NS/MPD2 | NS/MPD3 |
|---|
|
|
|
|
|---|
| Parameters | T0 | T1 | T0 | T1 | T0 | T1 |
|---|
| Age at
T0 | 2 months | | 10 days | | 1 month | |
| WBC (/μl) | 14,500 | 8,200 | 43,500 | 8,480 | 20,200 | 16,000 |
| Monocytes
(/μl) | 2,500 | 1,400 | 5,520 | 620 | 2,550 | 1,280 |
| CD34+
(/μl) | 58 | 150 | 1,374 | 13 | 205 | 45 |
| Annexin
V+ (%) | 2.4 | 7.5 | 0.2 | 0.8 | 1.6 | 3.0 |
| Hyper-response to
GM-CSF | +++ | ++ | +++ | No | No | +++ |
| Spontaneous CFU
growth | ++ | ++ | + | No | No | No |
| Circulating
dysplastic monocytes with myeloid dysplasia | No | +++ | ++ | No | + | No |
|
Hepatosplenomegaly | No | ++ | No | No | ++ | No |
|
Thrombocytopenia | No | +++ | ++ | No | No | No |
Discussion
Genetic diseases associated with a high tumor risk
are models for the study of carcinogenesis. A close correlation is
often observed between such genetic conditions and specific
acquired neoplastic diseases. NS and JMML were shown to be strictly
correlated to each other. Indeed, the same signal transduction
pathway (RAS/MAPK) is hyperactivated both in NS and in JMML, with
an involvement of the same genes. Moreover, NS patients presented
an increased risk of developing JMML or, more frequently, a
transient myeloproliferative disorder associated with Noonan
syndrome (NS/MPD).
Strictly correlated to the hyperactivation of the
RAS/MAPK pathway is the hypersensitivity to GM-CSF observed in JMML
(7).
In the present study, we conducted a molecular study
of a cohort of NS and JMML patients with a functional evaluation of
their circulating hematopoietic progenitors, and correlated the
results with the clinical-hematological course. In particular, our
aim was to evaluate the circulating CD34+ cell count and
apoptotic rate and to relate such findings with in vitro
colony growth (in the absence and with increasing concentrations of
GM-CSF), hematological features and the clinical history of each
patient. The analyses were performed on PB, even though the
biological variability of hematopoietic progenitor counts and
clonogenic assays in PB is higher than in bone marrow. A bone
marrow aspirate would have been unethical in NS patients without
any sign of a hematological disease.
Even though a constitutional GM-CSF hypersensitivity
has been suggested in NS (6,27), we
observed hypersensitivity to GM-CSF in only 6 of the 27 NS
patients. One of them developed a myeloproliferative disorder 12
months later (NS/MPD1) and 2 patients (NS/MPD2 and NS/MPD3) had a
transient myeloproliferative syndrome at the time of the study. In
patient NS/MPD3, a hyper-response to GM-CSF was observed during the
follow-up, but not at diagnosis. The other 3 NS patients with
GM-CSF hypersensitivity had normal hematological profiles. These
data suggest that the response to GM-CSF is variable in NS
patients.
PTPN11 mutations in JMML affect amino acids
differently from those involved in NS (5,28,29).
Somatic JMML-associated mutations are predicted to result in a
stronger SHP-2 gain of function than germ-line mutations described
in NS, and the leukemic transformation in NS seems related to
cooperating molecular lesions (5).
In our NS series, one patient (NS5) carried the Gly503Glu PTPN11
mutation, also described in JMML. Interestingly, this patient
showed a normal hematological profile without hypersensitivity to
GM-CSF, but his mother, who presented with a short stature and
typical facial appearance, as revealed by anamnestic data and
family photographs, died due to non-Hodgkin lymphoma.
In a previous study, we observed in normal subjects
a progressive decrease in circulating CD34+ cells and a
progressive increase in their apoptotic rate from the first months
of life to adult age (25). An
increase in circulating CD34+ cells has been described
in myelofibrosis with myeloid metaplasia in adults (30) and in RAEB in adults (31) and in children, associated with a low
apoptotic rate (25). Here, we
observed a similar behavior in the JMML and in the NS/MPD patients.
