Introduction
Malignant glioma has emerged as one of the most
common and deadly neurosurgical disorders in adults. Although the
etiology of gliomas remains largely unknown, hereditary disorders
of the central nervous system (CNS), such as neurofibromatoses and
the tuberous sclerosis complex, have been implicated as
predisposing risk factors (1). The
5-year survival rate of malignant glioma is less than 15% and most
patients die within 18 months following diagnosis (2), making this form of cancer particularly
devastating. The most common treatment strategy involves surgical
resection followed by radiotherapy. Surgical resection of glioma is
usually incomplete. Therefore, the residual or metastasized cancer
cells often necessitate subsequent radiotherapy. Although
radiotherapy can prolong the length of survival, its effect is
generally modest. Recent technical advances in radiotherapy, such
as intensity-modulated radiation therapy that avoids non-cancerous
tissue, have only marginally improved its clinical efficacy
(3–5). However, malignant gliomas are
frequently resistant to radiation therapy, which not only
complicates treatment but also impairs the survivor’s quality of
life.
Although the exact mechanism underlying glioma
radioresistance has remained elusive, several studies have
implicated glioma stem cells (GSCs) as the potential source of this
characteristic. In general, tumor stem-like cells (TSCs) have
become a recent focus of research aiming to understand treatment
response, and TSCs have been shown to play major roles in
determining sensitivity or resistance to chemotherapy and
radiotherapy. Bao et al(6)
characterized the subset of glioma cells (GCs) that survived
ionizing radiation (IR) and determined that these tumor cells
expressed the CD133 stem cell marker; in addition, the authors
showed that the CD133+ glioma cells were able to repair
radiation-induced DNA damage more effectively than the
CD133− glioma cells and underwent less apoptosis
following irradiation. However, a subsequent study by McCord et
al(7) demonstrated that a
portion of CD133+ TSCs could more sensitively respond to
radiation and showed that this radiosensitive subset of cells had a
different DNA damage response than the cells of the traditional
glioblastoma in vitro model. These results suggest that
CD133+ cells may represent an in vitro model
system useful for studying the detailed mechanisms that mediate the
glioblastoma radioresponse. To identify the radiosensitivity of
CD133+ GSCs, it is necessary to further compare the
radiosensitivity of CD133+ GSCs with established cell
lines by clonogenic analysis and comet assay.
Cell cycle checkpoint responses play essential roles
in DNA strand breaks (DSBs) repair by IR and thus induce cell cycle
arrest to repair damaged DNA. It is well-established that ataxia
telangiectasia mutated (ATM) protein is critical for initiation of
these checkpoint pathways (8). ATM
kinase and its downstream substrates, p53 and checkpoint kinase 2
(Chk2), have been implicated in the control of the G1 phase
checkpoint pathways (8–12). However, the roles of ATM and its
substrates in the radiosensitivity of GSCs are not clear. To
observe the effects of ATM in the radiosensitivity of GSCs, KU55933
as a specific and reversible inhibitor of ATM, was used in our
study. We found that ATM played an important role in the
radioresistance of GSCs.
Materials and methods
Cell culture
Two glioma cell lines were used in this study: U87
and U251. The glioma cell lines were grown in monolayer in
Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen Life
Technologies, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS)
(HyClone, Logan, UT, USA). The glioma cell lines were separated
into CD133+ and CD133− cells according to the
CD133/2-PE antibody (Miltenyi Biotec) as a surface marker by
fluorescence-activated cell sorting (FACS). Isolated
CD133+ cells were then cultured in neural stem cell
medium (NSC-M) supplemented with 20 ng/ml of basic fibroblast
growth factor (bFGF), 20 ng/ml epidermal growth factor (EGF) (both
from Peprotech, Inc., Rocky Hill, NJ, USA), 10 ng/ml leukemia
inhibitory factor (LIF) (Chemicon, Temecula, CA, USA) and a 1:50
dilution of B27 (Gibco-BRL, Carlsbad, CA, USA). The cells were
grown at 37°C in a humidified incubator with 5% CO2 in
air for ~7 days. The formed tumor spheres were harvested and used
for culture and the indicated assays.
Detection of CD133 and nestin expression
by immunofluorescence
Sterile coverslips were installed into 24-well
plates. GSCs were collected by centrifuging at 1,000 rpm. Then,
single-cell suspensions were plated at a 1×105/ml cell
density. Medium was supplemented with NSC-M in order to observe
expression of CD133 and nestin. After culturing in NSC-M for 7–8
days, the tumor spheres were formed, washed 3 times with
phosphate-buffered solution (PBS; 5 min each), and fixed by
incubating with 4% polyoxymethylene (for 30 min). After an
additional 3 PBS washes (5 min each), 0.5% Triton X-100 was added
and the sample was then immunoreacted with 10% FBS and 50 μl of
CD133 and nestin antibodies (1:50; Zhongshan Biotechnology, China)
by incubating at 4°C for 12 h. After an additional 3 PBS washes (5
min each), the TRITC and FITC secondary antibodies (1:50; Zhongshan
Biotechnology) were added and immunoreacted by incubating at 37°C
for 1 h. After a final 3 PBS washes (5 min each), DAPI (Beyotime
Institute of Biotechnology) staining was added and the
immunoreactivity was observed with a Leica rapid scanning laser
confocal microscope.
Detection of the percentage of CD133
cells by flow cytometry (FCM)
A total of 1×105 GCs and GSCs was
collected by centrifuging at 1,000 rpm. Then, the cells were mixed
with 20 μl of FcR blocking reagent, 10 μl of CD133/1-PE antibody,
or 10 μl of mouse IgG2b (all from Miltenyi Biotec). The mixture was
incubated at 4°C for 10 min. After adding 800 μl of PBS, the
treated cells were collected by centrifuging at 1,000 rpm and
washed with 500 μl PBS. The percentage of CD133+ cells
in the sample was determined by FCM analysis (Beckon-Dickinson,
Franklin Lakes, NJ, USA).
Detection of multipotency by
immunofluorescence
Sterile coverslips were installed into 24-well
plates. GSCs were collected by centrifuging at 1,000 rpm. Then,
single-cell suspensions were plated at a 1×104/ml cell
density. Medium was supplemented with 10% FBS in order to observe
the differentiation of GSCs. After 7–8 days, glial fibrillary
acidic protein (GFAP), mitogen-activated protein (MAP2) and myelin
basic protein (MBP) were examined by immunofluorescence. The
examining method was the same as for the detection of CD133 and
nestin. All antibodies of GFAP, MAP2 and MBP (1:50) were provided
by a commercial company (Zhongshan Biotechnology).
