Introduction
Endometrial cancer is the eighth leading cause of
cancer-related mortality among women (1). Approximately 80% of endometrial
carcinomas are endometrioid endometrial cancers (EECs) and are
generally considered as estrogen-dependent tumors (2). EECs frequently possess mutations in
PI3K (phosphatidylinositol 3-kinase) pathway genes including
PTEN, PIK3CA, K-ras and AKT1(3–7).
Cyclin D1, encoded by the CCND1 gene, is
located on chromosome 11q13 and plays a crucial role in cell cycle
regulation (8). Cyclin D1 interacts
with cyclin-dependent-kinase 4 and 6 (CDK4/6), and the complex
phosphorylates retinoblastoma protein (pRb) to promote cell cycle
progression from G1 to S phase (9–11).
Cyclin D1 is activated by upstream inputs, including the
Wnt-β-catenin and Ras/PI3K signaling pathways (11,12).
Degradation of cyclin D1 is regulated by GSK3β, a downstream target
of the PI3K pathway. The phosphorylation of cyclin D1 at Thr-286 by
GSK3β triggers its nuclear export and cytoplasmic proteolysis via
the 26S proteasome (13). A
polymorphism of CCND1 (G/A870), encoding cyclin D1b, was
reported to influence cancer risk and prognostic outcome (14). Cyclin D1b has a different
configuration at the C-terminal domain, which includes Thr286, and
is constitutively localized to the nucleus (15,16).
Thus, this polymorphism (G/A870) is suggested to increase the risk
of endometrial cancer, possibly through nuclear accumulation of
cyclin D1 (17). Overexpression of
cyclin D1 is a common event in various types of human cancers,
including those of the breast, lung, bladder, esophagus and
endometrium (13). However,
mutations of CCND1 have only been reported in endometrial
and esophageal cancer (18,19). In the present study, we detected a
novel CCND1 mutation in endometrial cancer, and analyzed
whether this T286I mutation causes gain-of-function.
Materials and methods
Tumor samples and genomic DNA
Surgical samples were obtained from 88 patients with
primary endometrial carcinoma who underwent resection of their
tumors at the University of Tokyo Hospital. All of the patients
provided informed consent for the collection and use of their
samples, and the use of tissues for the present study was approved
by the appropriate institutional ethics committees.
Histopathologically, 80 out of 88 cases (91%) were EECs (Table I). Genomic DNA was isolated from the
tumor sections or lymphocyte pellets as previously described
(7).
 | Table IClinicopathological background of all
cases (n=88). |
Table I
Clinicopathological background of all
cases (n=88).
| Variables | No. of patients
(%) |
|---|
| Median age (range)
in years | 55 (28–79) |
| FIGO stage |
| I | 39 (44) |
| II | 9 (10) |
| III | 31 (35) |
| IV | 9 (10) |
| Histological
subtype |
| Endometrioid
adenocarcinoma | 80 (91) |
| Grade 1 | 43 (49) |
| Grade 2 | 28 (32) |
| Grade 3 | 9 (10) |
| Adenosquamous
carcinoma | 3 (3) |
| Squamous cell
carcinoma | 1 (1) |
| Clear cell
carcinoma | 1 (1) |
| Mixed type | 3 (3) |
| Lymph
metastasis | 20 (23) |
| Lymph-vascular
invasion | 39 (44) |
| Muscle invasion
(>1/3) | 53 (60) |
PCR and sequencing
The primer sequences of exon 5 for the CCND1
gene were as follows: forward 5′-TGCTGGAGTCA AGCCTGCG-3′ and
reverse 5′-ACTGTCAGGGGAGCA CCTG-3′. The PCR products were sequenced
using the BigDye (Applied Biosystems, Foster City, CA, USA)
terminator method on an autosequencer. Mutations for PTEN
(exons 1–8), K-Ras (exons 1 and 2), PIK3CA (exons 9
and 20) and CTNNB1 (exon 3) were analyzed as previously
described (3,4,7,20).
