Introduction
It is well established that the human complement
system is a central part of the innate immune response. Complement
is known to play a major role as a first defense against microbes
(1,2), and also participates in diverse
physiological processes and contributes to various immune and
inflammatory diseases (3–6). There are three conventional mechanisms
of complement activation, known as the classical, lectin and
alternative pathways (7,8). Activation of complement causes
recruitment of immune cells; opsonization of coated cells; and
direct killing of affected cells through a membrane attack complex
(MAC). During complement activation, soluble proinflammatory
peptide fragments C3a and C5a are released from C3 and C5
molecules, respectively. C3a and C5a are referred to as
anaphylatoxins and exhibit a variety of biological activities in
the immune response (9–11).
There is increasing evidence for the contribution of
complement activation to cancer progression (12,13).
During carcinogenesis, tumor cells acquire genetic and epigenetic
alterations that promote their malignant growth. As a result, the
complement system can recognize these tumor cells. For decades,
complement has been recognized as an effector arm of the immune
system that contributes to the destruction of tumor cells (14,15);
however, cancer cells can resist the harmful effects of complement
by different extracellular and intracellular mechanisms. In fact,
new findings on the contribution of complement to tumor growth have
challenged the paradigm that complement always protects against
tumors. For example, some studies have revealed that the generation
of anaphylatoxins C3a and C5a in the tumor microenvironment leads
to significant tumor progression (12,13,16,17).
Nevertheless, the precise roles of C3a and C5a in human breast
cancer are still controversial.
Response gene to complement 32 (RGC-32) is a novel
gene whose expression is induced by complement (18–20).
The RGC-32 gene is abundantly expressed in the placenta, skeletal
muscle, kidney and liver. Upregulation of RGC-32 gene expression
has been demonstrated in a wide range of cell lines in response to
a variety of stimuli, such as sublytic C5b-9, steroid hormones and
growth factors (18,19,21).
It has been reported that RGC-32 plays an important role in
cellular proliferation and differentiation (18,22),
particularly in human pancreatic cancer and colon cancer (21,23).
Based on these findings, the role of the RGC-32 gene in C5a-induced
proliferation of breast cancer cells and its regulation by
signaling pathways warrant further investigation.
In the present study, we evaluated the role of C3a
and C5a in human breast cancer cells. We found that breast cancer
cells are able to enhance proliferation in response to C5a
stimulation compared with non-malignant breast epithelial cells.
Blockade of the C5a receptor (C5aR) strongly slowed the
proliferation of breast cancer cells exposed to C5a. Further
investigation revealed that the upregulation of the RGC-32 gene
could contribute to C5a-induced breast cancer cell proliferation.
Meanwhile, Akt activation was found to be necessary for C5a-induced
RGC-32 expression and breast cancer cell proliferation. These
results provide novel information concerning the relationship
between complement activation and breast cancer, which may
influence the development of future therapeutic strategies.
Materials and methods
Reagents
Polyclonal antibodies against RGC-32 were purchased
from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Monoclonal
antibodies against β-actin were purchased from Cell Signaling
Technology (Danvers, MA, USA). HRP-conjugated anti-rabbit IgG and
HRP-conjugated anti-mouse IgG antibodies, 20X LumiGLO®
reagent and 20X peroxide were purchased from Cell Signaling
Technology. RIPA lysis buffer was purchased from Thermo Scientific
(Fremont, CA, USA). Tbe Cell Counting Kit-8 (CCK-8) was from
Dojindo Laboratories (Kumamoto, Japan). The incision enzymes
HindIII and EcoRV as well as T4 DNA ligase were
purchased from Fermentas (Burlington, Ontario, Canada). The
QIAprep® Spin Miniprep kit was obtained from Qiagen
(Hilden, Germany). The mammalian expression vector of pcDNA3.1 and
Lipofectamine 2000 were from Invitrogen (Carlsbad, CA, USA). The
shRNA expression plasmids of pGPU6/GFP were purchased from
GenePharma (Shanghai, China). pGL3-basic vector and the dual
luciferase assay system were from Promega (Madison, WI, USA). High
Capacity cDNA reverse transcription kits and TaqMan®
Fast Advanced Master Mix were obtained from ABI (Foster, CA, USA).
