Introduction
Colorectal cancer (CRC) is the third most common
cancer and the leading cause of cancer-related mortality worldwide
(1), with a high potential for
tumor invasion and metastasis. Similar to many other solid
malignancies, metastasis of CRC involves proteolysis of the
extracellular matrix, alterations in tumor cell adhesion and
motility, and colonization in distant organs (2). In addition, formation of new blood
vessels is also indispensable for the persistence of metastastic
growth. Despite the obvious importance of metastasis, the molecular
mechanism underlying these processes remains unclear.
Growing evidence has recently supported the
cancer-related effects of microRNAs (miRNAs), a newly identified
class of small non-coding RNA molecules which function through
negatively regulating target gene expression. They can bind to
specific complementary sites within the 3′ untranslated regions
(3′UTRs) of their target mRNA, to inhibit translation or to induce
degradation, even to regulate mRNA transcription (3–5).
Recent studies have revealed the critical role of miRNAs in
regulating a variety of genes pivotal for invasion or metastasis
(6,7).
miR-126, which locates within intron 7 of the
epidermal growth factor-like domain 7 gene (EGFL7), is highly
expressed in endothelial cells. Two studies have reported its
crucial role in promoting embryonic angiogenesis by promoting
vascular endothelial growth factor (VEGF) signaling in zebrafish
(8) and in a mouse model (9). In contrast, miR-126 was identified to
be one of the dysregulated miRNAs in multiple cancer types. Its
downregulation was observed in breast, pancreatic and gastric
cancer (10–12). Low expression of miR-126 in
non-small cell lung cancer and renal cell carcinoma was
significantly correlated with reduced patient survival (13,14).
Through targeting oncogenic genes, such as SLC7A5, Crk and ADAM9,
miR-126 plays a role as a tumor suppressor to inhibit tumor cell
proliferation, invasion and the EMT process (11,15,16).
Conversely, some recent studies have presented an oncogenic role of
miR-126. They found that overexpression of miR-126 may contribute
to gastric carcinogenesis by inhibiting SOX2 expression (17), and was highly associated with
metastasis of prostate cancer (18). These contradictory results imply
that miR-126 may function by diverse mechanisms in different
physiological and pathological contexts.
VEGF has commonly been acknowledged as the most
prominent factor to promote tumor development (19). It operates to increase the
permeabilization of blood vessels and induces formation of new
blood vessels, to facilitate orthotopic and metastatic growth. In
CRC, VEGF signaling involved neovascularity represents a key
mediator of tumor initiation and dissemination (20).
Quite recently, studies have shown decreased
expression of miR-126 in CRC (21),
and that DNA methylation results in the silencing of miR-126 in
bladder and lung cancer (22,23).
Of specific note, in silico prediction indicates that
miR-126 has a conserved binding site within the 3′UTR of VEGF mRNA.
We thus hypothesized that overexpression of VEGF in CRC is, at
least partly, due to silencing of miR-126 caused by DNA
methylation.
Materials and methods
Tissue samples and cell lines
Twelve pairs of primary CRC and matched adjacent
normal colonic epithelium, and 62 primary CRC tissues were
collected. All samples were obtained from patients who underwent
surgical resection at Nanfang Hospital (Guangzhou, China) and were
snap-frozen in liquid nitrogen, and stored at −80°C for further
use.
HEK293 cell line and 6 human CRC cell lines,
including LoVo, HT29, SW480, SW620, SW1116 and HCT116, were
purchased from the American Type Culture Collection (Manassas, VA,
USA). The cells were maintained routinely in RPMI-1640 (Gibco-BRL,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Sigma,
St. Louis, MO, USA) and cultured at 37°C in a 5% CO2
atmosphere.
TaqMan real-time-PCR analysis of miR-126
expression
TaqMan real-time-PCR was performed to detect mature
miR-126 expression in tissue sample and cell lines. Total RNA was
extracted using TRIzol reagent (Invitrogen Life Technologies,
Carlsbad, CA, USA). Expression of mature miR-126 was determined by
the TaqMan miRNA assay (Applied Biosystems, Foster City, CA, USA).
Data were processed using the 2−ΔΔCt method. RNU6B
(Ambion, Austin, TX, USA) was used as an endogenous control.
Ectopic miR-126 expression
Enforced expression of miR-126 in LoVo and SW620
cells was achieved by transfection with pre-miR-126 (Ambion). Cells
were seeded in 6-well clusters or 96-well plates for 24 h and
transfected with 30 nM pre-miR-126 using Lipofectamine 2000
(Invitrogen Life Technologies) for 24 or 48 h. Scramble precursor
(Ambion) was used as a negative control. Transfected LoVo and SW620
cells were used in further functional assays or for RNA/protein
extraction.
Cell proliferation assay
Alamar blue assay (Invitrogen Life Technologies) was
conducted to measure cell proliferation. Cells were seeded in a
96-well plate at 0.5×104/well for 24 h, then transfected
with pre-miR-126 or scramble control. The transfected cells were
incubated for 24, 48 and 72 h, respectively. Ten microliters of
Alamar blue reagent was added to each well at 2 h before the end of
the incubation. Following the incubation, the absorbance of each
well at 570 nm (600 nm as reference wavelength) was determined
using a microplate reader.
Cell invasion and migration assays
The invasive potential of LoVo and SW620 cells was
evaluated using a cell invasion assay kit (Millipore, Billerica,
MA, USA) following the manufacturer’s instructions. Briefly,
transfected cells were resuspended in serum-free RPMI-1640 medium
at a density of 1.0×106/ml. Cell suspension (300 μl) and
500 μl RPMI-1640 containing 10% FBS were respectively added to each
insert and the matched lower chamber. After 48 h, non-invading
cells were removed using a cotton swab, and then the underside of
the insert was stained. Six random fields (at a magnification of
×100) for each insert were counted. For the migration assay, the
procedures were similar to those of the invasion assay, except that
200 μl cell suspension was cultured in each Transwell upper insert
(Corning Incorporated, Corning, NY, USA) for 48 h. For human
microvascular endothelial cell (HMVEC) migration,
1.0×105 transfected LoVo cells were seeded in the lower
chamber and incubated for 24 h. Then 5×104 HMVECs were
added in the matched upper insert, and co-cultured with LoVo cells
for 4 h. The underside of the insert was stained. Six random fields
(at a magnification of ×100) for each insert were counted.
Western blot analysis
Immunoblotting was performed to detect the
expression of VEGF in CRC cell lines after transfection. Protein
(30 μg) was loaded onto a SDS-PAGE gel, transferred onto a PVDF
membrane and subsequently probed with 1:1,000 diluted mouse
monoclonal VEGF-A antibody (Cell Applications, San Diego, CA, USA)
at 4°C overnight, followed by incubation with HRP-conjugated
secondary antibody. Signals were visualized using ECL substrates
(Millipore). β-actin was used as an endogenous protein for
normalization.
