Introduction
Progesterone (Pg) is known to play a pivotal role in
the reproductive cycle and pregnancy, in which the fetal-maternal
immune response is suppressed during pregnancy (1). Furthermore, it has been well
documented that Pg contributes to the control of T-cell-mediated
autoimmune reactions, in which the immune balance between
CD4+ T-helper Th1 and Th2 cells is regulated (2). Pg induces apoptosis in Th1 cells,
whereas Pg is metabolized into non-toxic 20α-hydroxyprogesterone by
20α-hydroxysteroid dehydrogenase for the escape of Th2 cells from
apoptosis (3). On the basis of a
common theory, it is believed that Pg binds to the classic
cytoplasmic/nuclear Pg receptor (PR) in order to modulate gene
expression, a process which is known as genomic effects (1). Genomic effects are not restricted to
immune-associated cells, but are also well known in various types
of cells including tumor cells (1,4–7).
Indeed, it has been demonstrated that Pg induces apoptosis in
diverse types of tumor cells, such as mesothelioma cells and
breast, cervical and ovarian cancer cells by activating the genomic
signaling pathway (4–7). Results of the investigation of the
antiproliferative action of Pg against tumor cells suggest that Pg
acts on non-target cells devoid of classic PR through a different
process known as non-genomic effects. In fact, a recent study using
a uterine cervix-derived cell line lacking classic PR demonstrated
that the arrest of the cell cycle at the G1/G0 phase as well as
apoptosis induction was responsible for Pg-mediated dose-dependent
growth inhibition (8).
Intensive research for identifying receptor
molecules for Pg to manifest rapid functioning has been conducted.
Recently, cell membrane-bound progesterone receptors (mPRs)
unrelated to the classic PR, which are composed of mPRα, mPRβ and
mPRγ, have received a great deal of attention (9–14). It
is believed that rapid responses to Pg are initiated by binding to
a respective receptor localized on the cell membrane (i.e., mPR)
coupled with inhibitory G-proteins (Gi) (9,12,14).
Indeed, progestin, a synthetic Pg, has been demonstrated to
activate a variety of signaling pathways through mPRα (14). It has been further demonstrated that
the binding of Pg to mPRα alters secondary messenger pathways
through activation of the pertussis toxin (PTX)-sensitive Gi
(2,13,14).
As mentioned above, Pg plays a pivotal
immunoregulatory role by regulating the balance between Th1 and Th2
cells. Moreover, Gamberucci et al(15) demonstrated that Pg inhibited
capacitative Ca2+ entry in Jurkat T lymphocytes in case
of the binding of Pg to membrane, and proposed an involvement of
non-genomic mechanism for the inhibitory effects of Pg. Recently,
mPRs including mPRα and mPRβ have been detected in human peripheral
blood leukocytes, T lymphocytes and Jurkat cells, suggesting a
potential novel mechanism for the immunoregulatory function of Pg
through activation of mPRs (16).
However, the effects of Pg on Jurkat cells have not yet been
investigated in detail in regard to antiproliferation as well as
apoptosis induction. In the present study, we investigated the
effects of Pg on a classic PR-negative Jurkat cell line, A3, and
its caspase-8 deficient mutant cell line, I9.2 (17,18).
We also used PTX, an inhibitor specific for Gi protein, to clarify
the involvement of mPRα in the non-genomic Pg signaling pathway,
since mPRα has been well documented in mediating Pg actions among
the mPRs (14,19,20).