Human C3a and C5a were from R&D Systems (Minneapolis, MN,
USA).
Cell culture
Established human malignant breast cancer cell lines
(MCF-7 and MDA-MB-231) as well as a human non-malignant breast
epithelial cell line (HBL-100) were purchased from the American
Tissue Culture Collection (ATCC, Rockville, MD, USA). Cells were
cultured in DMEM supplemented with 10% heat-inactivated fetal
bovine serum, penicillin (100 U/ml) and streptomycin (100 μg/ml).
Cells were incubated at 37°C in a 5% CO2 incubator.
Construction of the plasmids
To silence the human RGC-32 gene, different shRNA
sequences against RGC-32 mRNA (NM_014059.2) were designed. The
expression plasmids of RGC-32 shRNA were then constructed by using
the plasmids of pGPU6/GFP. The most effective shRNA expression
plasmids against the RGC-32 gene and the corresponding scrambled
shRNA expression plasmids as a negative control were chosen for
further functional experiments. In addition, pcDNA3.1/RGC-32 was
constructed by inserting the ORF of human RGC-32 cDNA (NM_014059.2)
into the plasmids of pcDNA3.1. The PCAF gene containing an HA tag
was amplified by polymerase chain reaction (PCR) from cDNA of
normal human breast epithelial cells. The PCR products and the
vector of pcDNA3.1 were digested with HindIII and
EcoRV, and then ligated by using T4 DNA ligase. Meanwhile,
the human RGC-32 promoter (−1600 nt ~ −1 nt before ATG) was
constructed into the pGL3-basic vector.
Cellular transfection
MCF-7 and MDA-MB-231 cell lines were transfected
using Lipofectamine 2000 according to the manufacturer’s
instructions (24).
Luciferase
MCF-7 and MDA-MB-231 cells at 80% confluency in
96-well plates were transfected with the pGL3-basic vector
containing a human RGC-32 promoter driving luciferase, using
Lipofectamine 2000. After 36 h of transfection, the cells were
stimulated with C5a for 9 h, and then lysed for luciferase assays
using the dual luciferase assay system.
Quantitative reverse
transcriptase-PCR
After isolation of total RNA from cells using TRIzol
reagent, an equal amount (2 μg) of total RNA from each sample was
synthesized as a first-strand cDNA using the High Capacity cDNA
reverse transcription kits. The cDNA was than amplified using
TaqMan® Fast Advanced Master Mix to detect the level of
RGC-32 mRNA. The 7300 Real-Time PCR system (ABI) was used for the
experiment. The reaction program consisted of an initial
denaturation step at 95°C for 10 min, denaturation at 95°C for 15
sec and annealing at 60°C for 60 sec for 40 cycles. The β-actin
gene was used as an internal control. The relative level of gene
expression was obtained by calculating the ratio of the cycle
numbers of the initial exponential amplification phase as
determined by the sequence detection system for RGC-32 and β-actin
using the 2−ΔΔCt formula. Each sample was assayed in
triplicate.
Western blot analysis
The proteins (40 μg) were subjected to 12% SDS
polyacrylamide gel electrophoresis and transferred onto PVDF
membranes (Millipore, Billerica, MA, USA) by a semi-dry
electrophoretic transfer cell (Bio-Rad). The membranes were
incubated for 1 h at room temperature in blocking buffer (5%
skimmed milk in TBS-T), and were then incubated with the antibodies
against RGC-32 and β-actin, respectively, overnight at 4°C. After
washing with TBST-T three times, the membranes were incubated with
HRP-conjugated anti-rabbit and anti-mouse antibodies for 1 h at
37°C. The bands were then visualized by the ECL detection system
(Bio-Rad) with 3 to 10 min exposure after washing the membranes.
Finally, the radiographic band density was measured using Quantity
One software (Bio-Rad). Control for protein loading was verified by
using β-actin as the internal standard. Similar results were
obtained in three independent experiments.
CCK-8 assay
MCF-7, MDA-MB-231 and HBL-100 cells were incubated
with CCK-8 for the final 3 h before detection. The formazan product
was then visualized at the absorbance of 450 nm, and the absorbance
was directly proportional to the number of living cells (25,26).
Each sample was assayed in triplicate.