Immunohistochemistry (IHC)
Immunohistochemical staining was performed according
to standard techniques using a Ventana Benchmark XT autostainer
(Ventana Medical Systems Inc., Tucson, AZ, USA). Anti-cyclin D1
rabbit monoclonal antibody (#N161987; Dako, Carpinteria, CA, USA)
was applied at a dilution of 1:100.
Cell lines and transfection
The human embryonic kidney HEK293T cell line was
obtained from the American Type Culture Collection (ATCC; Manassas,
VA, USA). The T286I cyclin D1 mutant vector was generated using the
QuickChange Lightning Site-Directed Mutagenesis kit (Stratagene, La
Jolla, CA, USA). Transfection was performed using Effectene
transfection reagent (Qiagen, Valencia, CA, USA).
Western blotting
HEK293T cells were lysed as previously described
(3). Preparations of nuclear and
cytoplasmic extracts were constructed using NucBuster Protein
Extraction kit (Merck, Darmstad, Germany). Western blotting was
performed as per standard protocols. The expression of proteins was
examined using antibodies against cyclin D1 (ab16663; Abcam,
Cambridge, UK), HA (#2367; Cell Signaling Technology, Inc.,
Beverly, MA, USA) and p84 (ab487; Abcam). Antibody against β-actin
(A2228; Sigma, St. Louis, MO, USA) was used as an internal control.
The secondary antibodies used were NA931V and NV934V (GE
Healthcare, Sydney, Australia). All antibodies were used at the
recommended concentrations as directed by the manufacturer. The
proteins were visualized using an ECL Western Blot Detection kit
(Amersham Biosciences, Piscataway, NJ, USA).
Immunocytochemistry
Immunocytochemistry was performed as previously
described (21). The primary
antibodies used were the same as those for western blotting. The
secondary antibodies were Alexa Fluor 488-conjugated donkey
anti-mouse IgG and Alexa Fluor 555-conjugated goat anti-rabbit IgG
(Invitrogen, Carlsbad, CA, USA). The primary and secondary
antibodies were used at a 1:100 dilution. All slides were briefly
counterstained with DAPI (1 μg/ml) for 5 min for nuclear
determination and analyzed by confocal fluorescence microscopy
(Carl-Zeiss MicroImaging Inc., Oberkochen, Germany).
Luciferase assays
HEK293T cells were transfected with wild-type (WT)
cyclin D1 or T286I cyclin D1 expression plasmids (0.4 μg) together
with a reporter plasmid (pRb-TA-luc) (0.25 μg). As an internal
control, cells were co-transfected with phRL CMV-Renilla
vector (Promega, Madison, WI, USA) (0.01 ng). Luciferase activity
was detected using the Microplate Luminometer LB96V (EG&G
Breatholod, Wildbad, Germany). Each experiment was repeated three
times.
Clonogenic cell survival assays
HEK293T cells were seeded in 6-well plates. After
transfection with WT- or T286I-cyclin D1, cells were incubated for
1 week with DMEM supplemented with G418 (1,000 μg/ml) (Invitrogen).
The colonies were stained with Giemsa.
Statistical analysis
Statistical analyses of data from luciferase and
clonogenic cell survival assays were carried out using the
Student’s t-test. P<0.05 was considered to indicate a
statistically significant result.
