Introduction
Pancreatic cancer is the fourth leading cause of
cancer-associated mortalities worldwide and is characterized by
poor diagnosis and prognosis, and lack of efficient treatment
(1). At the time of pancreatic
cancer diagnosis, most of the patients have unresectable disease
(2). Considerable efforts have been
made to develop novel therapeutic treatments for this disease, but
progress on this issue is limited. Application of gemcitabine alone
or in combination with other chemotherapy drugs provides moderate
benefit for survival, but is also accompanied by severe
side-effects (3–6). Genetic screening of pancreatic cancer
types determines various mutations in tumors, such as the most
frequently mutated genes, including TP53, SMAD4 and Kras (7,8).
However, the therapeutic agents targeting these molecules have not
achieved an optimum effect (9).
Thus, identifying new molecular mechanisms involved in pancreatic
tumorigenesis is necessary to provide novel insights for developing
new and potent therapeutic options.
In recent years, microRNAs (miRNAs) have been shown
to be involved in the regulation of a variety of human diseases
(10). miRNAs are a subset of
small, non-coding and endogenous RNA of 18–24 nucleotides in length
that regulate gene expression by directly targeting the
3′-untranslated region (UTR) of mRNA leading to translational
inhibition (11,12). Recently, it was demonstrated that
miRNAs are involved in a variety of cell processes, including
apoptosis, proliferation and differentiation (10). Aberrant expression of miRNAs and
their target genes have been identified in many cancer types, which
are associated with oncogenesis and development (13). Aberrant expression of miRNAs is also
found in pancreatic cancer (14).
Findings of recent studies have shown that miR-216a, a member of
the miR-216 family, is highly decreased in pancreatic cancer
(15–17). However, the mechanism and function
of miR-216a in pancreatic cancer remains to be determined.
Janus kinase 2 (JAK2)/signal transducer and
activator of transcription 3 (STAT3) signaling pathway were shown
to be involved in various types of cancer, including pancreatic
cancer (18). STAT3 is a latent
cytosolic transcription factor that is phosphorylated by JAK2,
leading to STAT3 dimerization and nuclear localization to activate
downstream genes (19,20). The JAK2/STAT3 signaling pathway is
involved in various cell processes, including proliferation and
apoptosis, immune evasion and resistance, as well as angiogenesis
in various types of cancer (21–23).
Targeting JAK2/STAT3 may provide novel insight for pancreatic
cancer therapy, such as lestaurtinib and INCB-18424, which are used
in clinical trials to act on JAK2 (24).
Using bioinformatic algorithms, we identified JAK2
as a putative target gene of miR-216a. Considering the decreased
expression level of miR-216a in pancreatic cancer, the interaction
between JAK2 and miR-216a may provide novel insights into the
understanding of pancreatic cancer tumorigenesis as well as
developing new therapeutic approaches. In the present study, the
dual luciferase report assay was applied to verify whether JAK2 was
a direct target of miR-216a. The effect of miR-216a overexpression
or silencing on JAK2 and the downstream gene protein expression was
determined in pancreatic cancer cells. The effect of miR-216a on
cell growth and apoptosis in vitro and in vivo was
also investigated. Our results showed that miR-216a targeting JAK2
negatively regulated pancreatic carcinogenesis, suggesting that
miR-216a may be used as a potential target for the treatment of
pancreatic tumorigenesis.
Materials and methods
Animals and cell lines
BALB/c nude mice weighing 25–30 g were purchased
from the Medical Experimental Animal Center (Guangdong, China) and
raised under standard conditions of room temperature and a
dark-light cycle with free access to water. The animal experiments
were approved and reviewed by the Institutional Animal Care and Use
Committee of the China Medical University. The human Capan-2 and
PANC-1 pancreatic cancer cell lines obtained from the Chinese
Academy of Sciences (Shanghai, China) were cultured as per
supplier’s instructions. In brief, cells were grown in Dulbecco’s
modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, CA, USA)
supplemented with 10% fetal bovine serum (Invitrogen) containing
penicillin (100 U/ml) and streptomycin (100 mg/ml). Human embryonic
kidney (HEK) 293 cells were cultured in DMEM supplemented with 10%
fetal bovine serum and penicillin/streptomycin. The cells were
cultured in an incubator at 37°C with 5% CO2.
Luciferase reporter assay
The JAK2 3′-UTR was amplified using the primers:
JAK2 3′-UTR forward, 5′-CCGCTCGAG AAGAAATGACCTTCATTCTGAGACCA-3′ and
reverse, 5′-AAGGAAAAAAGCGGCCGCTAAAGTAAGAAACTA TTTTCTTTTTAATCAA-3′.