Statistical analysis
All data are expressed as mean ± SD. All statistical
analysis was carried out using SPSS 11.5 software. One-way analysis
of variance (ANOVA), followed by post hoc Bonferroni test with
adjustment for multiple comparisons, was used to test the
significance of the differences between groups. A P-value <0.05
was considered to indicate a statistically significant
difference.
Results
C5a promotes the proliferation of human
breast cancer cells
In order to determine the role of C3a and C5a in the
proliferation of human breast cancer cells, two human breast cancer
cell lines (MCF-7 and MDA-MB-231) and a non-malignant breast
epithelial cell line (HBL-100) as a control were cultured in
vitro. Purified human C3a and C5a were then added to the human
breast cancer cell lines and the non-malignant breast epithelial
cell line, respectively, to evaluate their ability to induce the
proliferation of breast cancer cells. The results showed that C5a
stimulation enhanced the proliferative response of both human
breast cancer cell lines in vitro, but had no marked effect
on the proliferation of the non-malignant breast epithelial cell
line (Fig. 1A–C). No significant
increase in cellular proliferation in response to C3a was observed
in the breast cancer cell lines MCF-7 and MDA-MB-231 (data not
shown).
Blockade of C5aR slows the proliferation
of breast cancer cells
Since the breast cancer cell lines showed a
significant increase in proliferation in response to C5a
stimulation, the influence of C5a on the proliferation of breast
cancer cells was further studied using the anti-C5aR neutralizing
antibody as a C5aR antagonist. The result revealed that blockade of
C5aR with the anti-C5aR neutralizing antibody markedly reduced the