Detection of tumorigenicity by
transplantation of GSCs and GCs in vivo
Twelve Balb/C nude mice (4 weeks old; The
Experimental Animal Laboratories, Shanghai) were anesthetized with
i.p. ketamine and xylazine, and GCs and GSCs of 1×103,
1×104 and 1×105 were respectively implanted
into the mice into the left (GCs) and right (GSCs) subcutaneous
regions of the back, near lower extremity. Tumor growth was
monitored weekly. The mice were sacrificed at the 8th week after
implantation. The same experiment was repeated once with identical
conditions. All the animal experiments were performed in strict
accordance with the Institutional Animal Care guidelines.
Irradiation
The established cell lines and the CD133+
subset of cells were irradiated using a 6-MV X Rad source (SN4474;
Varian) to deliver doses of 2, 4, 6 and 8 Gy. The rate of the dose
was 300 cGy/min. The cells were harvested 4 h after
irradiation.
Clonogenic survival assay
Cell survival was measured using a standard
colony-forming efficiency assay. Briefly, GSCs were irradiated by
doses of 2, 4, 6 and 8 Gy X-rays, respectively and then
disaggregated into a single-cell suspension and diluted to a final
concentration of 1×103 cells/ml. A 2-ml cell suspension
was added to each well of 6-well culture plates. The plates were
incubated at 37°C in 5% CO2 for 2 weeks. Prior to colony
counting, cells were washed (PBS), fixed [0.0037% (v/v)
formaldehyde], and stained (0.1% w/v-crystal violet), rinsed
(dH2O) and dried. Colonies consisting of >50 cells
were counted as 1 surviving colony. Data were calculated as the
surviving fraction relative to the control plates. Survival assays
were repeated at least 3 times.
Single-cell gel electrophoresis (neutral
comet assay)
The single-cell gel electrophoresis assay was
carried out with the Comet Assay kit (Trevigen), according to the
manufacturer’s instructions. Briefly, 4 h after monolayer cultures
were irradiated by 2 Gy X-rays, single-cell suspensions of GCs and
GSCs were generated, washed with PBS, and mixed with low melting
agarose (1:10). The cell-agarose mixtures were pipetted onto the
comet assay slides. Cell lysis was carried out by incubating at 4°C
for 2.5–3 h, after which the cells were subjected to
electrophoresis for 20–25 min at 4°C. The resolved samples were
fixed and the DNA was visualized by staining with 5 μg/ml Goldview
(SBS Genetech) and observation with a confocal laser microscope.
Digital fluorescent images were obtained and used to calculate the
percentage of comet tails/100 cells.
Western blotting
Cells were collected on dry ice, and 1 ml total
protein extraction kit containing protease inhibitor was added.
Total protein was extracted after homogenization. Coomassie
brilliant blue staining was used to examine the protein
concentration. Subsequently, SDS-PAGE (10% gel) electrophoresis was
used to separate the proteins (50 mg of protein/sample), and
proteins were transferred to a polyvinylidene fluoride (PVDF)
membrane. The membrane was then incubated overnight with antibodies
diluted to 1:1,000 (5% w/v-BSA/TBST), followed by secondary
antibodies diluted to 1:1,000 (Zhongshan Biotechnology), incubated,
and then subjected to film development and further analysis.
Antibodies included: ATM and phos-ATM (S1981) (Epitomics); p53,
phos-p53 (S15), Chk2 and phos-Chk2 (T68) (Santa Cruz Biotechnology
Inc., Santa Cruz, CA, USA).
Drug treatment
To observe the effects of ATM on the
radiosensitivity of GSCs and GCs, KU55933 was added as an ATM
specific inhibitor into the culture solution at a concentration of
10 μM 4 h prior to IR. Then, the cells were irradiated by 2 Gy
X-rays and 4 h later were harvested followed by cell cycle phase
and cell apoptosis analyses by FCM.
Cell cycle phase and cell apoptosis
analyses by FCM
The distribution of cells in the various phases of
the cell cycle was determined by FCM. Single-cell suspensions of
GCs and GSCs (1×105 cells/ml) were washed twice with PBS
and fixed with 75% alcohol. After treatment with 500 μl of 1 g/l
RNase at 37°C for 30 min, the cells were collected for fixation
again, stained with propidium iodide (PI), and analyzed by FCM.
Single-cell suspensions of GCs and GSCs (1×105 cells/ml)
were aliquoted (100 μl) into microcentrifuge tubes and mixed with 5
μl of Annexin IV-FITC and 20 μg/ml PI. After 15 min of incubation
at room temperature, the percentage of apoptotic cells were
analyzed by FCM.
Statistical analysis
All statistical analyses were carried out using the
SPSS software package (version 17.0 SPSS). Data are expressed as
the means ± standard deviation (SD). The significance of intergroup
differences was analyzed by the t-test, followed by the
Student-Newman-Keuls test for multiple comparisons. A p-value of
<0.05 was considered statistically significant.
Results
Identification of GSCs: nestin and CD133
expression
After 24 h of culturing in NSC medium, isolated
CD133+ cells formed neurospheres composed of 3–5 cells.