Materials and methods
Cell culture
Human acute lymphocytic T-cell leukemia derived from
a lymphoblastoid cell line Jurkat clone A3, which is sensitive to
Fas-mediated apoptosis, and I9.2, which is a caspase-8-deficient
mutant of A3 (17,18) were obtained from the American Type
Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured
as previously described (6) in
RPMI-1640 medium without Phenol red (Sigma-Aldrich, St. Louis, MO,
USA) supplemented with 10% charcoal stripped fetal bovine serum
(FBS) (Biological Industries, Kibbutz Beit Haemek, Israel) and 10
mg/ml gentamicin (Life Technologies, Carlsbad, CA, USA) at 37°C in
a humidified atmosphere (5% CO2 in air). Peripheral
blood mononuclear cells (PBMNCs) were isolated from three healthy
volunteers using Histopaque-1077 (Sigma) according to the
manufacturer’s instructions, and cultured in RPMI-1640 medium
without Phenol red supplemented with 10% charcoal stripped FBS, 100
U/ml penicillin and 100 μg/ml streptomycin (Life Technologies) at
37°C in a humidified atmosphere (5% CO2 in air). For
experiments, the cell density was adjusted to 5×105
cells/ml prior to the treatments. The present study was approved by
the IRB Committee of Tokyo University of Pharmacy and Life
Sciences. An informed consent was obtained from all healthy
volunteers.
Treatment with Pg in the presence or
absence of Boc-D-FMK
Cells were seeded on flasks or plates at
1×105 cells/ml, followed by treatment with different
concentrations of Pg for different period of time as indicated. In
order to clarify the implication of the caspase cascade in the
effects of Pg, cells were preincubated with Boc-D-FMK, a pancaspase
inhibitor (Sigma-Aldrich), at the indicated concentrations for 1 h,
before the addition of Pg at a final concentration of 50 μM,
followed by an additional incubation for 24 h.
Cell viability assay
After treatment with various concentrations of Pg
(10–100 μM) in the presence or absence of Boc-D-FMK for the
designated time, cell viability was measured by
2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-((Phenyl-amino)-carboxyl)-2H-tetrazolium
hydroxide (XTT) assay (Sigma-Aldrich) as previously described
(21). Relative cell viability was
calculated as the ratios of the absorbance at 450 nm of each
treatment group against the respective control group.
Cell cycle analysis
After treatment with 50 μM Pg for 24 and 48 h, cell
cycle analysis was performed using a FACSCanto flow cytometer
(Becton-Dickinson, Mountain View, CA, USA) according to a method
reported previously with modifications (22). For staining cellular DNA, cells were
washed twice with PBS, fixed with 1% paraformaldehyde/PBS for 30
min, and then washed twice again with PBS, permeabilized in 70%
(v/v) cold ethanol and maintained at −20°C for at least 4 h. Then,
cell pellets were washed twice with PBS after centrifugation and
incubated with 0.25% Triton X-100 for 5 min on ice. After
centrifugation and washing with PBS, cells were resuspended in 500
μl of propidium iodide (PI)/RNase A/PBS (5 μg/ml PI and 0.1% RNase
A in PBS) and incubated for 30 min in the dark at room temperature.
A total of 10,000 events were acquired and Diva software
(Becton-Dickinson) and ModFit LT™ version 3.0 (Verity Software
House, Topsham, ME, USA) were used to calculate the number of cells
at each sub-G1, G0/G1, S and G2/M phase fraction.
Lactate dehydrogenase (LDH) assay
After treatment with 50 μM Pg for 48 and 72 h, LDH
leakage from cells was measured using the LDH-Cytotoxic Test Wako
kit (Wako Pure Chemical Industries, Osaka, Japan) according to the
method previously described with slight modifications (23). Briefly, after the cells were
centrifuged at 450 × g for 5 min at 4°C, 25 μl of culture
supernatants was collected and transferred into wells of a 96-well
plate, followed by the addition of 25 μl PBS to each well. After
discarding the remaining culture supernatants, the cells were lysed
with 200 μl of 0.1% Triton X-100. A portion of cell lysates (12.5
μl) was subsequently loaded into the 96-well plate, followed by the
addition of 37.5 μl to each well. LDH activities in both the
culture supernatants and the cell lysates were determined by adding
50 μl of ‘substrate solution’ from the kit, followed by incubation
at 37°C for 15 min. The reaction was stopped by the addition of 100
μl of ‘stopping solution’, and the absorbance at 550 nm was
measured with a microplate reader. Cell damage was calculated as a
percentage of LDH leakage from damaged cells using the following
formula: LDH leakage (%) = (Sup)/(Sup + Cell) × 100 where Sup and
Cell refer to the absorption of the culture supernatant and cell
lysate, respectively.