proliferation of the two breast cancer cell lines cultured with
human C5a (Fig. 2A and B). Taken
together, these findings (Figs. 1
and 2) indicate that anaphylatoxin
C5a promotes the proliferation of the human breast cancer cell
lines.
RGC-32 expression is induced in breast
cancer cells exposed to C5a
It is well accepted that RGC-32 is a novel gene
whose expression is induced by complement and other stimuli
(18–20). Given that C5a may contribute to the
proliferation of breast cancer cells, we further detected the
expression of the RGC-32 gene in breast cancer cells exposed to
C5a. The results showed that the expression of RGC-32 was markedly
elevated in the breast cancer cells in response to C5a stimulation
at both the mRNA and protein levels (Fig. 3A–D). The luciferase experiments
further demonstrated that transcription of the RGC-32 gene was
significantly increased in the breast cancer cells after
stimulation by C5a (Fig. 3E and F).
These findings indicate that C5a in vitro has the ability to
promote RGC-32 gene expression in human breast cancer cell
lines.
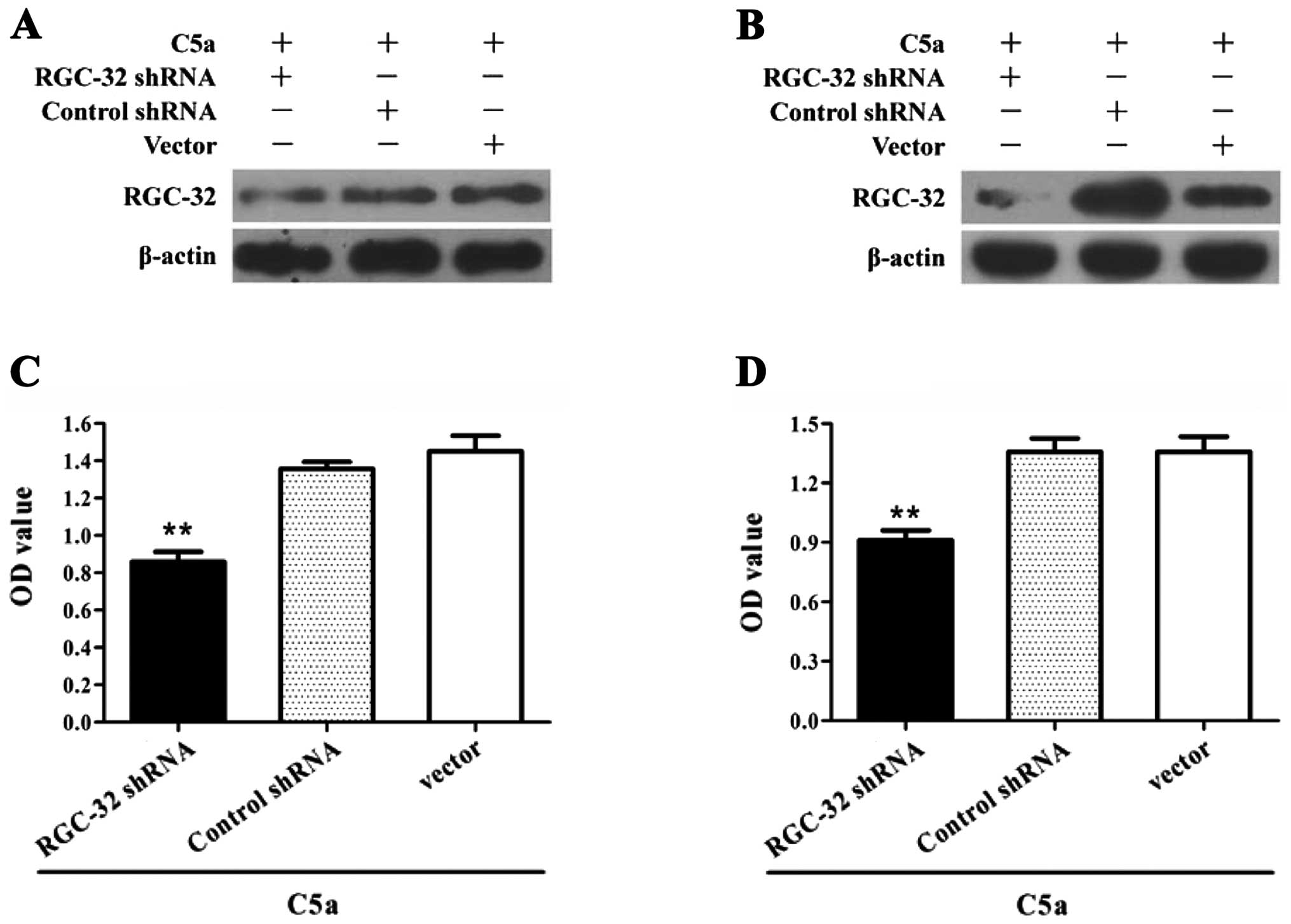
Expression of RGC-32 is required for
C5a-induced proliferation in breast cancer cells
It remains to be determined whether RGC-32
expression is necessary for the proliferation of breast cancer in
response to C5a stimulation. Thus, breast cancer cells were
transfected with RGC-32 shRNA-expressing plasmids, and then the
cellular proliferation in response to C5a stimulation was
determined by the CCK-8 assay. The results revealed that treatment
with RGC-32 shRNA obviously suppressed the proliferation of breast
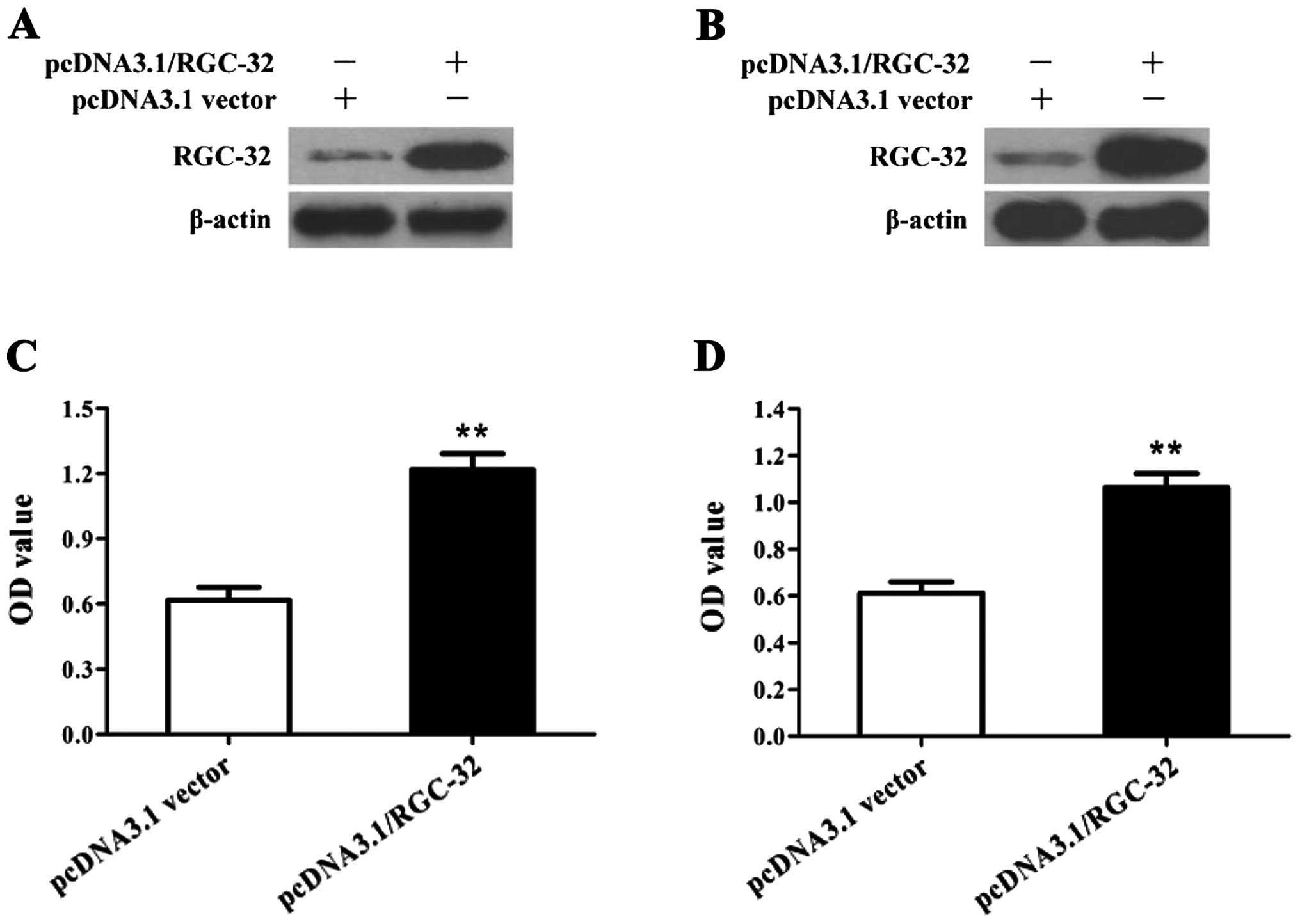
cancer cells in response to C5a stimulation (Fig. 4). Remarkably, overexpression of the
RGC-32 gene by plasmids of pcDNA3.1/RGC-32 promoted the