PB CD34+ cell counts in the majority of our NS patients
were normal, but the apoptotic CD34+ cell rate was
significantly lower than that in the controls, as in JMML and
NS/MPD. Previous studies have pointed to an increased proliferative
activity of hematopoietic progenitors in NS. Our results allow us
to identify NS as a disease with a lower-than-normal apoptotic
activity of circulating hematopoietic progenitors. This increase in
the survival of hematopoietic progenitors appears to be a hallmark
of NS patients.
Two out of 3 NS/MPD patients shared the same
functional pattern of JMML, characterized by high circulating
CD34+ cell counts with a very low apoptotic rate,
hypersensitivity to GM-CSF and spontaneous CFU-GM growth.
The present data represent the complex hematopoietic
functional profile of a series of NS patients and suggest that the
evaluation of the absolute CD34+ count and the apoptotic
rate as well as CFU-GM assay performed on PB could be a
non-invasive and repeatable method that can facilitate the
identification of NS patients with a higher risk of
myeloproliferative evolution for whom an intensified hematological
follow-up program is justified.
Acknowledgements
This study was partially supported by a grant from
the ‘Comitato per lo sviluppo dell’Ospedale Infantile di
Torino-ONLUS- io sto con il Regina Margherita’ and a grant from
Ricerca Sanitaria Finalizzata-Regione Piemonte. We thank Mr. Andrew
Martin Garvey for the editorial assistance.
References
|
1
|
Nora JJ, Nora AH, Sinha AK, Spangler RD
and Lubs HA: The Ullrich-Noonan syndrome (Turner phenotype). Am J
Dis Child. 127:48–55. 1974.PubMed/NCBI
|
|
2
|
Bader-Meunier B, Tchernia G, Miélot F, et
al: Occurrence of myeloproliferative disorder in patients with
Noonan syndrome. J Pediatr. 130:885–889. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fukuda M, Horibe K, Miyajima Y, Matsumoto
K and Nagashima M: Spontaneous remission of juvenile chronic
myelomonocytic leukemia in an infant with Noonan syndrome. J
Pediatr Hematol Oncol. 19:177–179. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Choong K, Freedman MH, Chitayat D, Kelly
EN, Taylor G and Zipursky A: Juvenile myelomonocytic leukemia and
Noonan syndrome. J Pediatr Hematol Oncol. 21:523–527. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kratz CP, Niemeyer CM, Castleberry RP, et
al: The mutational spectrum of PTPN11 in juvenile myelomonocytic
leukemia and Noonan syndrome/myeloproliferative disease. Blood.
106:2183–2185. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bastida P, García-Miñaúr S, Ezquieta B,
Dapena JL and Sanchez de Toledo J: Myeloproliferative disorder in
Noonan syndrome. J Pediatr Hematol Oncol. 33:e43–e45. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Emanuel PD, Bates LJ, Zhu SW, Castleberry
RP, Gualtieri RJ and Zuckerman KS: Selective hypersensitivity to
granulocyte-macrophage colony-stimulating factor by juvenile
chronic myeloid leukemia hematopoietic progenitors. Blood.
77:925–929. 1991.
|
|
8
|
Hasle H, Niemeyer CM, Chessells JM,
Baumann I, Bennett JM, Kerndrup G and Head DR: A pediatric approach
to the WHO classification of myelodysplastic and myeloproliferative
diseases. Leukemia. 17:277–282. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tartaglia M, Mehler EL, Goldberg R, et al:
Mutations in PTPN11, encoding the protein tyrosine phospatase
SHP-2, cause Noonan syndrome. Nat Genet. 29:465–468. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tartaglia M, Kalidas K, Shaw A, et al:
PTPN11 mutations in Noonan syndrome: molecular spectrum,
genotype-phenotype correlation, and phenotypic heterogeneity. Am J
Hum Genet. 70:1555–1563. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schubbert S, Zenker M, Rowe SL, et al:
Germline KRAS mutations cause Noonan syndrome. Nat Genet.
38:331–336. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Razzaque MA, Nishizawa T, Komoike Y, et
al: Germline gain-of-function mutations in RAF1 cause Noonan
syndrome. Nat Genet. 39:1013–1017. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Roberts AE, Araki T, Swanson KD, et al:
Germline gain-of-function mutations in SOS1 cause Noonan syndrome.