ELISA assay
An ELISA kit (Cusabio, China) was used to detect the
VEGF concentration in the cell supernatant following the
manufacturer’s instructions.
Luciferase reporter assay
The full-length 3′UTR of VEGF (1923 nt) containing
one miR-126 potential binding site was amplified by PCR using the
following primers: VEGF-A forward,
5′-CCGctcgagGCCGGGCAGGAGGAAGGAG-3′ and reverse,
5′ATAAGAATgcggccgcTGAGATCAGAATTAAATTCTTTAATAC-3′. The PCR product
was subcloned into a psiCHECK-2 vector (Promega Corporation,
Madison, WI, USA) immediately downstream to the luciferase gene
sequence. A psiCHECK-2 construct containing 3′UTR of VEGF with a
mutant seed sequence of miR-126 was also synthesized using the
primers: mutVEGF-A forward, 5′-AAG
AGAAAGTGTTTTATATATCGATCTTATTTAATATCCCTTTTTA-3′ and reverse,
5′-TAAAAAGGGATATTAAATAAGATCGATATATAAAACACTTTCTCTT-3′.
All constructs were verified by DNA sequencing.
HEK293 cells were plated in 24-well clusters, then co-transfected
with 500 ng constructs with or without miR-126 precursors. At 48 h
after transfection, luciferase activity was detected using a
dual-luciferase reporter assay system (Promega Corporation) and
normalized to Renilla activity.
In vitro and in vivo angiogenesis
assays
In vitro and in vivo angiogenesis
assays were conducted to determine the potential of miR-126 to
affect tumor vascularity. For the in vitro Matrigel tube
formation study, HMVECs were seeded in a 96-well plate pre-coated
with growth factor-reduced Matrigel (BD Biosciences). Conditioned
medium (CM) obtained from miR-126 precursor or scramble control
transfected cells was added, followed by incubated at 37°C for 12
h. The capacity of tube formation was assessed by counting the
tubes in 3 randomly chosen fields under an inverted microscope (at
a magnification of ×100). The tubes were defined as structures
formed by 2 identifiable HMVECs connecting at both ends. In
vivo, the chorioallantoic membrane (CAM) model was performed as
previously described (24).
5-Aza-2′-deoxycytidine (5-aza-CdR)
treatment
For the demethylation study, the 6 CRC cell lines
were treated with 5-Aza-CdR (Sigma) at a concentration of 3 μM for
72 h, replacing the medium and the drug every 24 h.
DNA methylation analysis
To establish the methylation status of miR-126 CpG
islands, we performed 2 types of PCR analysis of bisulfate-modified
genomic DNA (Active Motif, Carlsbad, CA, USA). First, the
methylation-specific PCR (MSP) of CRC cell lines was conducted
using methylated and unmethylated primers: M forward,
5′-TTTAAGTTATTTTTTTTAGGTTCGG-3′ and reverse,
5′-ATTATATAACCTCCTCCTAAAACGC-3′; U forward,
5′-TTTAAGTTATTTTTTTTAGGTTTGG-3′ and reverse,
5′-TTATATAACCTCCTCCTAAAACACC-3′.
PCR products were subjected to 2.5% agarose gel
electrophoresis and visualized by ethidium bromide staining and UV
transillumination. Second, bidirectional bisulfite sequencing (BSP)
was used to analyze the corresponding CpG islands. PCR products
were subcloned into the pMD®18-T vector (Takara, China),
3 candidate clones were selected and sequenced. The primers used
were as follows: forward, 5′-GTGTGGTTAGGGGTTGTGTT-3′ and reverse,
5′-CACACCCAATACTCAAAAAATTTC-3′.
Statistical analysis
All data from 3 independent experiments are
expressed as means ± SD and processed using SPSS 13.0. The
expression of miR-126 in CRC tissues and matched adjacent colonic
epithelium was compared by paired t-test. The difference between
the experimental groups and control was estimated by one-way ANOVA.
A P-value of <0.05 was considered to indicate a statistically
significant result.
Results
miR-126 is commonly downregulated in
CRC
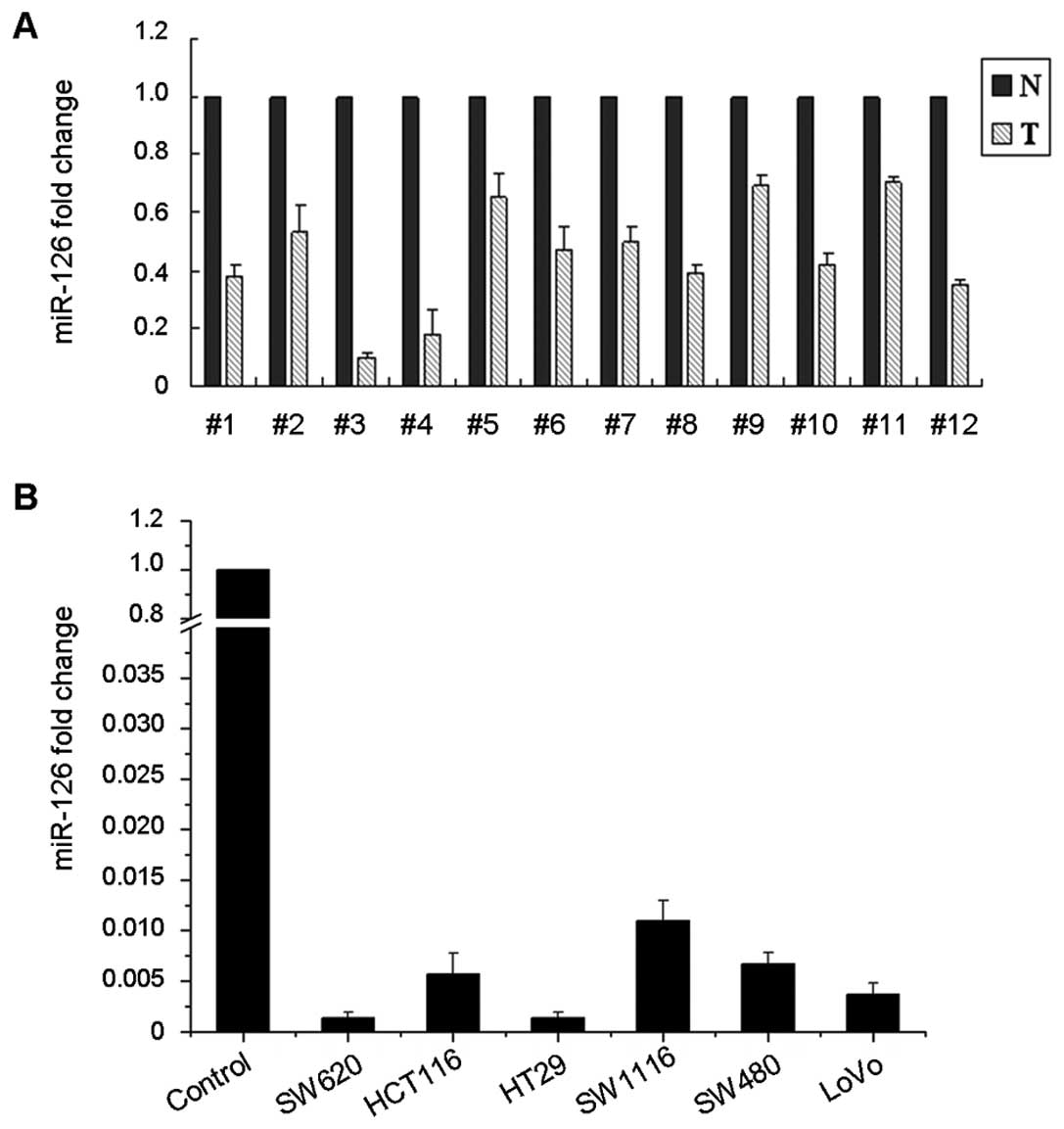
We performed real-time PCR using TaqMan probe to
detect the endogenous miR-126 level in primary CRC tissues and cell
lines. miR-126 was significantly decreased in all of the 12 CRC
tissues when compared to their matched adjacent normal colonic
tissues (Fig. 1A). We extended the
test to 6 human CRC cell lines: LoVo, SW620, SW480, SW1116, HT29
and HCT116. These 6 cell lines showed notable loss of miR-126,
whereas the control normal colonic mucosa pooled from 3 individuals
expressed a strong level of miR-126 (Fig. 1B).