proliferation of the breast cancer cells (Fig. 5), indicating that RGC-32 expression
is involved in C5a-induced proliferation of breast cancer cells.
These findings indicate that C5a-induced RGC-32 expression is
required for the proliferation of breast cancer cells.
Akt activation is necessary for
C5a-induced RGC-32 expression and breast cancer cell
proliferation
Akt is a widely recognized contributory factor in
the proliferation of cancer cells. Since Akt activation has been
reported to promote cell proliferation in response to complement
stimulation (27–29), and both C5a stimulation and RGC-32
gene expression were involved in the proliferation of breast cancer
cells (Fig. 5), the influence of
Akt activation on C5a-induced RGC-32 expression and breast cancer
cell proliferation was further explored in the present study. The
results revealed that inhibition of Akt activation with Ly294002
strongly reduced C5a-induced RGC-32 expression and breast cancer
cell proliferation (Fig. 6). It was
therefore confirmed that Akt activation contributes to C5a-induced
RGC-32 expression and breast cancer cell proliferation.
Discussion
Breast carcinogenesis is a multi-step process in
which normal breast cells acquire genetic and epigenetic
alterations that result in the expression of many cancer-associated
molecules (30–32). However, the mechanism by which
breast cancer cells undergo proliferation still remains
controversial.
Complement is known to play important roles as a
first defense against microbes, and also participates in diverse
physiological processes and contributes to various immune and
inflammatory diseases (4,33–35).
In addition, there is increasing evidence for the contribution of
complement activation to cancer progression (12,16).