Over the next 7 to 8 days, the neurospheres increased in size,
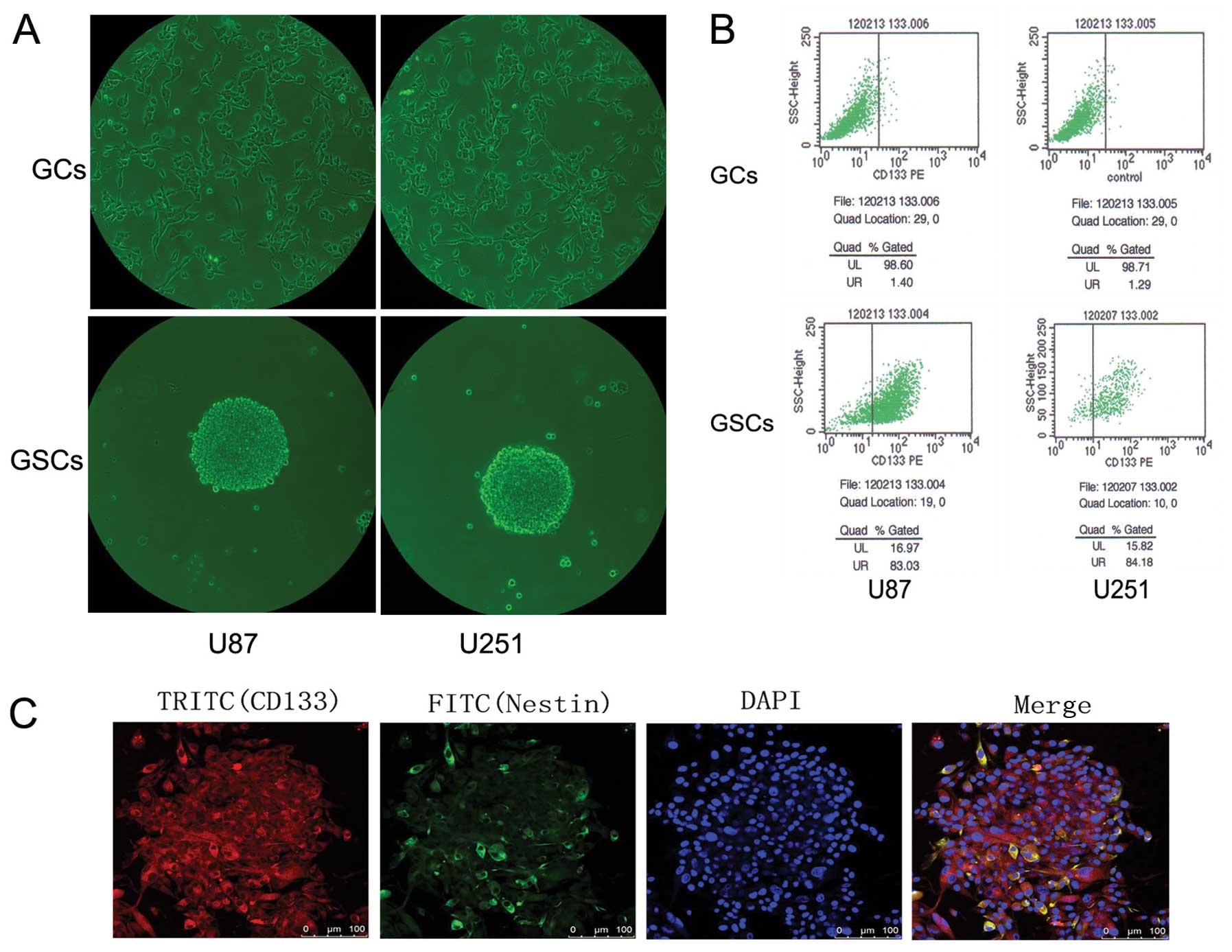
reaching an average of 200 μm (range, 100–300 μm) (Fig. 1A). CD133 and nestin expression was
detected in the neurospheres (Fig.
1B). The percentage of CD133+ cells was
significantly higher in the GSCs than in the GCs (83.03 vs. 1.4%
and 84.18 vs. 1.29%) (Fig. 1C).
Identification of GSCs: multipotency
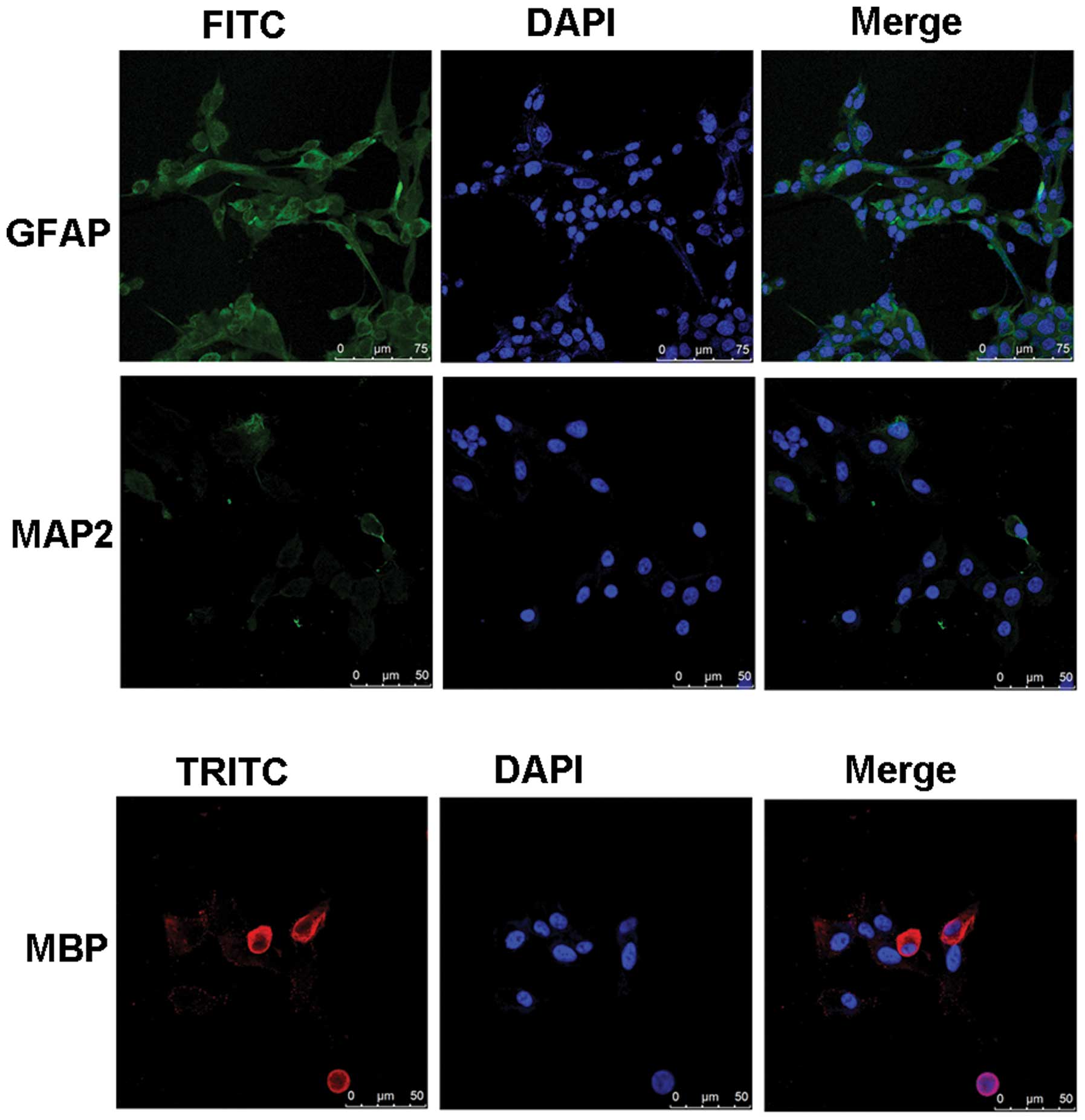
The pinning phenomenon of GSC neurospheres was
observed 5 h after exposure to the 10% FBS. By 24 h, the pod had
protruded outside of the neurosphere, and by 72 h, the neurospheres
had flattened. At day 7 of culture, the bottom of the culture
container was covered with GSCs. Most of the GSCs expressed the
astrocyte-specific GFAP marker, but only a small proportion of GSCs
expressed the neuron-specific MAP2 and the oligocyte-specific MBP
markers (Fig. 2).