miR-126 directly suppresses the
expression of VEGF at the post-transcriptional level
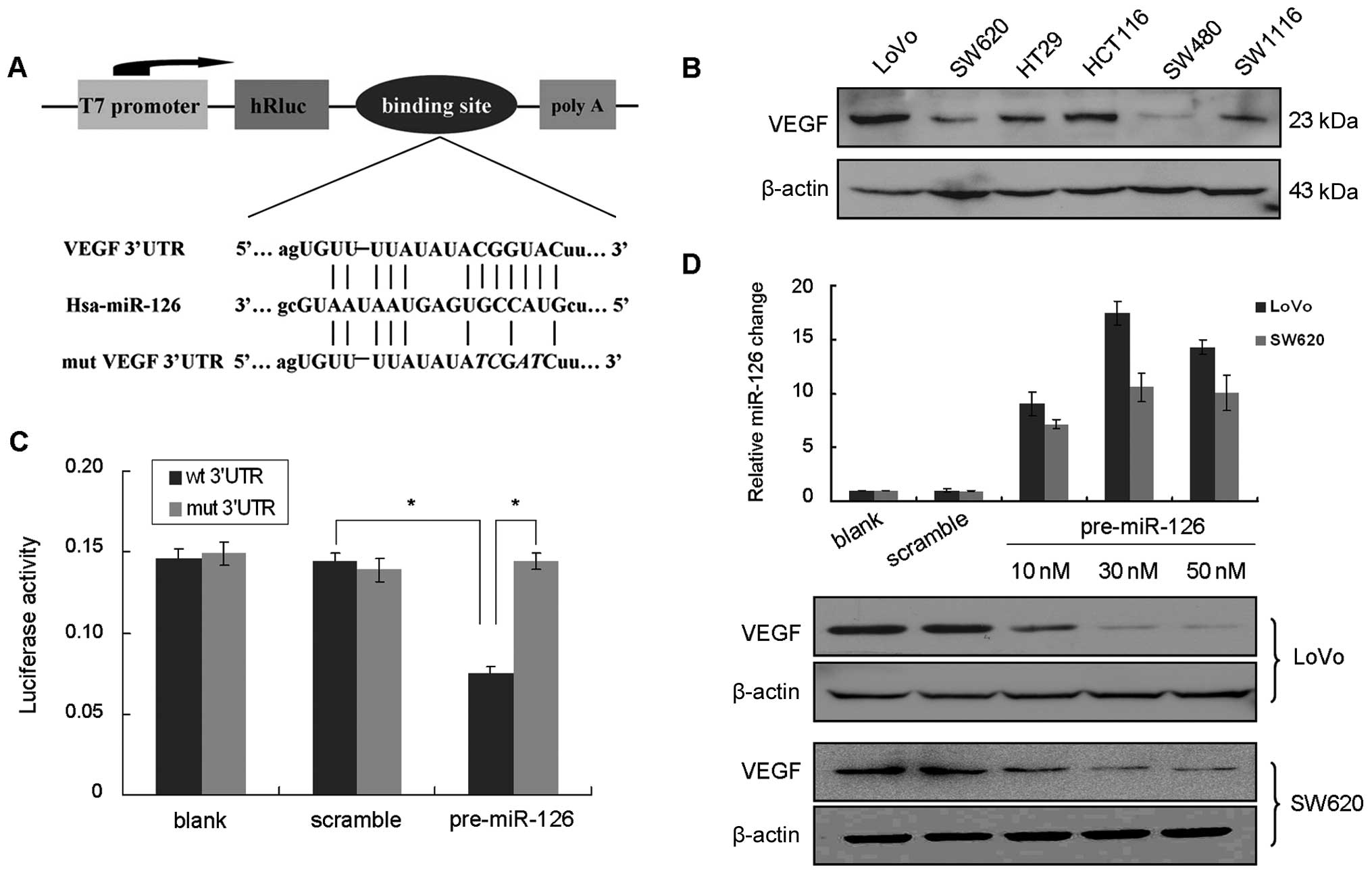
Using in silico prediction database, we
hypothesized that VEGF is a potential target gene of miR-126
(Fig. 2A). We initially tested the
VEGF expression profile in 6 CRC cell lines. All of the cell lines
exhibited notable expression of VEGF (Fig. 2B). LoVo and SW620 cells were
selected to verify our hypothesis. Both LoVo and SW620 cells were
transfected with pre-miR-126 to restore miR-126 expression.
Restoration of miR-126 in the cell lines significantly suppressed
VEGF protein expression (Fig. 2D).
However, no alteration in VEGF mRNA was observed by qPCR (data not
shown). This indicates that miR-126 may target VEGF at the
translational level.