Annexin V/PI analysis
TACS™ Annexin V-FITC apoptosis detection kit
(Trevigen, Gaithersburg, MD, USA) was used for the detection of
early apoptotic and late apoptotic/necrotic cells according to the
manufacturer’s instructions, Briefly, after treatment with 50 μM Pg
for 24 and 48 h, respectively, cells were washed twice with PBS.
Then, cells (1×106) were resuspended in 100 μl Annexin V
incubation reagent (10 mM HEPES pH 7.4, 150 mM NaCl, 5 mM KCl, 1 mM
MgCl2, 1.8 mM CaCl2, 5 μg/ml PI, Annexin
V-FITC). Cells were incubated in the dark for 15 min at room
temperature, followed by the addition of 400 μl binding buffer.
Fluorescence intensities of FITC and PI were measured by a
FACSCanto flow cytometer. A total of 30, 000 events were acquired,
and data were analyzed by Diva software. Annexin V(−)PI(−) cells,
Annexin V(+)PI(−) cells and Annexin V(+)PI(+) cells were defined as
viable cells, early apoptotic cells and late apoptotic/necrotic
cells, respectively.
Western blot analysis
Western blot analysis was carried out according to
the methods previously described (24). Briefly, after separation of proteins
on a SDS polyacrylamide gel electrophoresis, followed by
transferring to a nitrocellulose membrane, protein bands were
detected using the following primary antibodies and dilution
ratios: rabbit anti-mPRα (BioWorld Technology, Minneapolis, MN,
USA) at 1:1,000; rabbit anti-caspase-3 (Enzo Life Sciences,
Farmingdale, NY, USA) at 1:1,000; rabbit anti-caspase-8 (BD
Pharmingen, Franklin Lakes, NJ, USA) at 1:4,000; rabbit
anti-caspase-9 (Cell Signaling Technology, Danvers, MA, USA) at
1:1,000; mouse anti-PR (Sigma) at 1:1,000 and β-actin (Sigma) at
1:5,000. Blotted protein bands were detected with respective
horseradish peroxidase-conjugated secondary antibodies and an
enhanced chemiluminescence (ECL) Western blot analysis system (GE
Healthcare, Buckinghamshire, UK).
Determination of loss of mitochondrial
membrane potential (ΔΨm)
The ΔΨm was determined by flow cytometry after cell
loading with Rhodamine 123 as previously described with
modification (25). Cells were
seeded at 1×105 cells/ml, followed by pretreatment with
50 ng/ml PTX (List Biological Laboratories, Campbell, CA, USA) for
24 h. After washout of PTX, the cell density of PTX-treated and
control group cells (non-treated cells) were again adjusted to
1×105 cells/ml, followed by treatment with 50 μM Pg for
an additional 24-h incubation. After incubation with 10 μM
Rhodamine 123 in PBS for 15 min in the dark at room temperature,
the fluorescence intensities of Rhodamine 123 were measured by a
FACSCanto flow cytometer. A total of 30,000 events were acquired,
and data were analyzed by Diva software.
Statistical analysis
Experiments were independently repeated three times,
and the results are shown as the means ± standard deviation (SD) of
three assays. The Student’s t-test was applied and P<0.05 was
considered to indicate a statistically significant result.
Results
Antiproliferative effect of Pg on A3 and
I9.2 cell growth
Growth inhibition was observed in both A3 and I9.2
cells after treatment with Pg at concentrations ranging from 10 to
100 μM for 24 h and 48 h in a dose- and time-dependent manner
(Fig. 1A and B). The relative
growth inhibition rate in the A3 and I9.1 cells was ~50 and 70%
when treated with 50 μM Pg for 24 and 48 h, respectively (Fig. 1A and B). A similar pattern of growth
inhibition was observed in both cell lines regardless of caspase-8
status (Fig. 1A and B). In
contrast, almost no growth inhibition was observed in PBMNCs
following treatment with Pg, although a slight but significant
growth inhibition was observed in the cells treated with as high as
100 μM Pg for 48 h (Fig. 1C and D).