Cancer cells appear to be able to establish a convenient balance
between complement activation and inhibition, taking advantage of
complement initiation without suffering its deleterious effects.
Various studies have demonstrated that generation of anaphylatoxins
C3a and C5a in the tumor microenvironment leads to significant
tumor progression (12,16,17).
During the development of tumors, complement C3 and C5 activation
may support chronic inflammation, promote an immunosuppressive
micro-environment, induce angiogenesis, and activate cancer-related
signaling pathways. Little is known, however, concerning the roles
of C3a and C5a in breast cancer.
In the present study, we provide evidence for the
contribution of C5a to the proliferation of breast cancer cells. In
particular, we found that breast cancer cells were able to
proliferate in response to C5a stimulation much more efficiently
than non-malignant breast cells, indicating that breast cancer
cells are more sensitive to C5a stimulation than non-malignant
breast cells. In contrast, C3a did not significantly affect the
proliferation of breast cancer cells. Other effects of C3a on
breast cancer cells need to be determined to explore the potential
roles of C3a in breast cancer.
RGC-32 is a novel gene induced by complement and
other stimuli (18–20). It is well known that upregulation of
RGC-32 gene expression contributes to the proliferation of target
cells (18,19,21).
However, the precise role of the RGC-32 gene in C5a-induced
proliferation of breast cancer cells needs to be explored. Our
present study demonstrated that the expression of RGC-32 was
markedly elevated in breast cancer cells in response to C5a
stimulation. Further investigation revealed that gene transfer of
RGC-32 shRNA obviously diminished the proliferation of breast
cancer cells in response to C5a stimulation. Conversely,
overexpression of the RGC-32 gene by transfection of
pcDNA3.1/RGC-32 promoted the proliferation of breast cancer cells
without C5a stimulation. These findings indicate that C5a-induced
RGC-32 expression is required for the proliferation of breast
cancer cells.
Further experiments were carried out to discover the
possible mechanism by which C5a stimulates the expression of the
RGC-32 gene in breast cancer cells. The phosphoinositide 3-kinase
(PI3K)/Akt signaling pathway is one of the most frequently
dysregulated pathways in human cancers (36,37).
On the other hand, Akt activation has been reported to promote cell
proliferation in response to complement stimulation (27–29).
Thus, in a subsequent experiment, we sought to explore the role of
Akt activation in regulating RGC-32 expression and cellular
proliferation of breast cancer cells in response to C5a. The
results revealed that inhibition of Akt activation suppressed
RGC-32 expression and cellular proliferation of breast cancer cells
induced by C5a. Further studies need to be performed to discover
the possible regulatory mechanism by which Akt increases the
expression of the RGC-32 gene in breast cancer cells.
In conclusion, we evaluated the roles of C3a and C5a
in promoting the proliferation of breast cancer cells in
vitro. We found that breast cancer cells are able to undergo
proliferation in response to C5a stimulation rather than C3a. We
also found a significant increase in the expression of the RGC-32
gene in breast cancer cells induced by C5a. Further investigation
revealed that upregulation of the RGC-32 gene could contribute to
C5a-induced proliferation of breast cancer cells. Finally, Akt
activation was shown to be necessary for C5a-induced RGC-32
expression and breast cancer cell proliferation. On the basis of
these observations, we conclude that C5a promotes the proliferation
of breast cancer cells through Akt-activated RGC-32 gene
expression. These findings provide novel information concerning the
relationship between complement activation and breast cancer, which
may influence the development of future therapeutic strategies.
References
|
1
|
Tong HH, Lambert G, Li YX, et al: Deletion
of the complement C5a receptor alleviates the severity of acute
pneumococcal otitis media following influenza A virus infection in
mice. PloS One. 9:e951602014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Courtois A, Berthou C, Guezennec J,
Boisset C and Bordron A: Exopolysaccharides isolated from
hydrothermal vent bacteria can modulate the complement system. PloS
One. 9:e949652014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wende E, Laudeley R, Bleich A, et al: The
complement anaphylatoxin C3a receptor (C3aR) contributes to the
inflammatory response in dextran sulfate sodium (DSS)-induced
colitis in mice. PloS One. 8:e622572013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Elvington M, Blichmann P, Qiao F, et al: A
novel protocol allowing oral delivery of a protein complement
inhibitor that subsequently targets to inflamed colon mucosa and
ameliorates murine colitis. Clin Exp Immunol. 500–508. 2014.