Results
T286I CCND1 mutation in endometrial
cancer
Clinicopathological characteristics are examined and
are presented in Table I. Briefly,
sequencing of the CCND1 gene identified somatically acquired
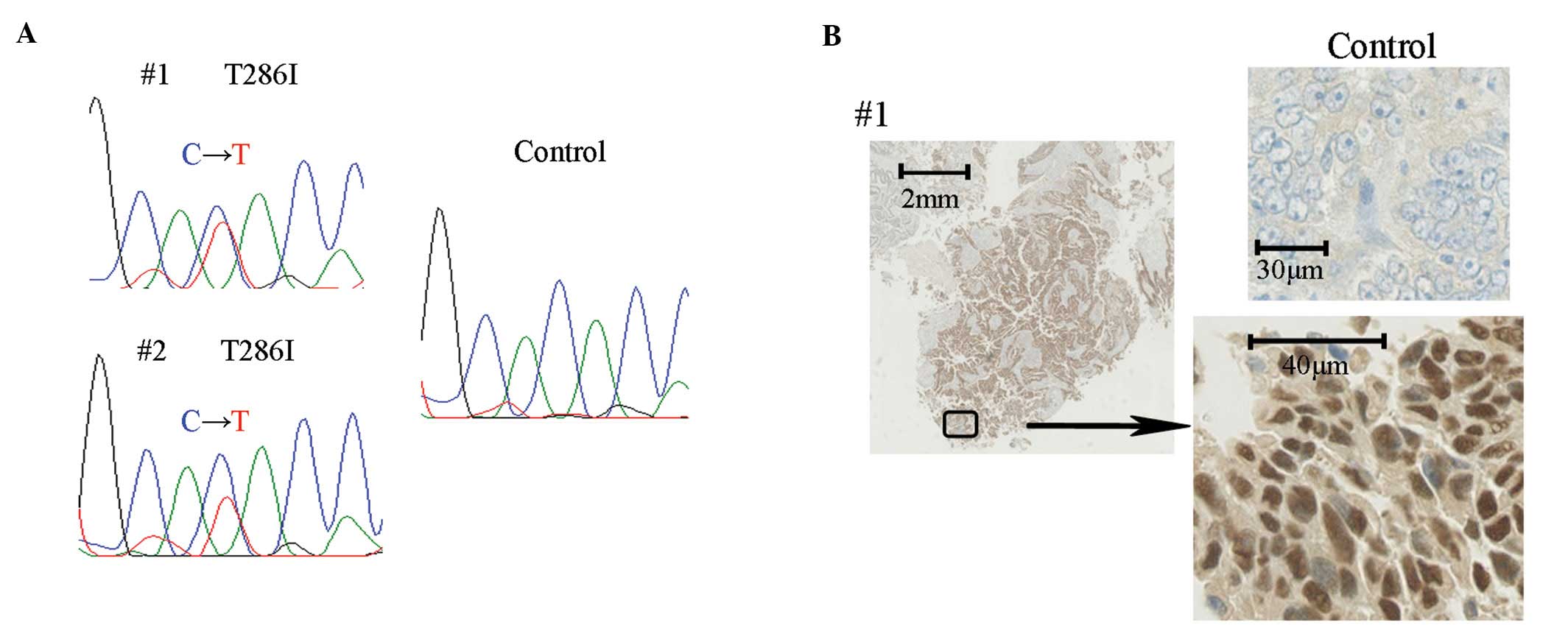
(non-germline) mutations in 2 out of 88 (2.3%) endometrial cancer
specimens (Fig. 1A). Both mutations
were a substitution of amino acid 286 from threonine to isoleucine
(T286I). These 2 patients died of the disease within 11 and 26
months of diagnosis, respectively. One case was stage IV
endometrial carcinoma (grade 3), whereas the other was mixed
adenocarcinoma with stage IIIc. Immunohistochemical staining of
these 2 tumors showed accumulation of cyclin D1 in the nucleus
(Fig. 1B).