To further confirm whether the inhibition of VEGF is
due to the interaction between miR-126 and the putative binding
site in 3′UTR of VEGF mRNA, we constructed a luciferase reporter
vector with the putative VEGF 3′UTR binding site for miR-126 and
its mutant version by site direct mutagenesis (Fig. 2A). We transfected the luciferase
reporter vector alone or together with pre-miR-126 or scramble
control into HEK293 cells. A significant decrease in luciferase
activity was noted when the VEGF 3′UTR vector was co-transfected
with pre-miR-126, compared to the mutant vector, whereas the
miR-126-mediated suppression of luciferase activity was abolished
in the mutant VEGF 3′UTR vector (Fig.
2C). These results confirm a direct interaction of miR-126 on
VEGF 3′UTR.
miR-126 inhibits CRC cell growth,
migration and invasion
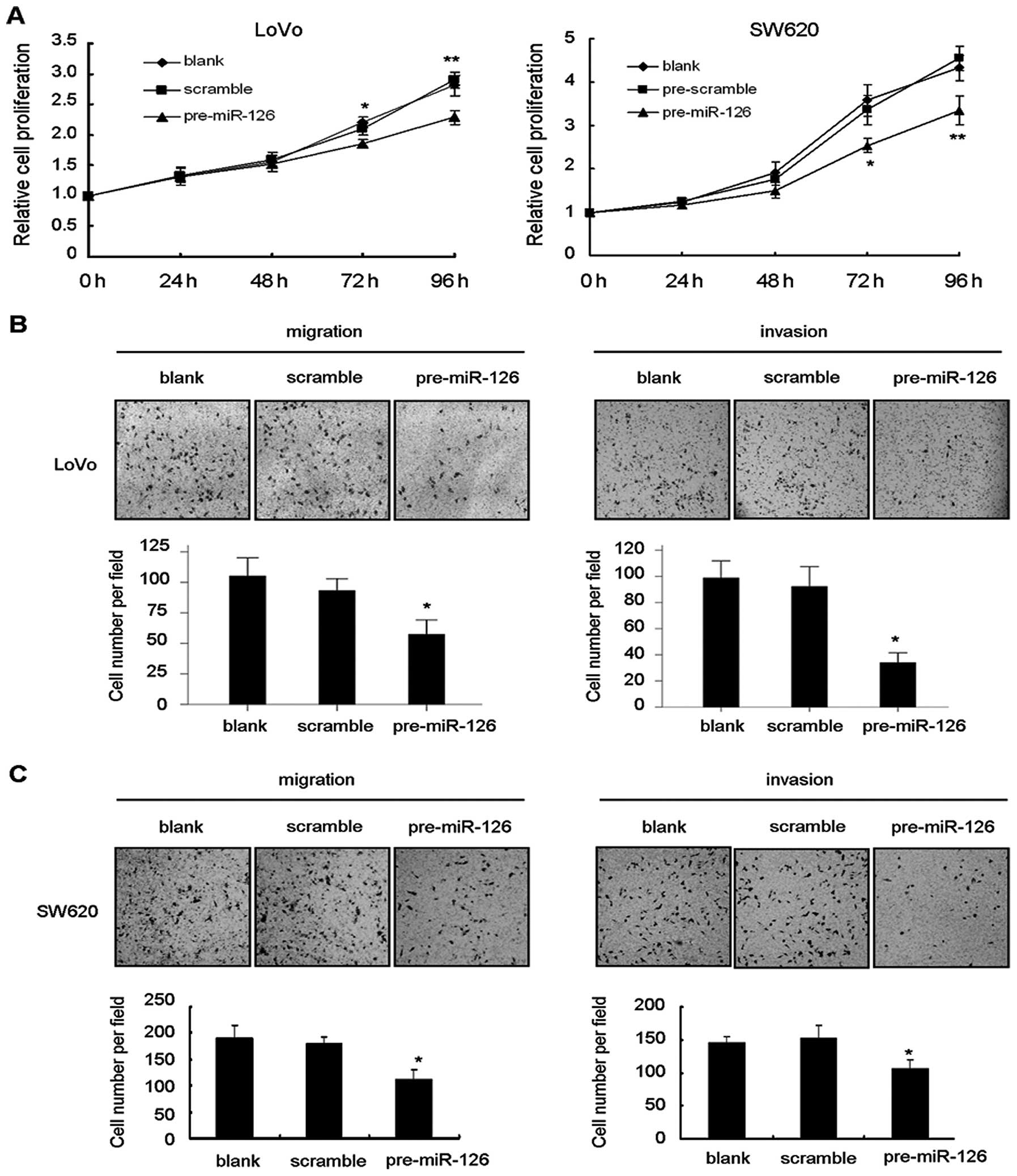
We performed gain of function assays to validate
whether miR-126 regulates cellular processes, including cell
growth, migration and invasion. Restoration of miR-126 in LoVo and
SW620 cells significantly inhibited cell growth (Fig. 3A). In the Transwell migration assay,
restoration of miR-126 in LoVo cells apparently impaired cell
migration and invasion when compared to the blank and scramble
control (Fig. 3B). Similar results
were also observed in parallel assays using SW620 cells (Fig. 3C).