Furthermore, flow cytometric analysis showed that almost no cell
cycle arrest was observed in both A3 and I9.2 cells, although there
were trends in a decrease in the number of cells in the G2/M phase
in both cell lines after treatment with 50 μM Pg for 24 or 48 h
(Fig. 1E). More importantly, a
significant increase in the number of cells in sub-G1 phase was
observed, indicating apoptosis induction in both A3 and I9.2 cells
treated with Pg (Fig. 1E).
Pg-mediated LDH release and apoptosis
induction in A3 and I9.2 cells
The release of LDH provides an accurate measure of
the cell membrane integrity and cell viability. After treatment
with 50 μM Pg for 48 and 72 h, LDH leakage analysis was thus
performed to examine whether Pg treatment affects cell membrane
integrity. A time-dependent LDH leakage was observed in both A3 and
I9.2 cells irrespective of caspase-8 deficiency (Fig. 2A). Furthermore, after treatment with
50 μM for 24 and 48 h, a significant decrease in the number of
viable cells was observed concomitantly with a prominent increase
in the number of apoptotic cells in both the A3 and I9.2 cell lines
(Fig. 2B).
Pg-mediated caspase activation in A3 and
I9.2 cells
After treatment with 50 μM Pg for 24 and 48 h,
western blot analysis was conducted to determine the activation of
caspase-8, -9 and -3. As expected, neither the proform nor the
cleaved form of caspase-8 was observed in caspase-8-deficient I9.2
cells, whereas its cleaved form was observed in A3 cells after Pg
treatment, indicating the activation of caspase-8 in these cells
(Fig. 3A). Furthermore, an
increased amount of the cleaved form of caspase-9 and caspase-3 was
observed in both A3 and I9.2 cells (Fig. 3A). These results clearly indicate
that caspase cascade activation is responsible for Pg-mediated