CCND1 mutation at T286I coexists with
triple mutations of K-Ras, PIK3CA and PTEN
Mutational analysis of K-Ras, PIK3CA
and PTEN was performed in both samples with the CCND1
mutation at T286I. Case no. 1 possessed overlapping mutations in
K-Ras at G13D, in PIK3CA at H1047R and in PTEN
(there was a deletion of 19 bp at codons 135–141). Case no. 2 also
possessed these three mutations; in K-Ras at G12D, in
PIK3CA at G1007R and in PTEN at R130G and deletion of
‘A’ in codon 267 (Table II).
| Table IIClinicopathological background and
mutation status of the two cases harboring the CCND1
mutation. |
Table II
Clinicopathological background and
mutation status of the two cases harboring the CCND1
mutation.
| Case no. | Age (years) | Histology | Grade | Stage | Survival
(months) | Mutation
status |
|---|
|
|---|
| CCND1 | K-Ras | PIK3CA | PTEN |
|---|
| 1 | 52 | Endomerioid | 3 | 4 | 11 | T286I | G13D | H1047R | Del 135–141 (Del 19
bp, stop 140) |
| 2 | 55 | Mixed | 1 | 3c | 26 | T286I | G12D | G1007R | R130G, Del 267 (Del
A, stop 275) |
T286I mutation induces constitutive
nuclear cyclin D1 accumulation
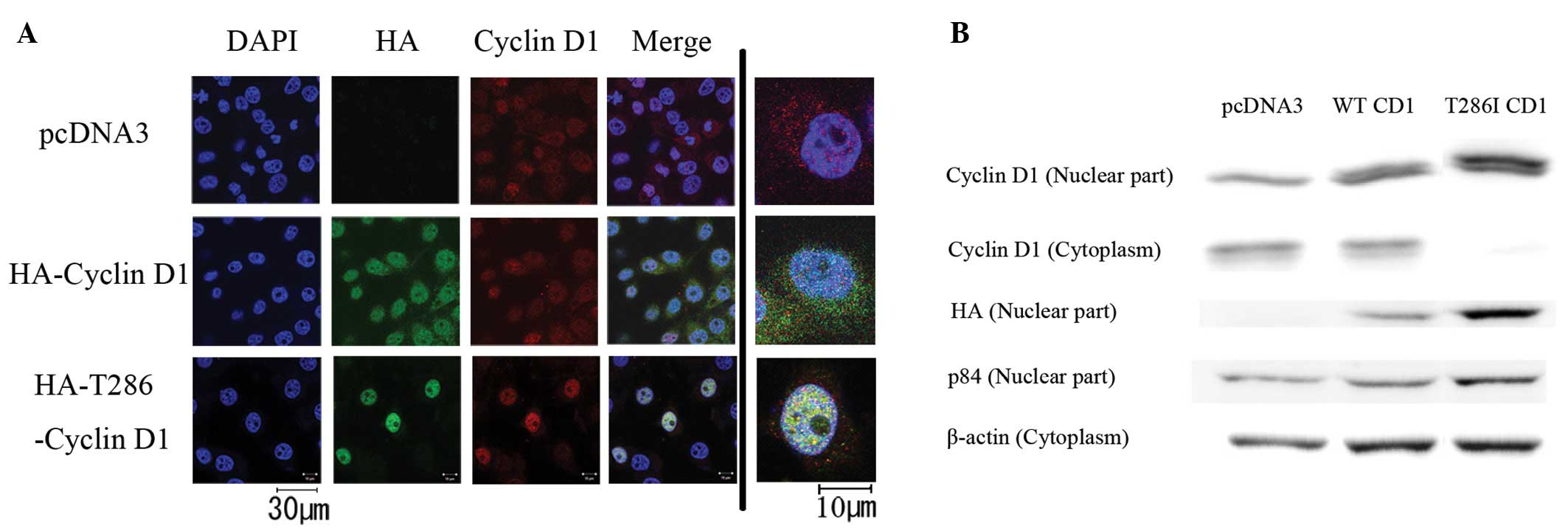
We introduced mutant cyclin D1 (T286I) into HEK293T
cells, which do not possess CCND1 mutations. We transfected
three types of pcDNA3 plasmids: pcDNA3 as a control (pcDNA3-CT),
HA-tagged WT cyclin D1 (CD1-WT) and HA-tagged T286I mutant cyclin
D1 (CD1-T286I). Immunocytochemistry indicated that endogenous
cyclin D1 was localized to both the nucleus and cytoplasm in the
pcDNA3-CT-transfected cells (Fig.