miR-126 exerts anti-angiogenenic effects
on CRC
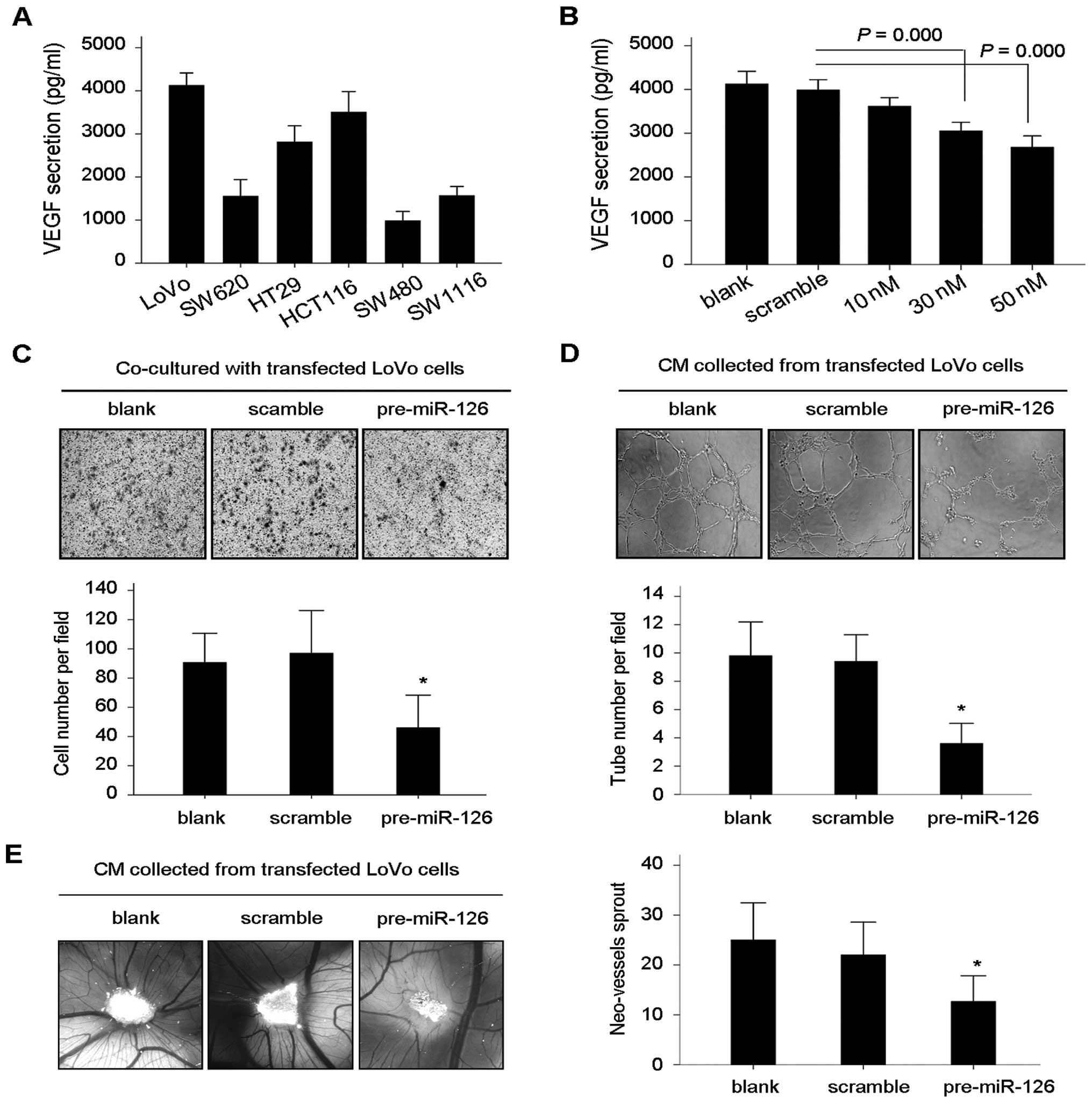
We investigated whether miR-126 regulates tumor
vasculature via mediation of VEGF expression. First, ELISA assay
was performed to detect VEGF secretion by CRC cell lines (Fig. 4A) and the concentration of VEGF in
conditioned medium (CM) obtained from pre-miR-126-transfected LoVo
cells (Fig. 4B). Restoration of
miR-126 resulted in decreased VEGF secretion by LoVo cells to the
culture medium (Fig. 4B). When
human microvascular endothelial cells (HMVECs) were co-cultured
with pre-miR-126-transfected LoVo cells in the Transwell system,
the migration of HMVECs was significantly inhibited (Fig. 4C). Correspondingly, in the
endothelial tube formation assay, we observed the reduced
spontaneous ability of HMVECs to form capillary tubes in the
presence of CM obtained from pre-miR-126-transfected LoVo cells,
when compared with tube formation in the presence of CM from the
blank and scramble controls (Fig.
4D). For further validation of the anti-angiogenic effect of
miR-126 in vivo, we performed chick embryo chorioallantoic
membrane assay (CAM) using gelatin sponge loaded with the different
CM as mentioned. The neo-vessel formation was significantly
inhibited in the presence of CM from the pre-miR-126-transfected
LoVo cells (Fig. 4E).
miR-126 is epigenetically silenced in
CRC
To validate whether miR-126 is silenced by DNA
methylation, we examined the methylation status of EGFL7, the host
gene of miR-126, in primary CRC tissues and cell lines. The CpG
island status within the EGFL7 promoter region, along with the
localization of PCR products, is presented in Fig. 5A. Clinicopathologic features of CRC
patients are listed in Table I.
Notably, there was no significant association observed between
methylation status and TNM stage. Using methylation-specific PCR
(MSP), all of the CRC cell lines showed extensively methylation of
the EGFL7 promoter (Fig. 5B).
Bisulfate sequencing (BSP) results exhibited extensive methylation
throughout the promoter region of these cell lines (Fig. 5C). We treated the same panel of CRC
cell lines with 5-aza-CdR. Compared to the untreated cells,
enhanced expression of miR-126 and downregulation of VEGF were
observed in the demethylated cells (Fig. 5D). These results suggest that
promoter methylation resulted in the silencing of miR-126 in CRC,
and may be partly responsible for the high VEGF expression in
CRC.
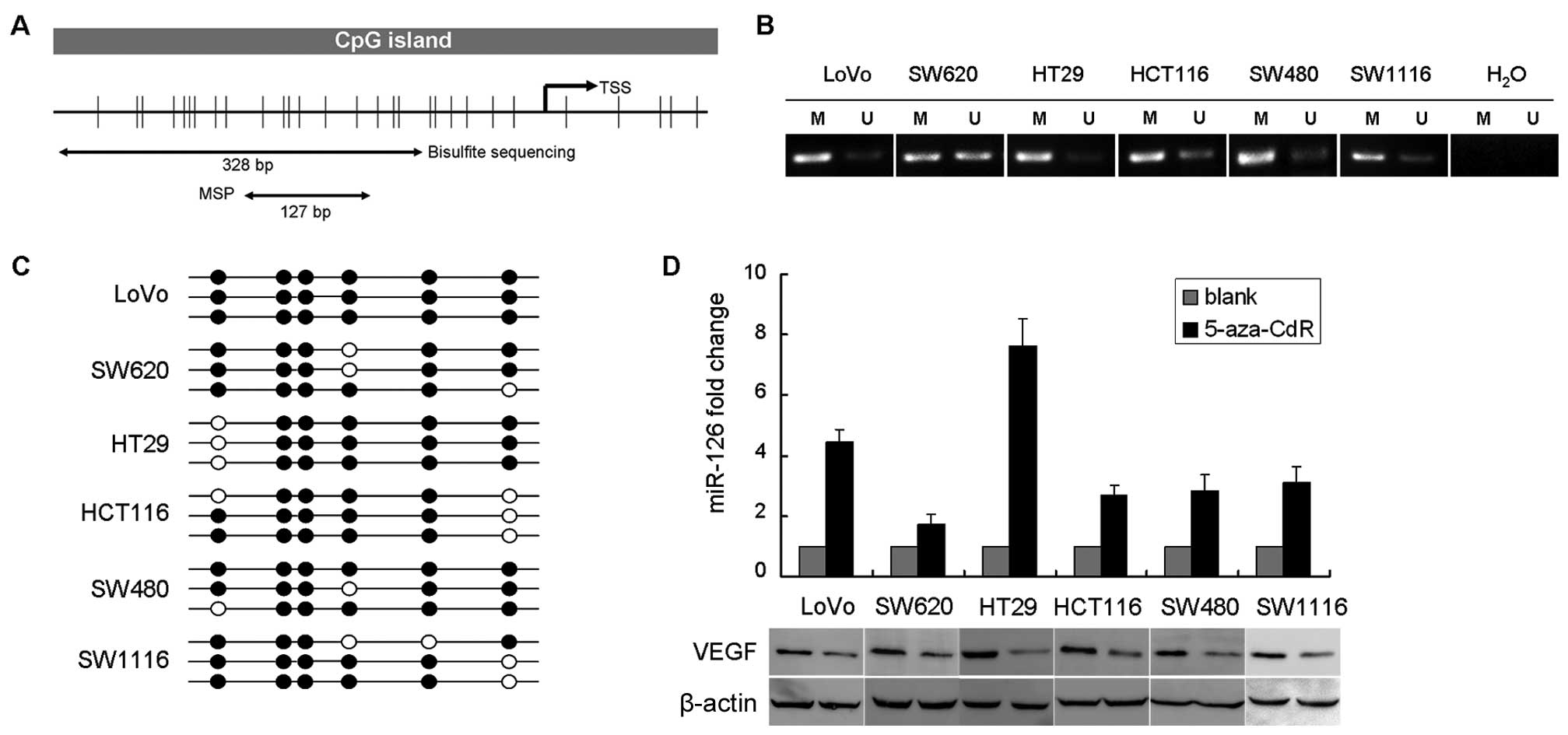 | Figure 5Analysis of the methylation status of
miR-126 CpG islands. (A) miR-126 is an intronic miRNA located
within intron 7 of the EGFL7 gene. Top, location of miR-126 within
the EGFL7 gene. Bottom, map of the EGFL7 CpG island within the
promoter region, with the positions of PCR products used for
methylation analysis. Vertical tick marks, CpG sites; E, exon; TSS,
putative transcribe start site. (B) Methylation status of the EGFL7
promoter region in 6 CRC cell lines by MSP. M, methylated; U,
unmethylated. (C) Bisulfate sequencing of CpG islands within the
EGFL7 promoter region in 6 CRC cell lines. Each circle indicates a
CpG dinucleotide. Black circle, methylated CpG; open circle,
unmethylated CpG. Three clones were sequenced for each cell line.
(D) miR-126 fold change was determined by TaqMan real-time PCR and
VEGF expression was determined by western blotting in 6 CRC cell
lines following 5-aza-CdR treatment. miRNA, microRNA; EGFL7,
epidermal growth factor-like domain 7; CRC, colorectal cancer; MSP,
methylation-specific PCR; VEGF, vascular endothelial growth factor;
5-aza-CdR, 5-aza-2′-deoxycytidine. |
 | Table IClinicopathological features of the
CRC patients analyzed for miR-126 methylation. |
Table I
Clinicopathological features of the
CRC patients analyzed for miR-126 methylation.
|
Characteristics | miR-126 methylated
n=52 | miR-126
unmethylated n=10 | Total n=62 | P-value |