Identification of GSCs:
tumorigenicity
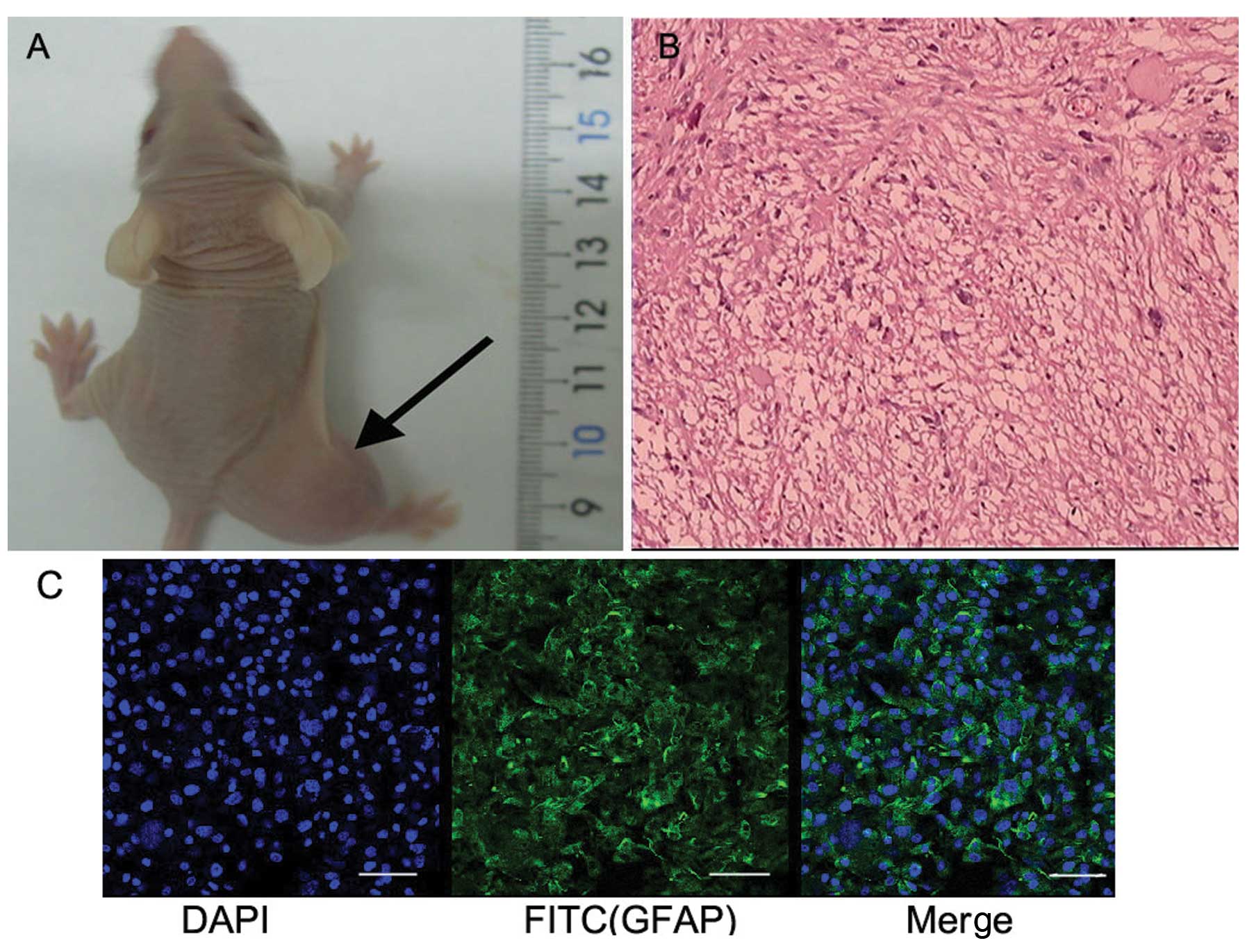
Our study showed that transplanted tumors formed
in vivo after implanting 1,000–100,000 GSCs subcutaneously
into nude mice, while at least 100,000 GCs were implanting to form
transplanted tumors in vivo. The transplanted glioblastoma
was characterized by H&E staining and immunohistochemistry for
GFAP in the transplanted tumor tissue sections. These results
indicated that GSCs more easily formed tumors than GCs (Fig. 3).