2A). Exogenous CD1-WT was more clearly expressed in the nucleus
than that in the cytoplasm. In the CD1-T286I-transfected cells,
cyclin D1 was strongly accumulated in the nucleus, and endogenous
cyclin D1 was undetectable in the cytoplasm. In western blot
experiments, both nuclear and cytoplasmic expression of cyclin D1
was observed in the pcDNA3-CT- and CD1-WT-transfected cells
(Fig. 2B). However, either
endogenous or exogenous cyclin D1 was predominantly expressed in
the nucleus in the D1-T286I- transfected cells. These results
suggest that the CCND1 mutation at T286I prevents nuclear
export of cyclin D1.
T286I mutation reduces Rb expression and
promotes cell proliferation
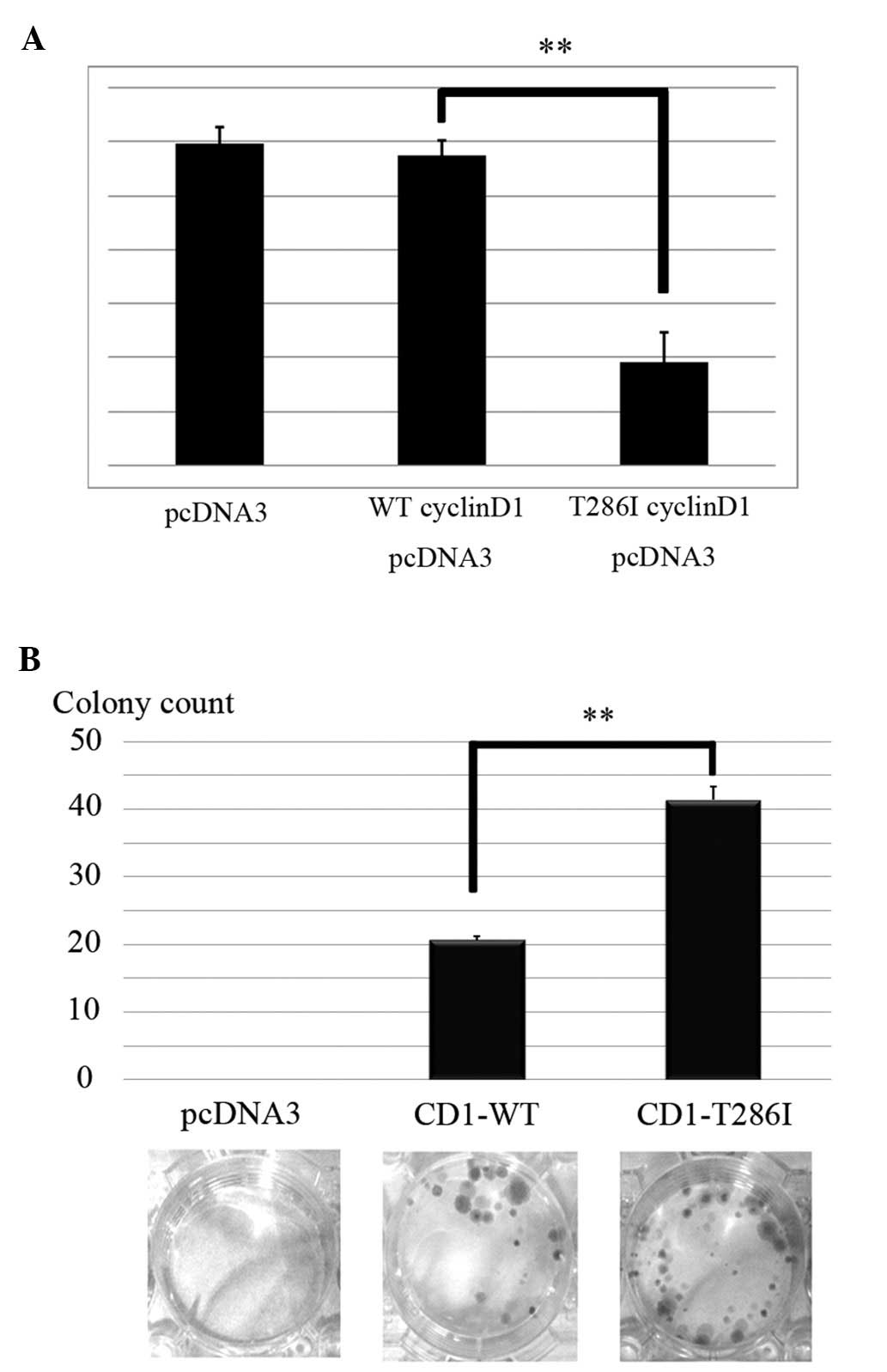
As nuclear cyclin D1 and pRb expression are
antagonistic for cell cycle progression, we transiently transfected
cyclin D1 vectors and examined pRb transcriptional activity using
luciferase assays. No significant difference was observed in pRb
transcriptional activity by introduction of CD1-WT, when compared
with pcDNA3-CT. However, CD1-T286I clearly suppressed the
transcriptional activation of pRb (P=0.002) (Fig. 3A).
Finally, to investigate the effects of T286I on cell
proliferation, we performed clonogenic cell survival assays. No
colonies were observed in the pcDNA3-CT-transfected cells, whereas
colonies were observed in the CD1-WT cells (Fig. 3B). In addition, CD1-T286I further
increased the number of colonies in HEK293T cells (P=0.007)
(Fig. 3B). These results suggest
that accumulation of mutant cyclin D1 (T286I) in the nucleus
promotes cell cycle proliferation.
Discussion
Phosphorylation-dependent nuclear export of cyclin
D1 at Thr286 is suggested to be critical for prevention of aberrant
cell proliferation in vitro(22). This is the first report of
CCND1 mutations at Thr286 in endometrial cancer. We
identified this mutation (T286I) in two tumors (2.3%) in