apoptosis induction. Moreover, the addition of a pan-caspase
inhibitor, Boc-D-FMK, significantly suppressed Pg-triggered
cytocidal effects in both cells, reconfirming the involvement of
caspase cascade activation (Fig.
3B).
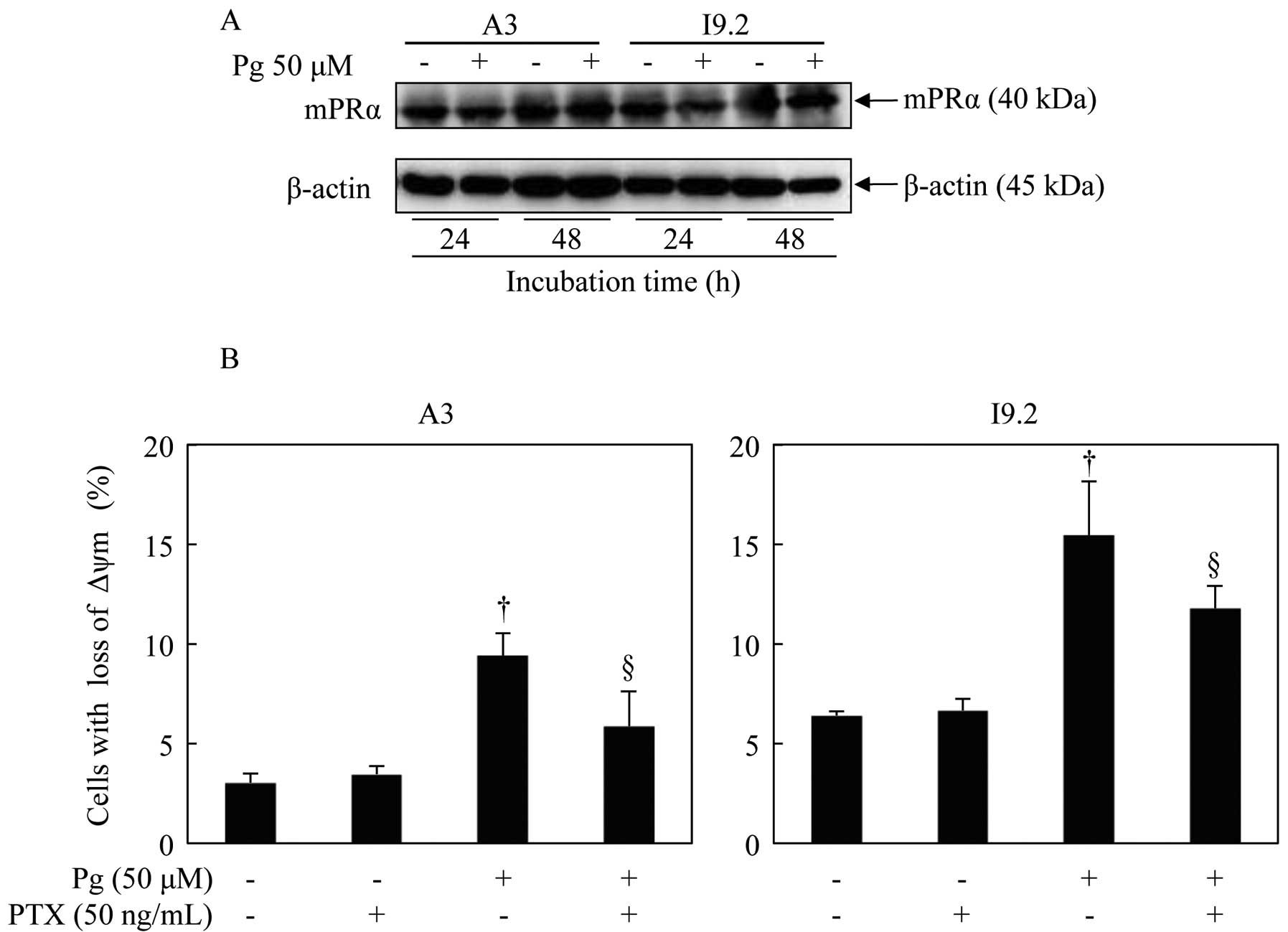
Contribution of mPRα to Pg-induced loss
of mitochondrial membrane potential (ΔΨm) in A3 and I9.2 cells
Western blot analysis was performed to identify the
presence of mPRα. In both cell types, the expression of mPRα was
observed, and its expression level was not affected by the
administration of 50 μM Pg for 24 and 48 h (Fig. 4A). Furthermore, the expression of
classic PR (PR-A and PR-B) was not detected in both cell lines
(data not shown), as has been previously reported (17,18).
After treatment with 50 μM Pg for 24 h, Rhodamine
123, a cell-permeant cationic fluorescent dye, was used to assay
ΔΨm in both A3 and I9.2 cells. The administration of Pg resulted in
a significant increase in cell populations with decreased ΔΨm
(decrease in fluorescence intensity) (Fig. 4B). However, the cell populations
with decreased ΔΨm were notably restored by pretreatment with PTX,
an inhibitor specific for Gi protein directly coupled to mPRα
(Fig. 4B).
Discussion
In the present study, we demonstrated that
Pg-mediated growth suppression was observed in both A3 and I9.2
cells in a dose- and time-dependent manner. Notably, no significant
difference in the degree of growth suppression between A3 and I9.2
(caspase-8-deficient) cells was observed, suggesting that the
growth suppression was caspase-8 independent. As expected, much
less cytotoxicity was observed in PBMNCs when treated with
concentrations of Pg, which showed significant cytotoxicity in both
cell lines. These results reconfirmed a high tolerance of Pg for
clinical application.