Clonogenic survival assay after
irradiation
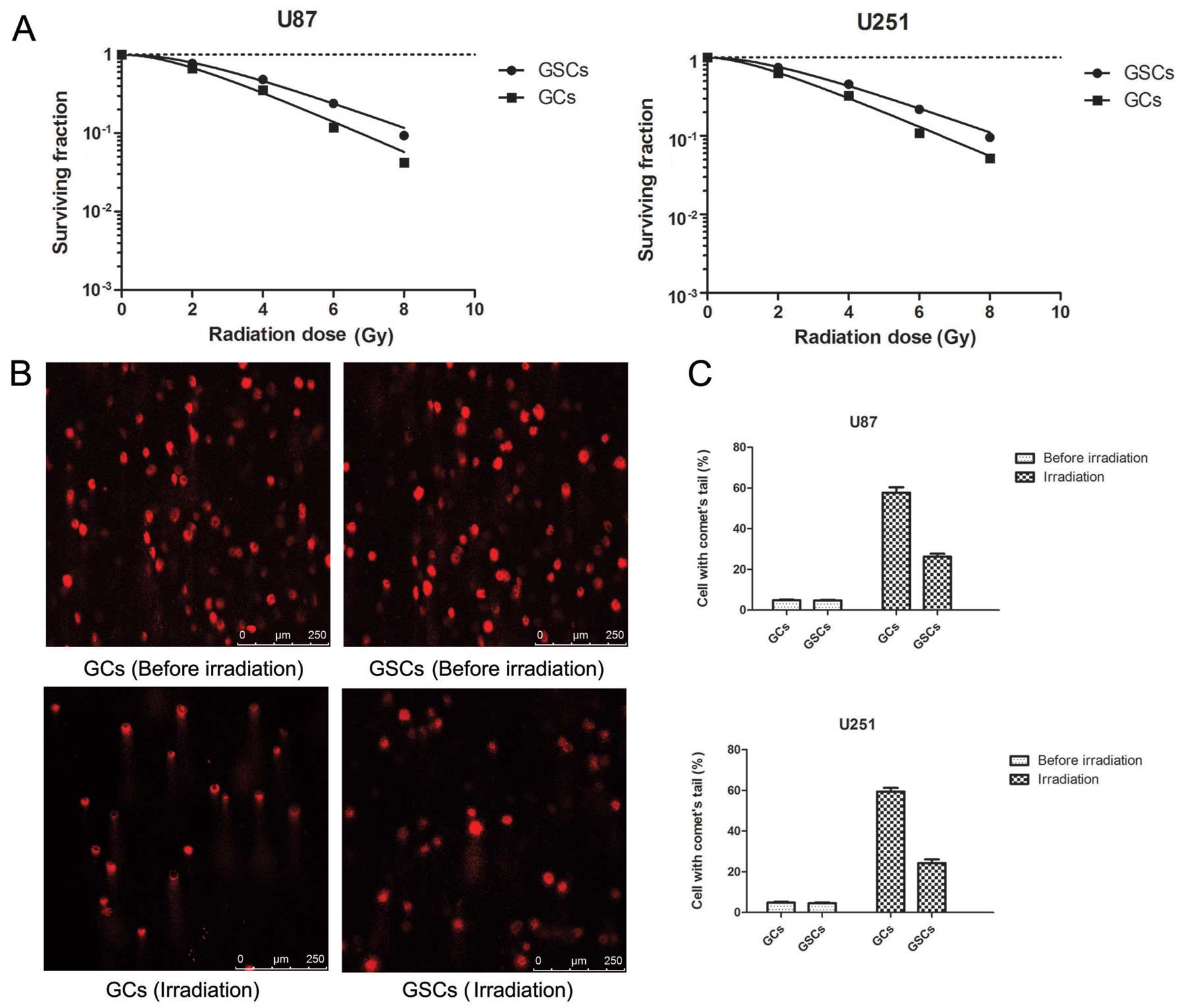
To compare the intrinsic radiosensitivities of GSCs
and established GC lines, it was necessary to perform clonogenic
analyses under optimal growth conditions for GSCs and GCs. The
surviving fraction of GSCs was higher than that of GCs at doses of
radiation such as 2, 4, 6 and 8 Gy (p<0.05) (Fig. 4A).
Single-cell gel electrophoresis (neutral
comet assay)
Before irradiation, none of the GSCs or GCs formed
comet tails in the single-cell gel electrophoresis. However, 4 h
after monolayer cultures were irradiated by 2 Gy X-rays, part of
the cells of both cell types showed comet tails. GCs appeared to be
more sensitive to irradiation as the number of cells with comet
tails was significantly greater than the number of cells with comet
tails in the GSCs (p<0.05) (Fig. 4B
and C).
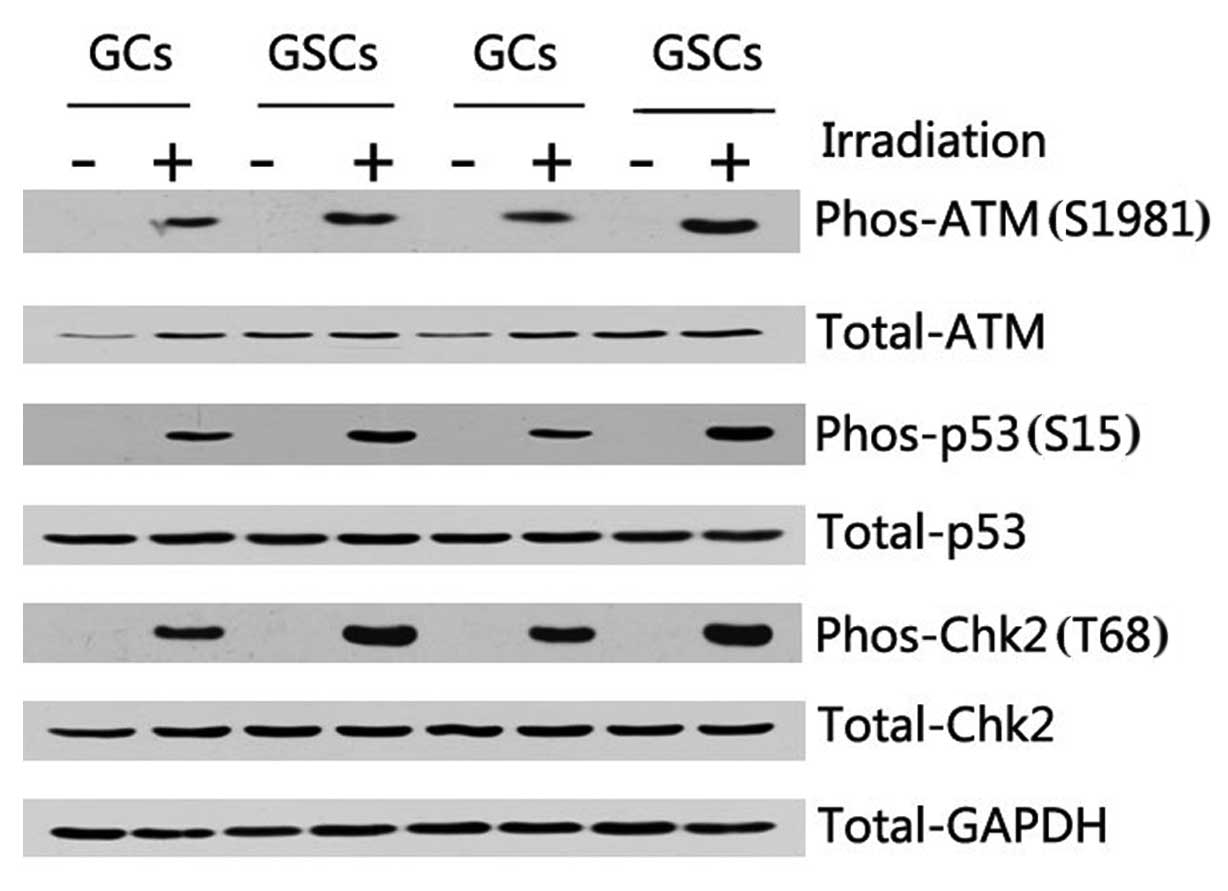
Cell cycle checkpoint proteins: ATM, p53
and Chk2
The ATM signaling pathway is conserved from the
human to the mouse and plays important roles in DNA damage
checkpoint responses. In human GCs and GSCs, ATM kinase activity
was induced by IR and there were strong increases in the
phosphorylation of p53 (S15) and Chk2 (T68) relative to cells
without IR (Fig. 7). In addition,
activation of the phosphorylation of these checkpoint proteins was
significantly higher in GSCs than in GCs, indicating that
CD133+ cells showed more radioresistance in response to
DNA damage by preferential checkpoint activation.