View Article : Google Scholar
|
|
5
|
Hundgeburth LC, Wunsch M, Rovituso D, et
al: The complement system contributes to the pathology of
experimental autoimmune encephalomyelitis by triggering
demyelination and modifying the antigen-specific T and B cell
response. Clin Immunol. 146:155–164. 2013. View Article : Google Scholar
|
|
6
|
Laudisi F, Spreafico R, Evrard M, et al:
Cutting edge: the NLRP3 inflammasome links complement-mediated
inflammation and IL-1β release. J Immunol. 191:1006–1010.
2013.PubMed/NCBI
|
|
7
|
Ma R, Cui Z, Hu SY, et al: The alternative
pathway of complement activation may be involved in the renal
damage of human anti-glomerular basement membrane disease. PloS
One. 9:e912502014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Osthoff M, Brown KD, Kong DC, Daniell M
and Eisen DP: Activation of the lectin pathway of complement in
experimental human keratitis with Pseudomonas aeruginosa.
Mol Vis. 20:38–45. 2014.PubMed/NCBI
|
|
9
|
Cravedi P, Leventhal J, Lakhani P, Ward
SC, Donovan MJ and Heeger PS: Immune cell-derived C3a and C5a
costimulate human T cell alloimmunity. Am J Transplant.
13:2530–2539. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
van der Touw W, Cravedi P, Kwan WH,
Paz-Artal E, Merad M and Heeger PS: Cutting edge: Receptors for C3a
and C5a modulate stability of alloantigen-reactive induced
regulatory T cells. J Immunol. 190:5921–5925. 2013.PubMed/NCBI
|
|
11
|
Lara-Astiaso D, Izarra A, Estrada JC, et
al: Complement anaphylatoxins C3a and C5a induce a failing
regenerative program in cardiac resident cells. Evidence of a role
for cardiac resident stem cells other than cardiomyocyte renewal.
Springerplus. 1:632012. View Article : Google Scholar
|
|
12
|
Kanmura S, Uto H, Sato Y, et al: The
complement component C3a fragment is a potential biomarker for
hepatitis C virus-related hepatocellular carcinoma. J
Gastroenterol. 45:459–467. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Habermann JK, Roblick UJ, Luke BT, et al:
Increased serum levels of complement C3a anaphylatoxin indicate the
presence of colorectal tumors. Gastroenterology. 131:1020–1029.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stecker JR, Savage AA, Bruno JG, Garcia DM
and Koke JR: Dynamics and visualization of MCF7 adenocarcinoma cell
death by aptamer-C1q-mediated membrane attack. Nucleic Acid Ther.
22:275–282. 2012.PubMed/NCBI
|
|
15
|
Baig NA, Taylor RP, Lindorfer MA, et al:
Complement dependent cytotoxicity in chronic lymphocytic leukemia:
ofatumumab enhances alemtuzumab complement dependent cytotoxicity
and reveals cells resistant to activated complement. Leuk Lymphoma.
53:2218–2227. 2012. View Article : Google Scholar
|
|
16
|
Nitta H, Wada Y, Kawano Y, et al:
Enhancement of human cancer cell motility and invasiveness by
anaphylatoxin C5a via aberrantly expressed C5a receptor (CD88).