It has been demonstrated that G0/G1 cell cycle
arrest and apoptosis induction contributed to Pg-mediated growth
inhibition in C-4I cells, a cervical cell line lacking classic PR
(8). However, almost no arrest at a
particular cell cycle phase was observed, despite there were trends
in a decrease in the number of G2/M phase cells in both the A3 and
I9.2 cells. This discrepancy between C-4I and A3 as well as I9.2
cells might be due to a cell type-specific event. Notably, a
significant accumulation of cells in the sub-G1 phase was observed
after treatment with Pg, suggesting that Pg-mediated growth
suppression is attributed to apoptosis induction rather than cell
cycle arrest. Furthermore, apoptosis induction as evidenced by
Annexin V analysis strengthened our hypothesis. Apart from
apoptosis induction, secondary necrosis probably also contributed
to Pg-mediated growth inhibition in both cells types since the
disruption of membrane integrity after treatment with Pg for 48 and
72 h was clearly demonstrated by the release of LDH.
In order to clarify the molecular details of the
apoptosis pathway, we focused on the activation of caspases
including caspase-9, -8 and -3, which are key players in two
principal signaling pathways of apoptosis induction, called the
intrinsic and extrinsic pathway (26,27).
The intrinsic mechanism of apoptosis involves a disruption of the
mitochondrial cell membrane, resulting in the loss of ΔΨm
associated with cytochrome c release, followed by the
activation of capase-9 and caspase-3 (26–28).
In contrast, the extrinsic pathway induced by death receptors, such
as Fas and tumor necrosis factor receptor, is responsible for the
activation of caspase-8 accompanied by the activation of caspase-3
(27,28). In the present study, we demonstrated
that the activation of capase-9, -8 and -3 was observed in A3 cells
after Pg treatment, suggesting that not only the intrinsic pathway
but also the extrinsic pathway was involved in Pg-mediated
apoptosis induction in the cells. To the best of our knowledge,
this is the first report showing the involvement of the intrinsic
pathway in Pg-mediated apoptosis in A3 cells, although Fas-mediated
apoptosis induction through caspase-8 has been well documented in
these cells (17,18). Therefore, an experiment focused on
the activation of Bid, a pro-apoptotic Bcl-2 family protein and
responsible for the crosstalk between intrinsic and extrinsic
pathway (29,30), is needed to clarify the relationship
between the two principal pathways. Furthermore, the activation of
caspase-9 and -3 was observed in caspase-8-deficient I9.2 cells,
suggesting that the intrinsic pathway, instead of the extrinsic
pathway, is involved in Pg-mediated apoptosis induction in the
cells. Collectively, these results suggest that the activation of
the caspase cascade associated with the intrinsic and/or extrinsic
pathway is responsible for Pg-mediated apoptosis in A3 as well as
in I9.2 cells. In fact, suppression of apoptosis by the addition of
a pan-caspase inhibitor, Boc-D-FMK, also supported our hypothesis.
We also demonstrated that exposure to Pg induced loss of ΔΨm in
both cells, which strongly supports our hypothesis of the
involvement of the intrinsic pathway.
Since the lack of classic PR has been demonstrated
in Jurkat cells (16,31), in order to clarify the molecular
mechanisms of Pg action, the involvement of mPRα was investigated,
a molecular of which the function and regulation has been best
characterized among the mPRs (19,20,32).
In the present study, the expression of mPRα was observed, and its
expression level was not affected by the administration of Pg.
Importantly, pretreatment with PTX, an inhibitor specific for Gi
protein activation (13,16,33),
significantly restored the decreased ΔΨm resulting from exposure to
Pg. Collectively, these results suggest that mPRα contributes to
Pg-mediated apoptosis induction in Jurkat cells devoid of classic
PR.
In conclusion, growth suppression accompanied by
apoptosis induction of Jurkat cells devoid of classic PR by Pg, was
mediated through mitochondrial membrane disruption followed by the
activation of the caspase cascade, as a result of the activation of
non-genomic effects. Results of the present study offer a novel
concept of Pg actions towards its clinical application for patients
with lymphocytic T cell leukemia. Furthermore, the present study
provides novel insight into the function of Pg as a regulator of
the immune response.
Acknowledgements
The present study was supported, in part, by grants
from the Ministry of Education, Culture, Sports, Science and
Technology and by the Promotion and the Mutual Aid Corporation for
Private Schools of Japan.