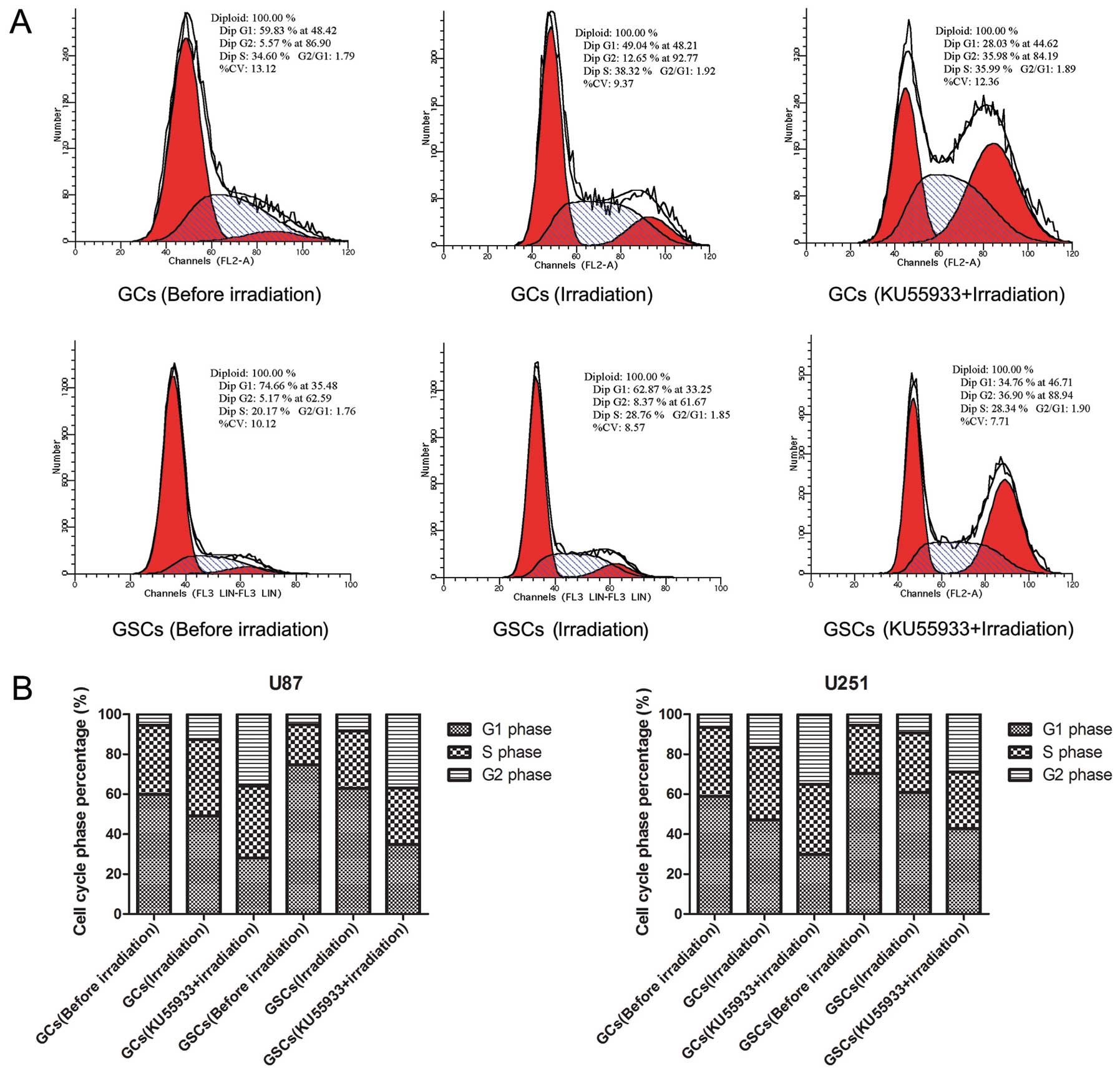
Irradiation-induced effects on cell cycle
distribution and cell apoptosis
Prior to irradiation, there was no difference
between the amount of GCs and GSCs in the G2 phase (p>0.05).
After 2 Gy irradiation, both cell types had a higher percentage of
cells in the G2 phase (p<0.05). Furthermore, GCs responded with
a higher percentage of cells in the G2 phase when compared with the
GSCs (p<0.05). In response to IR, GCs and GSCs treated with
KU55933 had an enhanced proportion of cells in the G2 phase and a
decreased proportion of cells in the G1 phase relative to the
untreated cells (p<0.05) (Fig.
5). Prior to irradiation, there was no significant difference
between the percentage of cells undergoing apoptosis (p>0.05).
After 2 Gy irradiation, both cell types consisted of a higher
number of apoptotic cells, and the apoptotic rate of the GCs was
significantly higher than that of the GSCs (p<0.05). In response
to IR, GCs and GSCs treated with KU55933 all resulted in an
enhanced apoptotic rate relative to the untreated cells (p<0.05)
(Fig. 6).