|---|
| Age (years), mean ±
SD | 57.83±13.76 | 57.1±11.61 | 57.71±13.35 | 0.156a |
| Age, ≥60 years | 25 (48.1) | 4 (40) | 29 (46.8) | 0.902b |
| Female, n (%) | 27 (51.9) | 5 (50) | 32 (51.6) | 0.911b |
| Proximal site, n
(%) | 8 (15.4) | 2 (20) | 10 (16.1) | 1b |
| Poorly
differentiation, n (%) | 8 (15.4) | 1 (10) | 9 (14.5) | 1b |
| Mucinous
adenocarcinoma, n (%) | 3 (5.8) | 2 (20) | 5 (8.1) | 0.379b |
| TNM, n (%) | | | | 0.348b |
| I | 9 (17.3) | 4 (40) | 13 (21.0) | |
| II | 15 (28.8) | 2 (20) | 17 (27.4) | |
| III | 23 (44.2) | 4 (40) | 27 (43.5) | |
| IV | 5 (9.6) | 0 (0) | 5 (8.1) | |
Discussion
The majority of deaths from tumors result from
complications caused by metastasis. Therefore, targeting metastatic
disease is a pivotal anticancer strategy. miRNAs have been
implicated in the regulation of cellular processes which are
deregulated in tumors, including proliferation, apoptosis,
differentiation, cell migration and invasion (25), and even tumor angiogenesis (26). Recent studies have identified
various miRNAs that may promote (27,28) or
inhibit (29,30) tumor invasion and metastasis,
providing potential therapeutic targets for anti-metastatic
strategy. miR-126 is well known as one of the angiogenesis
regulatory miRNAs that are termed angiomiRs (31). Emerging evidence indicates that
miR-126 plays a regulatory role in tumor progression. Tavazoie
et al(10) reported that
miR-126 inhibits overall tumor growth and proliferation, and
suppresses metastatic colonization and angiogenesis by blocking
endothelial recruitment in breast cancer (32). In pancreatic cancer, restoration of
miR-126 results in reduced cellular migration, invasion, and
induction of the epithelial marker E-cadherin via suppression of
ADAM9 (11). Enhanced expression of
miR-126 was also reported to increase the sensitivity of lung
cancer cells to anticancer agents (33). Based on these findings, we
hypothesized that miR-126 may be involved in CRC metastatic
processes. The most significant finding of our study was that
restoration of miR-126 directly suppressed VEGF expression
consequently inhibiting cell invasion and tumor angiogenesis in
CRC. In addition, DNA methylation was responsible, at least in
part, for the silencing of miR-126 expression in CRC.
Our results showed that miR-126 was commonly
downregulated in 6 CRC cell lines and the 12 CRC patient tissues,
which is consistent with a previous study (21). On the contrary, Otsubo et
al(17) reported miR-126 was
highly expressed in cultured and primary gastric cancer cells. This
implies that the regulatory role of miR-126 may be specific to
tumor context. Using a luciferase reporter, we revealed that
miR-126 directly binds to a specific complementary site within the
3′ untranslated region of VEGF mRNA. In the gain of function
assays, we restored miR-126 expression in metastatic LoVo cells and
found that re-expression of miR-126 suppressed VEGF expression
post-transcriptionally. Moreover, restoration of miR-126 impaired
cell growth, migration and invasion capability. It is essential for
VEGF to regulate tumor progression through interaction with its
tyrosine kinase receptors (VEGFRs) (34). Particularly, activation of
VEGF/VEGFR1 signaling was found to lead to significant induction of
cell motility and invasiveness of CRC cells (35). Therefore, our data indicated that
miR-126 inhibited cell migration and invasion through inhibition of
VEGF expression. Since tumor cell-released VEGF contributes to
tumor vasculature via stimulation of the sprouting and
proliferation of endothelial cells (26), we investigated the involvement of
miR-126 in tumor angiogenesis of CRC. Expectedly, due to the
decreased secretion of VEGF by CRC cells, the migration and
sprouting of HMVECs were impaired in vitro, and neo-vessel
formation decreased in vivo. Therefore, these findings
suggest that miR-126 is involved in metastatic processes.