References
|
1
|
Conneely OM, Mulac-Jericevic B, DeMayo F,
Lydon JP and O’Malley BW: Reproductive functions of progesterone
receptors. Recent Prog Horm Res. 57:339–355. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hughes GC: Progesterone and autoimmune
disease. Autoimmun Rev. 11:A502–A514. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Matsuzaki J, Tsuji T, Imazeki I, Ikeda H
and Nishimura T: Immunosteroid as a regulator for Th1/Th2 balance:
its possible role in autoimmune diseases. Autoimmunity. 38:369–375.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ahmad N and Kumar R: Steroid hormone
receptors in cancer development: a target for cancer therapeutics.
Cancer Lett. 300:1–9. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hellberg D: Sex steroids and cervical
cancer. Anticancer Res. 32:3045–3054. 2012.PubMed/NCBI
|
|
6
|
Horita K, Inase N, Miyake S, Formby B,
Toyoda H and Yoshizawa Y: Progesterone induces apoptosis in
malignant mesothelioma cells. Anticancer Res. 21:3871–3874.
2001.PubMed/NCBI
|
|
7
|
Lanari C, Wargon V, Rojas P and Molinolo
AA: Antiprogestins in breast cancer treatment: are we ready? Endocr
Relat Cancer. 19:R35–R50. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bertelsen EL, Endresen PC, Orbo A and
Sager G: Non-genomic cell growth inhibition by progesterone. cell
cycle retardation and induction of cell death. Anticancer Res.
24:3749–3755. 2004.PubMed/NCBI
|
|
9
|
Falkenstein E, Tillmann HC, Christ M,
Feuring M and Wehling M: Multiple actions of steroid hormones - a
focus on rapid, nongenomic effects. Pharmacol Rev. 52:513–556.
2000.PubMed/NCBI
|
|
10
|
Zhu Y, Hanna RN, Schaaf MJ, Spaink HP and
Thomas P: Candidates for membrane progestin receptors - past
approaches and future challenges. Comp Biochem Physiol C Toxicol
Pharmacol. 148:381–389. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hammes SR and Levin ER: Minireview: recent
advances in extranuclear steroid receptor actions. Endocrinology.
152:4489–4495. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ndiaye K, Poole DH, Walusimbi S, Cannon
MJ, Toyokawa K, Maalouf SW, Dong J, Thomas P and Pate JL:
Progesterone effects on lymphocytes may be mediated by membrane
progesterone receptors. J Reprod Immunol. 95:15–26. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dressing GE, Goldberg JE, Charles NJ,
Schwertfeger KL and Lange CA: Membrane progesterone receptor
expression in mammalian tissues: a review of regulation and
physiological implications. Steroids. 76:11–17. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thomas P: Characteristics of membrane
progestin receptor alpha (mPRα) and progesterone membrane receptor
component 1 (PGMRC1) and their roles in mediating rapid progestin
actions. Front Neuroendocrinol. 29:292–312. 2008.
|
|
15
|
Gamberucci A, Giunti R and Benedetti A:
Progesterone inhibits capacitative Ca2+ entry in Jurkat
T lymphocytes by a membrane delimited mechanism, independently of
plasma membrane depolarization. Cell Calcium. 36:175–180.
2004.PubMed/NCBI
|
|
16
|
Dosiou C, Hamilton AE, Pang Y, Overgaard
MT, Tulac S, Dong J, Thomas P and Giudice LC: Expression of
membrane progesterone receptors on human T lymphocytes and Jurkat
cells and activation of G-proteins by progesterone. J Endocrinol.