Clin Cancer Res. 19:2004–2013. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Corrales L, Ajona D, Rafail S, et al:
Anaphylatoxin C5a creates a favorable microenvironment for lung
cancer progression. J Immunol. 189:4674–4683. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fosbrink M, Cudrici C, Tegla CA, et al:
Response gene to complement 32 is required for C5b-9 induced cell
cycle activation in endothelial cells. Exp Mol Pathol. 86:87–94.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vlaicu SI, Cudrici C, Ito T, et al: Role
of response gene to complement 32 in diseases. Arch Immunol Ther
Exp. 56:115–122. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Badea TC, Niculescu FI, Soane L, Shin ML
and Rus H: Molecular cloning and characterization of RGC-32, a
novel gene induced by complement activation in oligodendrocytes. J
Biol Chem. 273:26977–26981. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vlaicu SI, Tegla CA, Cudrici CD, et al:
Epigenetic modifications induced by RGC-32 in colon cancer. Exp Mol
Pathol. 88:67–76. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li F, Luo Z, Huang W, et al: Response gene
to complement 32, a novel regulator for transforming growth
factor-beta-induced smooth muscle differentiation of neural crest
cells. J Biol Chem. 282:10133–10137. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhu L, Qin H, Li PY, et al: Response gene
to complement-32 enhances metastatic phenotype by mediating
transforming growth factor beta-induced epithelial-mesenchymal
transition in human pancreatic cancer cell line BxPC-3. J Exp Clin
Cancer Res. 31:292012. View Article : Google Scholar
|
|
24
|
Yunus MA, Chung LM, Chaudhry Y, Bailey D
and Goodfellow I: Development of an optimized RNA-based murine
norovirus reverse genetics system. J Virol Methods. 169:112–118.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li M, Han S, Zhang G, Wang Y and Ji Z:
Antiproliferative activity and apoptosis-inducing mechanism of
L-securinine on human breast cancer MCF-7 cells. Pharmazie.
69:217–223. 2014.PubMed/NCBI
|
|
26
|
Gong S, Tao Z, Liu X and Gan L: An
underlying prognosis predictor of hepatocellular carcinoma:
Oncoprotein 18. Biomed Rep. 2:85–88. 2014.PubMed/NCBI
|
|
27
|
Qiu W, Zhang Y, Liu X, et al: Sublytic
C5b-9 complexes induce proliferative changes of glomerular
mesangial cells in rat Thy-1 nephritis through TRAF6-mediated
PI3K-dependent Akt1 activation. J Pathol. 226:619–632. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tang H, Amara U, Tang D, Barnes MA,
McDonald C and Nagy LE: Synergistic interaction between C5a and
NOD2 signaling in the regulation of chemokine expression in RAW
264.7 macrophages. Adv Biosci Biotechnol. 4:30–37. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Curci C, Castellano G, Stasi A, et al:
Endothelial-to-mesenchymal transition and renal fibrosis in
ischaemia/reperfusion injury are mediated by complement
anaphylatoxins and Akt pathway. Nephrol Dial Transplant.
29:799–808. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Byler S, Goldgar S, Heerboth S, et al:
Genetic and epigenetic aspects of breast cancer progression and
therapy. Anticancer Res. 34:1071–1077. 2014.PubMed/NCBI
|
|
31
|
Faria JA, Correa NC, de Andrade C, et al:
SET domain-containing protein 4 (SETD4) is a newly identified
cytosolic and nuclear lysine methyltransferase involved in breast
cancer cell proliferation. J Cancer Sci Ther. 5:58–65. 2013.
|
|
32
|
Pei R, Wang P, Zhou Y, et al: Association
of BRCA1 K1183R polymorphism with survival in BRCA1/2-negative
Chinese familial breast cancer. Clin Lab. 60:47–53. 2014.PubMed/NCBI
|
|
33
|
Unnewehr H, Rittirsch D, Sarma JV, et al:
Changes and regulation of the C5a receptor on neutrophils during
septic shock in humans. J Immunol. 190:4215–4225. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu L, Zhang Y, Duan X, et al: C3a, C5a
renal expression and their receptors are correlated to severity of
IgA nephropathy. J Clin Immunol. 34:224–232. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Müller MC, Stroo I, Wouters D, et al: The
effect of C1-inhibitor in a murine model of transfusion-related
acute lung injury. Vox Sang. 107:71–75. 2014.PubMed/NCBI
|
|
36
|
Qin L, Ren Y, Chen AM, et al: Peroxisome
proliferator-activated receptor γ ligands inhibit VEGF-mediated
vasculogenic mimicry of prostate cancer through the AKT signaling
pathway. Mol Med Rep. 10:276–282. 2014.
|
|
37
|
Singel SM, Cornelius C, Zaganjor E, et al:
KIF14 promotes AKT phosphorylation and contributes to
chemoresistance in triple-negative breast cancer. Neoplasia.
16:247–256. 2014. View Article : Google Scholar : PubMed/NCBI
|