DNA methylation and associated silencing of
tumor-suppressor genes is a molecular hallmark of human tumors
(36). Recently, this phenomenon
has been extended to miRNAs with tumor-suppressor features, which
are downregulated in multiple tumors (37–39).
Saito and Jones (40) reported that
DNA methylation induced downregulation of EGFL7, and that
demethylation treatment with 5-aza-CdR simultaneously resulted in
restoration of miR-126 and its host gene, EGFL7, since as an
intronic miRNA, miR-126 tends to co-express with EGFL7. In the
present study, we observed extensive promoter methylation in 52 out
of 62 primary CRC tissues and 6 cell lines. In addition, 5-aza-CdR
treatment of the CRC cell lines restored miR-126 expression and
thereby led to a decrease in VEGF expression. Our results suggest
that silencing of miR-126 by promoter methylation is an important
mechanism underlying dysregulation of VEGF expression in CRC.
Tumor metastasis is defined as a consecutive process
including local invasion, intravasation, cell survival in the
circulatory system, extravasation, and colonization in a secondary
site (41). Additionally,
angiogenesis is also essential for tumor metastasis. Hurst et
al(42) recently proposed a
novel category of cancer-related miRNAs termed metastamiRs that are
associated with metastatic processes. For instance, miR-21 is a
mastermind of metastasis and promotes cell survival, migration,
invasion, intravasation and metastasis (43,44),
whereas the miR-200 family is delinquent and its absence
contributes to the EMT phenotype (45). These metastamiRs are potential
cancer prognostic markers and therapeutic targets for metastatic
cancers. Our findings provide evidence for the role of miR-126 as a
metastamiR through targeting VEGF.
In conclusion, we revealed the inhibitory effects of
miR-126 on VEGF, and partly elucidated the potential mechanism by
which miR-126 is implicated in CRC metastasis. miR-126 may be a
potential target for the therapeutic strategy against CRC.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (30971520) and the Science and
Technology Planning Project of Guangdong Province
(2010B031600098).
Abbreviations:
|
VEGF
|
vascular endothelial growth factor
|
|
UTR
|
untranslated region
|
|
miRNA
|
microRNA
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
References
|
1
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar
|
|
2
|
Steeg PS: Metastasis suppressors alter the
signal transduction of cancer cells. Nat Rev Cancer. 3:55–63. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Winter J, Jung S, Keller S, Gregory RI and
Diederichs S: Many roads to maturity: microRNA biogenesis pathways
and their regulation. Nat Cell Biol. 11:228–234. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev Cancer. 6:259–269. 2006.
View Article : Google Scholar
|
|
5
|
Place RF, Li LC, Pookot D, Noonan EJ and
Dahiya R: MicroRNA- 373 induces expression of genes with
complementary promoter sequences. Proc Natl Acad Sci USA.
105:1608–1613. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lim LP, Lau NC, Garrett-Engele P, Grimson
A, Schelter JM, Castle J, Bartel DP, Linsley PS and Johnson JM:
Microarray analysis shows that some microRNAs downregulate large
numbers of target mRNAs. Nature. 433:769–773. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dalmay T and Edwards DR: MicroRNAs and the
hallmarks of cancer. Oncogene. 25:6170–6175. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fish JE, Santoro MM, Morton SU, Yu S, Yeh
RF, Wythe JD, Ivey KN, Bruneau BG, Stainier DY and Srivastava D:
miR-126 regulates angiogenic signaling and vascular integrity. Dev
Cell. 15:272–284. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang S, Aurora AB, Johnson BA, Qi X,
McAnally J, Hill JA, Richardson JA, Bassel-Duby R and Olson EN: The
endothelial-specific microRNA miR-126 governs vascular integrity
and angiogenesis. Dev Cell. 15:261–271. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tavazoie SF, Alarcón C, Oskarsson T, Padua
D, Wang Q, Bos PD, Gerald WL and Massagué J: Endogenous human
microRNAs that suppress breast cancer metastasis. Nature.
451:147–152. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hamada S, Satoh K, Fujibuchi W, Hirota M,
Kanno A, Unno J, Masamune A, Kikuta K, Kume K and Shimosegawa T:
MiR-126 acts as a tumor suppressor in pancreatic cancer cells via
the regulation of ADAM9. Mol Cancer Res. 10:3–10. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Feng R, Chen X, Yu Y, Su L, Yu B, Li J,
Cai Q, Yan M, Liu B and Zhu Z: miR-126 functions as a tumour
suppressor in human gastric cancer. Cancer Lett. 298:50–63. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Donnem T, Fenton CG, Lonvik K, Berg T,
Eklo K, Andersen S, Stenvold H, Al-Shibli K, Al-Saad S, Bremnes RM
and Busund LT: MicroRNA signatures in tumor tissue related to
angiogenesis in non-small cell lung cancer. PLoS One. 7:e296712012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiao LR, Frampton AE, Jacob J, Pellegrino
L, Krell J, Giamas G, Tsim N, Vlavianos P, Cohen P, Ahmad R, Keller
A, Habib NA, Stebbing J and Castellano L: MicroRNAs targeting
oncogenes are down-regulated in pancreatic malignant transformation
from benign tumors. PLoS One. 7:e320682012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Miko E, Margitai Z, Czimmerer Z, Várkonyi
I, Dezso B, Lányi A, Bacsó Z and Scholtz B: miR-126 inhibits
proliferation of small cell lung cancer cells by targeting SLC7A5.