Discussion
Radiobiologists or radiation oncologists have known
for many years that gliomas respond poorly to radiation therapy
(13), although no experimental
system has yet provided definitive evidence to establish whether
this property of radioresistance is inherent or acquired. It has
been shown that glioblastoma cells expressing the stem cell marker
CD133 have a higher radioresistant quality than CD133−
tumor cells (6). This finding has
become the basis for the tumor stem cell theory of malignant glioma
pathogenesis and prognosis. Subsequent studies have shown that GSCs
are capable of initiating and reforming the original cancer after
isolation from the primary tumor tissue (14), and to have a more robust resistance
to chemotherapy and radiotherapy than non-tumorigenic cells.
CD133 is a glycoprotein that is specifically
localized to cellular protrusions and has been detected in a wide
range of human cancer and stem cells, including glioblastoma,
hematopoietic stem cells, endothelial progenitor cells, neuronal
stem cells and glial stem cells. Another marker that is often
detected in neural stem cells associated with tumors is nestin, an
intermediate filament protein that is expressed mostly in nerve
stem cells. GSCs in our study expressed both CD133 and nestin, and
at significantly higher levels than in GCs. GSC neurospheres formed
secondary spheres by cell passage. This finding suggests that GSCs
are capable of self-renewal (15,16).
GSCs can be induced to differentiate into cells expressing GFAP,
MAP2 and MBP, suggesting that GSCs are multipotent and capable of
differentiating into different cell types (17). Furthermore, GSCs were more capable
at initiating tumors in nude mice than GCs.
Radiosensitivity is a critical determining factor of
a patient’s response to radiotherapy and prognosis of malignant
glioma (18). In our study, we
found that more GSCs than GCs survived following different doses of
irradiation. Irradiation is believed to kill tumor cells by
inducing irreparable DNA damage and the subsequent apoptosis
mechanism. Tumor cells that are actively replicating their DNA may
produce different results in gel electrophoresis than their
quiescent counterparts, and this difference may be interpreted as
inherent sensitivity. Therefore, we used the comet assay, which is
a gel electrophoresis-based method, to measure DNA damage in tumor
cells (19,20). We found almost no comet tail
formation in the unirradiated GCs or GSCs. However, GCs formed
significantly more comet tails than GSCs after irradiation,
suggesting that GSCs have a more robust trend for
radioresistance.
In a previous study, McCord et al(7) demonstrated that a portion of
CD133+ TSCs from human glioblastoma surgical specimens
was more radiosensitive. In contrast, our study and the results of
Bao et al(6) demonstrated
that CD133+ cells were more resistant to radiation than
CD133− cells or established GCs. The discrepancy may be
explained by the different radiosensitivity of different regions
even in the same surgical specimen (21,22).
DNA damage by IR activates the DNA damage response (DDR) an
intricate signal transduction pathways that involves DNA repair and
activation of cell cycle checkpoints (23). Loss of function of critical proteins
from DDR pathways can cause cells to have enhanced sensitivity to
DNA damage (24). The ATM kinase is
an important component of these DDR pathways, and cells deficient
for ATM (A-T) display hypersensitivity to DSBs by IR (25). The activated ATM is essential for
the activation of downstream effector kinases, such as Chk2 and
p53, which mainly contribute to the proper regulation of IR-induced
cell cycle arrest in the G1 phase (26). Transient inhibition of ATM kinase is
sufficient to enhance cellular sensitivity to ionizing radiation.
Our study indicates that both GSCs and GCs showed a marked increase
in the phosphorylation of ATM, p53 and Chk2 checkpoint proteins in
response to DNA damage relative to cells prior to IR. Furthermore,
activation of the phosphorylation of these checkpoint proteins was
significantly greater in GSCs than in GCs. The data presented here
showed that the preferential checkpoint activation may contribute
to the resistance of CD133+ glioma cells.
Based on these phenomena, specific inhibition of ATM
function in combination with current radio-therapeutic treatment
may lead to enhanced cancer cell killing. KU55933 is capable of
sensitizing many types of human cancer cell lines to IR (27,28).
In fact, ATM and its phosphorylated downstream substrates
participate in the proper regulation of IR-induced arrest in the
G1, S, and G2 phases of the cell cycle. The distribution of the
cell cycle in the established cell lines used in our study was
mainly in the G1 phase. Fibroblasts in A-T patients showed a
defective cell cycle checkpoint; p53-dependent G1 arrest did not
occur (29) and therefore resulted
in a prolonged G2 phase. In our study, although both GSCs and GCs
untreated by KU55933 showed an increasing percentage of cells in
the G2 phase in response to IR, GCs responded with more cells in
the G2 phase than GSCs due to higher expression of ATM and its
downstream substrates in GSCs than in GCs. However, both GSCs and
GCs pretreated by KU55933 showed a significantly increased
percentage of cells in the G2 phase in response to IR relative to
untreated cells. There was no difference between GSCs and GCs when
pretreated with KU55933. The results showed that inhibition of ATM
led to loss of G1 phase arrest and therefore prolonged the G2 phase
in GSCs and GCs; G2 phase is the most susceptible phase in the
entire cell cycle in response to IR. ATM also functions in the
regulation of apoptosis. Our study demonstrated that inhibition of
ATM kinase induced increased apoptosis in both GSCs and GCs in
response to IR.
In summary, we isolated and identified GSCs from
malignant glioma cells. GSCs were capable of self-renewal and
multiple differentiation and displayed a higher capability of tumor
initiation in nude mice when compared with GCs. We also
demonstrated that GSCs were more resistant to radiation than GCs
due to high expression of phosphorylated cell cycle checkpoint
proteins. Therefore, GSCs contributed to malignant glioma
radioresistance and tumor repopulation. Most importantly, we found
that inhibition of ATM significantly reduced the radioresistance of
GSCs and GCs. Therefore therapies targeted to checkpoint proteins
in preclinical and clinical development may provide an effective
method by which to disrupt the resistance mechanism with the aim of
improving malignant glioma control with radiation treatment.
Acknowledgements
We acknowledge grant support from the National
Natural Science Foundation of China (nos. 81172387 and
30973074).