association with accumulation of cyclin D1 protein in the
nucleus.
Previously, Moreno-Bueno et al(19) reported the existence of CCND1
mutations at P287T, P287S and a 12 bp in-frame deletion (289–292)
in endometrial cancer. Benzeno et al(18) showed that these mutations
specifically disrupt phosphorylation-dependent nuclear export of
cyclin D1 and contribute to the genesis and progression of
neoplastic growth. However, the function of the CCND1
mutation at T286I (CD1-T286I) has not been well characterized. Our
results showed that CD1-T286I prevents the nuclear export of cyclin
D1 and significantly promotes cell proliferation. These findings
suggest that this CCND1 (T286I) mutation might promote tumor
progression in certain endometrial carcinomas.
The functions of the CCND1 mutation in
vivo were reported by Gladden et al(23). They showed that transgenic mice
expressing mutant cyclin D1 (T286A) induced oncogenic nuclear
retention of cyclin D1. This report is compatible with our data
that a missense mutation at Thr286 disrupts nuclear export of
cyclin D1. The clinical outcome of patients with cyclin D1
mutations were not reported in previous studies (18,19).
However, cyclin D1 overexpression is frequently observed in various
types of cancers, and the prognostic impact of the overexpression
was distinct among tumor types. Cyclin D1 overexpression is
suggested to be induced by chromosomal translocation, gene
amplification and protein stabilization (24,25).
The prognosis of patients with cyclin D1 overexpression is poor in
ovarian cancer (26) and favorable
in colorectal cancer (27). These
contradictory results may be caused by additional genetic
alterations in each cancer type. Indeed, expression of wild-type
cyclin D1 in murine fibroblasts or lymphocytes does not promote
neoplastic transformation without co-expression of a cooperating
oncogene such as Myc or Ras (28–31).