196:67–77. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Juo P, Kuo CJ, Yuan J and Blenis J:
Essential requirement for caspase-8/FLICE in the initiation of the
Fas-induced apoptotic cascade. Curr Biol. 8:1001–1008. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Juo P, Woo MS, Kuo CJ, Signorelli P,
Biemann HP, Hannun YA and Blenis J: FADD is required for multiple
signaling events downstream of the receptor Fas. Cell Growth
Differ. 10:797–804. 1999.PubMed/NCBI
|
|
19
|
Thomas P, Tubbs C, Detweiler C, Das S,
Ford L and Breckenridge-Miller D: Binding characteristics, hormonal
regulation and identity of the sperm membrane progestin receptor in
Atlantic croaker. Steroids. 70:427–433. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu Y, Rice CD, Pang Y, Pace M and Thomas
P: Cloning, expression, and characterization of a membrane
progestin receptor and evidence it is an intermediary in meiotic
maturation of fish oocytes. Proc Natl Acad Sci USA. 100:2231–2236.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Imai M, Kikuchi H, Denda T, Ohyama K,
Hirobe C and Toyoda H: Cytotoxic effects of flavonoids against a
human colon cancer derived cell line, COLO 201: a potential natural
anti-cancer substance. Cancer Lett. 276:74–80. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Darzynkiewicz Z, Bruno S, Del Bino G,
Gorczyca W, Hotz MA, Lassota P and Traganos F: Features of
apoptotic cells measured by flow cytometry. Cytometry. 13:795–808.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yoshino Y, Yuan B, Kaise T, Takeichi M,
Tanaka S, Hirano T, Kroetz DL and Toyoda H: Contribution of
aquaporin 9 and multidrug resistance-associated protein 2 to
differential sensitivity to arsenite between primary cultured
chorion and amnion cells prepared from human fetal membranes.
Toxicol Appl Pharmacol. 257:198–208. 2011. View Article : Google Scholar
|
|
24
|
Yuan B, Ohyama K, Bessho T and Toyoda H:
Contribution of inducible nitric oxide synthase and
cyclooxygenase-2 to apoptosis induction in smooth chorion
trophoblast cells of human fetal membrane tissues. Biochem Biophys
Res Commun. 341:822–827. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Johnson LV, Walsh ML and Chen LB:
Localization of mitochondria in living cells with rhodamine 123.
Proc Natl Acad Sci USA. 77:990–994. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ryter SW, Kim HP, Hoetzel A, Park JW,
Nakahira K, Wang X and Choi AM: Mechanisms of cell death in
oxidative stress. Antioxid Redox Signal. 9:49–89. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yuan B, Yoshino Y, Kaise T and Toyoda H:
Application of arsenic trioxide therapy for patients with
leukaemia. Biological Chemistry of As, Sb and Bi. Sun HZ: John
Wiley & Sons, Ltd; New York: pp. 263–292. 2011
|
|
28
|
Earnshaw WC, Martins LM and Kaufmann SH:
Mammalian caspases: structure, activation, substrates, and
functions during apoptosis. Annu Rev Biochem. 68:383–424. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kantari C and Walczak H: Caspase-8 and
bid: caught in the act between death receptors and mitochondria.
Biochim Biophys Acta. 1813:558–563. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Viswanath V, Wu Y, Boonplueang R, Chen S,
Stevenson FF, Yantiri F, Yang L, Beal MF and Andersen JK: Caspase-9
activation results in downstream caspase-8 activation and bid
cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced
Parkinson’s disease. J Neurosci. 21:9519–9528. 2001.PubMed/NCBI
|
|
31
|
Bamberger CM, Else T, Bamberger AM, Beil
FU and Schulte HM: Dissociative glucocorticoid activity of
medroxyprogesterone acetate in normal human lymphocytes. J Clin
Endocrinol Metab. 84:4055–4061. 1999.PubMed/NCBI
|
|
32
|
Xie M, Zhu X, Liu Z, Shrubsole M, Varma V,
Mayer IA, Dai Q, Chen Q and You S: Membrane progesterone receptor
alpha as a potential prognostic biomarker for breast cancer
survival: a retrospective study. PloS One. 7:e351982012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Karteris E, Zervou S, Pang Y, Dong J,
Hillhouse EW, Randeva HS and Thomas P: Progesterone signaling in
human myometrium through two novel membrane G protein-coupled
receptors: potential role in functional progesterone withdrawal at
term. Mol Endocrinol. 20:1519–1534. 2006. View Article : Google Scholar
|