References
|
1
|
Grossman SA and Batara JF: Current
management of glioblastoma multiforme. Semin Oncol. 31:635–644.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nieder C, Andratschke N, Wiedenmann N,
Busch R, Grosu AL and Molls M: Radiotherapy for high-grade gliomas.
Does altered fractionation improve the outcome? Strahlenther Onkol.
180:401–407. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reya T, Morrison SJ, Clarke MF and
Weissman IL: Stem cells, cancer, and cancer stem cells. Nature.
414:105–111. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Marx J: Cancer research. Mutant stem cells
may seed cancer. Science. 301:1308–1310. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Clarke MF, Dick JE, Dirks PB, Eaves CJ,
Jamieson CH, Jones DL, Visvader J, Weissman IL and Wahl GM: Cancer
stem cells - perspectives on current status and future directions:
AACR Workshop on cancer stem cells. Cancer Res. 66:9339–9344. 2006.
View Article : Google Scholar
|
|
6
|
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q,
Hjelmeland AB, Dewhirst MW, Bigner DD and Rich JN: Glioma stem
cells promote radioresistance by preferential activation of the DNA
damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McCord AM, Jamal M, Williams ES,
Camphausen K and Tofilon PJ: CD133+ glioblastoma
stem-like cells are radiosensitive with a defective DNA damage
response compared with established cell lines. Clin Cancer Res.
15:5145–5153. 2009.
|
|
8
|
Tichý A, Vávrová J, Pejchal J and Rezácová
M: Ataxia-telangiectasia mutated kinase (ATM) as a central
regulator of radiation-induced DNA damage response. Acta Medica
(Hradec Kralove). 53:13–17. 2010.PubMed/NCBI
|
|
9
|
Bower JJ, Zhou Y, Zhou T, Simpson DA,
Arlander SJ, Paules RS, Cordeiro-Stone M and Kaufmann WK: Revised
genetic requirements for the decatenation G2 checkpoint: the role
of ATM. Cell Cycle. 9:1617–1628. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Banin S, Moyal L, Shieh S, Taya Y,
Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y
and Ziv Y: Enhanced phosphorylation of p53 by ATM in response to
DNA damage. Science. 281:1674–1677. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Matsuoka S, Huang M and Elledge SJ:
Linkage of ATM to cell cycle regulation by the Chk2 protein kinase.
Science. 282:1893–1897. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jiang H, Reinhardt HC, Bartkova J,
Tommiska J, Blomqvist C, Nevanlinna H, Bartek J, Yaffe MB and
Hemann MT: The combined status of ATM and p53 link tumor
development with therapeutic response. Genes Dev. 23:1895–1909.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ignatova TN, Kukekov VG, Laywell ED,
Suslov ON, Vrionis FD and Steindler DA: Human cortical glial tumors
contain neural stem-like cells expressing astroglial and neuronal
markers in vitro. Glia. 39:193–206. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Singh SK, Clarke ID, Terasaki M, Bonn VE,
Hawkins C, Squire J and Dirks PB: Identification of a cancer stem
cell in human brain tumors. Cancer Res. 63:5821–5828.
2003.PubMed/NCBI
|
|
15
|
Kondo T, Setoguchi T and Taga T:
Persistence of a small subpopulation of cancer stem-like cells in
the C6 glioma cell line. Proc Natl Acad Sci USA. 101:781–786. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li G, Chen Z, Hu YD, Wei H, Li D, Ji H and
Wang DL: Autocrine factors sustain glioblastoma stem cell
self-renewal. Oncol Rep. 21:419–424. 2009.PubMed/NCBI
|
|
17
|
Li GH, Wei H, Lv SQ, Ji H and Wang DL:
Knockdown of STAT3 expression by RNAi suppresses growth and induces
apoptosis and differentiation in glioblastoma stem cells. Int J
Oncol. 37:103–110. 2010.PubMed/NCBI
|
|
18
|
Blazek ER, Foutch JL and Maki G: Daoy
medulloblastoma cells that express CD133 are radioresistant
relative to CD133− cells, and the CD133+
sector is enlarged by hypoxia. Int J Radiat Oncol Biol Phys.
67:1–5. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Diehn M and Clarke MF: Cancer stem cells
and radiotherapy: new insights into tumor radioresistance. J Natl
Cancer Inst. 98:1755–1757. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Singh NP, McCoy MT, Tice RR and Schneider
EL: A simple technique for quantitation of low levels of DNA damage
in individual cells. Exp Cell Res. 175:184–191. 1988. View Article : Google Scholar
|
|
21
|
Ng WL, Yan D, Zhang X, Mo YY and Wang Y:
Over-expression of miR-100 is responsible for the low-expression of
ATM in the human glioma cell line: M059J. DNA Repair. 9:1170–1175.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Allalunis-Turner MJ, Barron GM, Day RS
III, Dobler KD and Mirzayans R: Isolation of two cell lines from a
human malignant glioma specimen differing in sensitivity to
radiation and chemotherapeutic drugs. Radiat Res. 134:349–354.
1993. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Choi S, Gamper AM, White JS and Bakkenist
CJ: Inhibition of ATM kinase activity does not phenocopy ATM
protein disruption: implications for the clinical utility of ATM
kinase inhibitors. Cell Cycle. 9:4052–4057. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kennedy RD and D’Andrea AD: DNA repair
pathways in clinical practice: lessons from pediatric cancer
susceptibility syndromes. J Clin Oncol. 24:3799–3808. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shiloh Y: ATM and related protein kinases:
safeguarding genome integrity. Nat Rev Cancer. 3:155–168. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Canman CE, Wolff AC, Chen CY, Fornace AJ
Jr and Kastan MB: The p53-dependent G1 cell cycle checkpoint
pathway and ataxia-telangiectasia. Cancer Res. 54:5054–5058.
1994.PubMed/NCBI
|
|
27
|
Rainey MD, Charlton ME, Stanton RV and
Kastan MB: Transient inhibition of ATM kinase is sufficient to
enhance cellular sensitivity to ionizing radiation. Cancer Res.
68:7466–7474. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
White JS, Choi S and Bakkenist CJ:
Transient ATM kinase inhibition disrupts DNA damage-induced sister
chromatid exchange. Sci Signal. 3:ra442010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kastan MB, Onyekwere O, Sidransky D,
Vogelstein B and Craig RW: Participation of p53 protein in the
cellular response to DNA damage. Cancer Res. 51:6304–6311.
1991.PubMed/NCBI
|