In the present study, both patients with mutant cyclin D1 (T286I)
had poor prognosis as they harbored coexisting triple mutations of
K-Ras, PTEN and PIK3CA. Although the sample
size was extremely small, our data may suggest that activation of
both Ras/MAPK and PI3K pathways cooperate with mutant cyclin D1 and
promote tumor aggressiveness in certain endometrial carcinomas.
Further study is warranted to clarify the significance of
CCND1 mutations in endometrial cancer.
Acknowledgements
We thank Keiko Shoji, Yuichiro Miyamoto, Yoko
Matsumoto, Takahide Arimoto, Kensuke Tomio, Satoko Kojima and Reiko
Kurikawa for their support and assistance.
References
|
1
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar
|
|
2
|
McMeekin DS, Filiaci VL, Thigpen JT,
Gallion HH, Fleming GF and Rodgers WH: The relationship between
histology and outcome in advanced and recurrent endometrial cancer
patients participating in first-line chemotherapy trials: a
Gynecologic Oncology Group study. Gynecol Oncol. 106:16–22. 2007.
View Article : Google Scholar
|
|
3
|
Oda K, Stokoe D, Taketani Y and McCormick
F: High frequency of coexistent mutations of PIK3CA and
PTEN genes in endometrial carcinoma. Cancer Res.
65:10669–10673. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oda K, Okada J, Timmerman L, et al: PIK3CA
cooperates with other phosphatidylinositol 3′-kinase pathway
mutations to effect oncogenic transformation. Cancer Res.
68:8127–8136. 2008.
|
|
5
|
Shoji K, Oda K, Nakagawa S, et al: The
oncogenic mutation in the pleckstrin homology domain of AKT1 in
endometrial carcinomas. Br J Cancer. 101:145–148. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Murayama-Hosokawa S, Oda K, Nakagawa S, et
al: Genome-wide single-nucleotide polymorphism arrays in
endometrial carcinomas associate extensive chromosomal instability
with poor prognosis and unveil frequent chromosomal imbalances
involved in the PI3-kinase pathway. Oncogene. 29:1897–1908. 2010.
View Article : Google Scholar
|
|
7
|
Ikeda Y, Oda K, Nakagawa S, et al:
Genome-wide single nucleotide polymorphism arrays as a diagnostic
tool in patients with synchronous endometrial and ovarian cancer.
Int J Gynecol Cancer. 22:725–731. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: a changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kato J, Matsushime H, Hiebert SW, Ewen ME
and Sherr CJ: Direct binding of cyclin D to the retinoblastoma gene
product (pRb) and pRb phosphorylation by the cyclin D-dependent
kinase CDK4. Genes Dev. 7:331–342. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lundberg AS and Weinberg RA: Functional
inactivation of the retinoblastoma protein requires sequential
modification by at least two distinct cyclin-cdk complexes. Mol
Cell Biol. 18:753–761. 1998.
|
|
11
|
Tetsu O and McCormick F: β-Catenin
regulates expression of cyclin D1 in colon carcinoma cells. Nature.
398:422–426. 1999.
|
|
12
|
Albanese C, Johnson J, Watanabe G, et al:
Transforming p21ras mutants and c-Ets-2 activate the cyclin D1
promoter through distinguishable regions. J Biol Chem.
270:23589–23597. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Musgrove EA, Caldon CE, Barraclough J,
Stone A and Sutherland RL: Cyclin D as a therapeutic target in
cancer. Nat Rev Cancer. 11:558–572. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Knudsen KE, Diehl JA, Haiman CA and
Knudsen ES: Cyclin D1: polymorphism, aberrant splicing and cancer
risk. Oncogene. 25:1620–1628. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Solomon DA, Wang Y, Fox SR, et al: Cyclin
D1 splice variants. Differential effects on localization, RB
phosphorylation, and cellular transformation. J Biol Chem.