Nat Genet. 39:70–74. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tartaglia M, Pennacchio LA, Zhao C, et al:
Gain-of-function SOS1 mutations cause a distinctive form of Noonan
syndrome. Nat Genet. 39:75–79. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ferrero GB, Baldassarre G, Delmonaco AG,
et al: Clinical and molecular characterization of 40 patients with
Noonan syndrome. Eur J Med Genet. 51:566–572. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cordeddu V, Di Schiavi E, Pennacchio LA,
et al: Mutation of SHOC2 promotes aberrant protein N-myristoylation
and causes Noonan-like syndrome with loose anagen hair. Nat Genet.
41:1022–1026. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sarkozy A, Carta C, Moretti S, et al:
Germline BRAF mutations in Noonan, LEOPARD, and
cardiofaciocutaneous syndromes: molecular diversity and associated
phenotypic spectrum. Hum Mutat. 30:695–702. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cirstea IC, Kutsche K, Dvorsky R, et al: A
restricted spectrum of NRAS mutations causes Noonan syndrome. Nat
Genet. 42:27–29. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Martinelli S, De Luca A, Stellacci E, et
al: Heterozygous germline mutations in the CBL tumor-suppressor
gene cause a Noonan syndrome-like phenotype. Am J Hum Genet.
87:250–257. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Alonso A, Sasin J, Bottini N, et al:
Protein tyrosine phosphatases in the human genome. Cell.
117:699–711. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Andersen JN, Jansen PG, Echwald SM, et al:
A genomic perspective on protein tyrosine phosphatases: gene
structure, pseudogenes, and genetic disease linkage. FASEB J.
18:8–30. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen B, Bronson RT, Klaman LD, et al: Mice
mutant for Egfr and Shp-2 have defective cardiac semilunar
valvulogenesis. Nat Genet. 24:296–299. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Qu CK, Yu WM, Azzarelli B, Cooper S,
Broxmeyer HE and Feng GS: Biased suppression of hematopoiesis and
multiple developmental defects in chimeric mice containing Shp-2
mutant cells. Mol Cell Biol. 18:6075–6082. 1998.PubMed/NCBI
|
|
24
|
Saxton TM, Henkemeyer M, Gasca S, et al:
Abnormal mesoderm patterning in mouse embryos mutant for the SH2
tyrosine phosphatase Shp-2. EMBO J. 16:2352–2364. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Timeus F, Crescenzio N, Doria A, et al:
Flow cytometric evaluation of circulating CD34+cell
counts and apoptotic rate in children with acquired aplastic anemia
and myelodysplasia. Exp Hematol. 33:597–604. 2005.PubMed/NCBI
|
|
26
|
Sutherland DR, Anderson L, Keeney M, Nayar
R and Chin-Yee I: The ISHAGE guidelines for CD34+cell
determination by flow cytometry. International Society of
Hematotherapy and Graft Engineering. J Hematother. 5:213–226. 1996.
View Article : Google Scholar
|
|
27
|
Lavin VA, Hamid R, Patterson J, Alford C,
Ho R and Yang E: Use of human androgen receptor gene analysis to
aid the diagnosis of JMML in female Noonan syndrome patients.
Pediatr Blood Cancer. 51:298–302. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schubbert S, Lieuw K, Rowe SL, et al:
Functional analysis of leukaemia-associated PTPN11 mutations in
primary haematopoietic cells. Blood. 106:311–317. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jongmans M, Sistermans EA, Rikken A, et
al: Genotypic and phenotypic characterization of Noonan syndrome:
new data and review of the literature. Am J Med Genet A.
134A:165–170. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Barosi G, Viarengo G, Pecci A, Rosti V,
Piaggio G, Marchetti M and Frassoni F: Diagnostic and clinical
relevance of the number of circulating CD34(+) cells in
myelofibrosis with myeloid metaplasia. Blood. 98:3249–3255. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sullivan SA, Marsden KA, Lowenthal RM,
Jupe DM and Jones ME: Circulating CD34+cells: an adverse
prognostic factor in the myelodysplastic syndromes. Am J Haematol.
39:96–101. 1992.
|