FEBS Lett. 585:1191–1196. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Crawford M, Brawner E, Batte K, Yu L,
Hunter MG, Otterson GA, Nuovo G, Marsh CB and Nana-Sinkam SP:
MicroRNA-126 inhibits invasion in non-small cell lung carcinoma
cell lines. Biochem Biophys Res Commun. 373:607–612. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Otsubo T, Akiyama Y, Hashimoto Y, Shimada
S, Goto K and Yuasa Y: MicroRNA-126 inhibits SOX2 expression and
contributes to gastric carcinogenesis. PLoS One. 6:e166172011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Watahiki A and Wang Y, Morris J, Dennis K,
O’Dwyer HM, Gleave M, Gout PW and Wang Y: MicroRNAs associated with
metastatic prostate cancer. PLoS One. 6:e249502011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ferrara N and Kerbel RS: Angiogenesis as a
therapeutic target. Nature. 438:967–974. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Grothey A and Galanis E: Targeting
angiogenesis: progress with anti-VEGF treatment with large
molecules. Nat Rev Clin Oncol. 6:507–518. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li XM, Wang AM, Zhang J and Yi H:
Down-regulation of miR-126 expression in colorectal cancer and its
clinical significance. Med Oncol. 28:1054–1057. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Saito Y, Friedman JM, Chihara Y, Egger G,
Chuang JC and Liang G: Epigenetic therapy upregulates the tumor
suppressor microRNA-126 and its host gene EGFL7 in human
cancer cells. Biochem Biophys Res Commun. 379:726–731. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Watanabe K, Emoto N, Hamano E, Sunohara M,
Kawakami M, Kage H, Kitano K, Nakajima J, Goto A, Fukayama M,
Nagase T, Yatomi Y, Ohishi N and Takai D: Genome structure-based
screening identified epigenetically silenced microRNA associated
with invasiveness in non-small-cell lung cancer. Int J Cancer.
130:2580–2590. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Roccaro AM, Hideshima T, Raje N, Kumar S,
Ishitsuka K, Yasui H, Shiraish N, Ribatti D, Nico B, Vacca A,
Dammacco F, Richardson PG and Anderson KC: Bortezomib mediates
antiangiogenesis in multiple myeloma via direct and indirect
effects on endothelial cells. Cancer Res. 66:184–191. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kloosterman WP and Plasterk RH: The
diverse functions of microRNAs in animal development and disease.
Dev Cell. 11:441–450. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Weis SM and Cheresh DA: Tumor
angiogenesis: molecular pathways and therapeutic targets. Nat Med.
17:1359–1370. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gaziel-Sovran A, Segura MF, Di Micco R,
Collins MK, Hanniford D, Vega-Saenz de Miera E, Rakus JF, Dankert
JF, Shang S, Kerbel RS, Bhardwaj N, Shao Y, Darvishian F, Zavadil
J, Erlebacher A, Mahal LK, Osman I and Hernando E:
MiR-30b/30d regulation of GalNAc transferases enhances
invasion and immunosuppression during metastasis. Cancer Cell.
20:104–118. 2011. View Article : Google Scholar
|
|
28
|
Yang CH, Yue J, Pfeffer SR, Handorf CR and
Pfeffer LM: MicroRNA-21 regulates the metastatic behavior of B16
melanoma cells. J Biol Chem. 286:39172–39178. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fang JH, Zhou HC, Zeng C, Yang J, Liu Y,
Huang X, Zhang JP, Guan XY and Zhuang SM: MicroRNA-29b suppresses
tumor angiogenesis, invasion, and metastasis by regulating matrix
metalloproteinase 2 expression. Hepatology. 54:1729–1740. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu Y, Zhao F, Wang Z, Song Y, Luo Y, Zhang
X, Jiang L, Sun Z, Miao Z and Xu H: MicroRNA-335 acts as a
metastasis suppressor in gastric cancer by targeting Bcl-w and
specificity protein 1. Oncogene. 31:1398–1407. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang S and Olson EN: AngiomiRs - key
regulators of angiogenesis. Curr Opin Genet Dev. 19:205–211. 2009.
View Article : Google Scholar
|
|
32
|
Png KJ, Halberg N, Yoshida M and Tavazoie
SF: A microRNA regulon that mediates endothelial recruitment and
metastasis by cancer cells. Nature. 481:190–194. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu X, Li H, Long L, Hui L, Chen H, Wang
X, Shen H and Xu W: miR-126 enhances the sensitivity of non-small
cell lung cancer cells to anticancer agents by targeting vascular
endothelial growth factor A. Acta Biochim Biophys Sin. 44:519–526.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stoeltzing O, Liu W, Reinmuth N, Parikh A,
Ahmad SA, Jung YD, Fan F and Ellis LM: Angiogenesis and
antiangiogenic therapy of colon cancer liver metastasis. Ann Surg
Oncol. 10:722–733. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fan F, Wey JS, McCarty MF, Belcheva A, Liu
WB, Bauer TW, Somcio RJ, Wu Y, Hooper A, Hicklin DJ and Ellis LM:
Expression and function of vascular endothelial growth factor
receptor-1 on human colorectal cancer cells. Oncogene.
24:2647–2653. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Esteller M: Epigenetics in cancer. N Engl
J Med. 358:1148–1159. 2008. View Article : Google Scholar
|
|
37
|
Lujambio A, Calin GA, Villanueva A, Ropero
S, Sánchez-Céspedes M, Blanco D, Montuenga LM, Rossi S, Nicoloso
MS, Faller WJ, Gallagher WM, Eccles SA, Croce CM and Esteller M: A
microRNA DNA methylation signature for human cancer metastasis.
Proc Natl Acad Sci USA. 105:13556–13561. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Toyota M, Suzuki H, Sasaki Y, Maruyama R,
Imai K, Shinomura Y and Tokino T: Epigenetic silencing of
microRNA-34b/c and B-cell translocation gene 4
is associated with CpG island methylation in colorectal cancer.
Cancer Res. 68:4123–4132. 2008.PubMed/NCBI
|
|
39
|
Huang YW, Liu JC, Deatherage DE, Luo J,
Mutch DG, Goodfellow PJ, Miller DS and Huang TH: Epigenetic
repression of microRNA-129-2 leads to overexpression of SOX4
oncogene in endometrial cancer. Cancer Res. 69:9038–9046. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Saito Y and Jones PA: Epigenetic
activation of tumor suppressor microRNAs in human cancer cells.
Cell Cycle. 5:2220–2222. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nguyen DX, Bos PD and Massagué J:
Metastasis: from dissemination to organ-specific colonization. Nat
Rev Cancer. 9:274–284. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hurst DR, Edmonds MD and Welch DR:
Metastamir: the field of metastasis-regulatory microRNA is
spreading. Cancer Res. 69:7495–7498. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang P, Zou F, Zhang X, Li H, Dulak A,
Tomko RJ Jr, Lazo JS, Wang Z, Zhang L and Yu J: microRNA-21
negatively regulates Cdc25A and cell cycle progression in colon
cancer cells. Cancer Res. 69:8157–8165. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Asangani IA, Rasheed SA, Nikolova SA,
Leupold JH, Colburn NH, Post S and Allgayer H: MicroRNA-21 (miR-21)
post-transcriptionally downregulates tumor suppressor Pdcd4 and
stimulates invasion, intravasation and metastasis in colorectal
cancer. Oncogene. 27:2128–2136. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gregory PA, Bert AG, Paterson EL, Barry
SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y and Goodall GJ:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View Article : Google Scholar : PubMed/NCBI
|