278:30339–30347. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu F, Gladden AB and Diehl JA: An
alternatively spliced cyclin D1 isoform, cyclin D1b, is a nuclear
oncogene. Cancer Res. 63:7056–7061. 2003.PubMed/NCBI
|
|
17
|
Kang S, Kim JW, Park NH, Song YS, Kang SB
and Lee HP: Cyclin D1 polymorphism and the risk of endometrial
cancer. Gynecol Oncol. 97:431–435. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Benzeno S, Lu F, Guo M, et al:
Identification of mutations that disrupt phosphorylation-dependent
nuclear export of cyclin D1. Oncogene. 25:6291–6303. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Moreno-Bueno G, Rodriguez-Perales S,
Sanchez-Estevez C, et al: Cyclin D1 gene (CCND1) mutations
in endometrial cancer. Oncogene. 22:6115–6118. 2003. View Article : Google Scholar
|
|
20
|
Minaguchi T, Yoshikawa H, Oda K, et al:
PTEN mutation located only outside exons 5, 6, and 7 is an
independent predictor of favorable survival in endometrial
carcinomas. Clin Cancer Res. 7:2636–2642. 2001.PubMed/NCBI
|
|
21
|
Tanikawa M, Wada-Hiraike O, Nakagawa S, et
al: Multifunctional transcription factor TFII-I is an activator of
BRCA1 function. Br J Cancer. 104:1349–1355. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alt JR, Cleveland JL, Hannink M and Diehl
JA: Phosphorylation-dependent regulation of cyclin D1 nuclear
export and cyclin D1-dependent cellular transformation. Genes Dev.
14:3102–3114. 2000. View Article : Google Scholar
|
|
23
|
Gladden AB, Woolery R, Aggarwal P, Wasik
MA and Diehl JA: Expression of constitutively nuclear cyclin D1 in
murine lymphocytes induces B-cell lymphoma. Oncogene. 25:998–1007.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hall M and Peters G: Genetic alterations
of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human
cancer. Adv Cancer Res. 68:67–108. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bali A, O’Brien PM, Edwards LS, Sutherland
RL, Hacker NF and Henshall SM: Cyclin D1, p53, and
p21Waf1/Cip1 expression is predictive of poor clinical
outcome in serous epithelial ovarian cancer. Clin Cancer Res.
10:5168–5177. 2004.
|
|
27
|
Ogino S, Nosho K, Irahara N, et al: A
cohort study of cyclin D1 expression and prognosis in 602 colon
cancer cases. Clin Cancer Res. 15:4431–4438. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bodrug SE, Warner BJ, Bath ML, Lindeman
GJ, Harris AW and Adams JM: Cyclin D1 transgene impedes lymphocyte
maturation and collaborates in lymphomagenesis with the myc gene.
EMBO J. 13:2124–2130. 1994.PubMed/NCBI
|
|
29
|
Lovec H, Grzeschiczek A, Kowalski MB and
Moroy T: Cyclin D1/bcl-1 cooperates with myc genes in the
generation of B-cell lymphoma in transgenic mice. EMBO J.
13:3487–3495. 1994.PubMed/NCBI
|
|
30
|
Lovec H, Sewing A, Lucibello FC, Muller R
and Moroy T: Oncogenic activity of cyclin D1 revealed through
cooperation with Ha-ras: link between cell cycle control and
malignant transformation. Oncogene. 9:323–326. 1994.PubMed/NCBI
|
|
31
|
Uchimaru K, Endo K, Fujinuma H, Zukerberg
L, Arnold A and Motokura T: Oncogenic collaboration of the cyclin
D1 (PRAD1, bcl-1) gene with a mutated p53 and an activated ras
oncogene in neoplastic transformation. Jpn J Cancer Res.
87:459–465. 1996. View Article : Google Scholar : PubMed/NCBI
|