The mutated JAK2 3′-UTR was cloned using the primers: mut-JAK2
3′-UTR forward, 3′-GTA TCTATTTGTGGTGAATGATCGATCTAAATGGAACTA
TCTCCAAA-5′ and reverse, 5′-TTTGGAGATAGTTCC
ATTTAGATCGATCATTCACCACAAATAGATAC-3′ with a site-directed
mutagenesis kit (Haoran Bio, Shanghai, China), in which the
miR-216a binding site AGAGATT was mutated to CTCGCTC. The PCR
products were then sub-cloned into a pGL3 Luciferase Reporter
Vector (Promega, Madison, WI, USA) downstream of the luciferase
gene. To examine whether JAK2 was a target of miR-216a, HEK293
cells were seeded into 96-well plates and co-transfected with 100
ng pGL3 vector containing JAK2 3′-UTR or mutated forms with or
without 20 nmol/l miR-216a mimic (GenePharma, Shanghai, China)
using Lipofectamine (Invitrogen). The transfected cells were
cultured for 48 h, and luciferase activity was measured using the
Dual-Luciferase Reporter assay system (Promega).
Quantitative polymerase chain reaction
(RT-qPCR)
For mRNA analysis, total RNA from cells was
extracted using TRIzol (Invitrogen) according to the manufacturer’s
instructions. Complementary DNA was synthesized by using M-MLV
reverse transcriptase (Clontech, Palo Alto, CA, USA) according to
the standard protocol. miRVana kits (Ambion Inc., Austin, TX, USA)
and the TaqMan microRNA Reverse Transcription kit (Applied
Biosystems, Foster City, CA, USA) were used to measure miRNAs as
per the manufacturer’s instructions. Relative quantification of the
gene expression was performed using the 2−ΔΔCt method as
previously described (25). U6
(miRNA) or GAPDH (mRNA) was validated as the internal control.
Western blot analysis
Cell lysates from different experiments were
prepared using RIPA buffer, and the protein concentration was
measured using the Bradford assay. Protein (20 μg) was separated by
12% SDS-PAGE and transferred to nitro-cellulose membranes
(Amersham, Little Chalfont, UK). The membrane was then blocked with
2% non-fat dry milk at room temperature and incubated in primary
antibodies at 4°C overnight. Anti-phospho-JAK2 (D4A8) antibody,
anti-total JAK2 (D2E12) antibody, anti-phospho-STAT3 (D3A7) and
anti-total STAT3 (D3Z2G) at a 1:1,000 diltuion were purchased from
Cell Signaling Technology (Danvers, MA, USA). Anti-cleaved
caspase-3 antibody at a 1:500 dilution, anti-survivin antibody at a
1:600 dilution, anti-XIAP antibodies at a 1:800 dilution and
anti-GAPDH antibody at a 1:1,000 dilution were purchased from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The membrane was
subsequently incubated in peroxidase-conjugated secondary antibody
(Boster Corporation, Wuhan, Hubei, China) at a 1:1,000 dilution for
1 h. The protein bands were visualized using an enhanced
chemiluminescence detection system (Amersham).
Cell viability assay
Cell growth and viability were analyzed by an MTT
assay according to standard protocols. Briefly, the cells were
seeded in 96-well plates and miR-216a or anti-miR-216a mimic at a
final concentration of 20 nmol/l was added and incubated for 24,
48, 72 and 96 h. The previously applied medium was discarded, and
fresh medium containing MTT [5 mg/ml diluted in phosphate-buffered
saline (PBS)] was added and incubated for 4 h. Dimethyl sulfoxide
was used to dissolve the formazan for 15 min. Absorbance at 490 nm
was measured using an ELISA reader (BioTek, Winooski, VT, USA).
Apoptosis assay
Cell apoptosis was detected using Annexin V/PI
apoptosis kit (Invitrogen) according to the manufacturer’s
instructions. Briefly, the cells were harvested, washed with
ice-cold PBS, and resuspended in binding buffer. Subsequently, the
cells were incubated with 10 μl of Annexin V stock solution at 4°C
for 30 min. PI (5 μl) was then added and incubated for 5 min. The
cells were analyzed by a FACS analyzer (BD Biosciences, San Jose,
CA, USA).
Xenograft tumor growth
The PANC-1 pancreatic cancer cell line was
transfected with lentiviral vectors containing miR-216a or
anti-miR-216a (GenePharma). The transfected cells
(1×107) were suspended in PBS and injected
subcutaneously into the left groin. The length and width of the
tumor were measured every 5 days, and the volume was calculated as:
length × width 2 × 3/6. At 40 days after cell inoculation, the mice
were anesthetized by subcutaneous injection of sodium pentobarbital
(40 mg/kg). The tumor weight was assessed, and the total RNA and
protein were extracted for subsequent analysis.
Statistical analysis
Data were presented as the means ± standard
deviation (SD). Comparisons between two groups were determined by
the Student’s t-test, and among groups using one-way ANOVA followed
by Bonferroni post-hoc test. P<0.05 was considered to indicate a
statistically significant result. The data were analyzed using SPSS
version 11.5 (SPSS Inc., Chicago, IL, USA).
Results
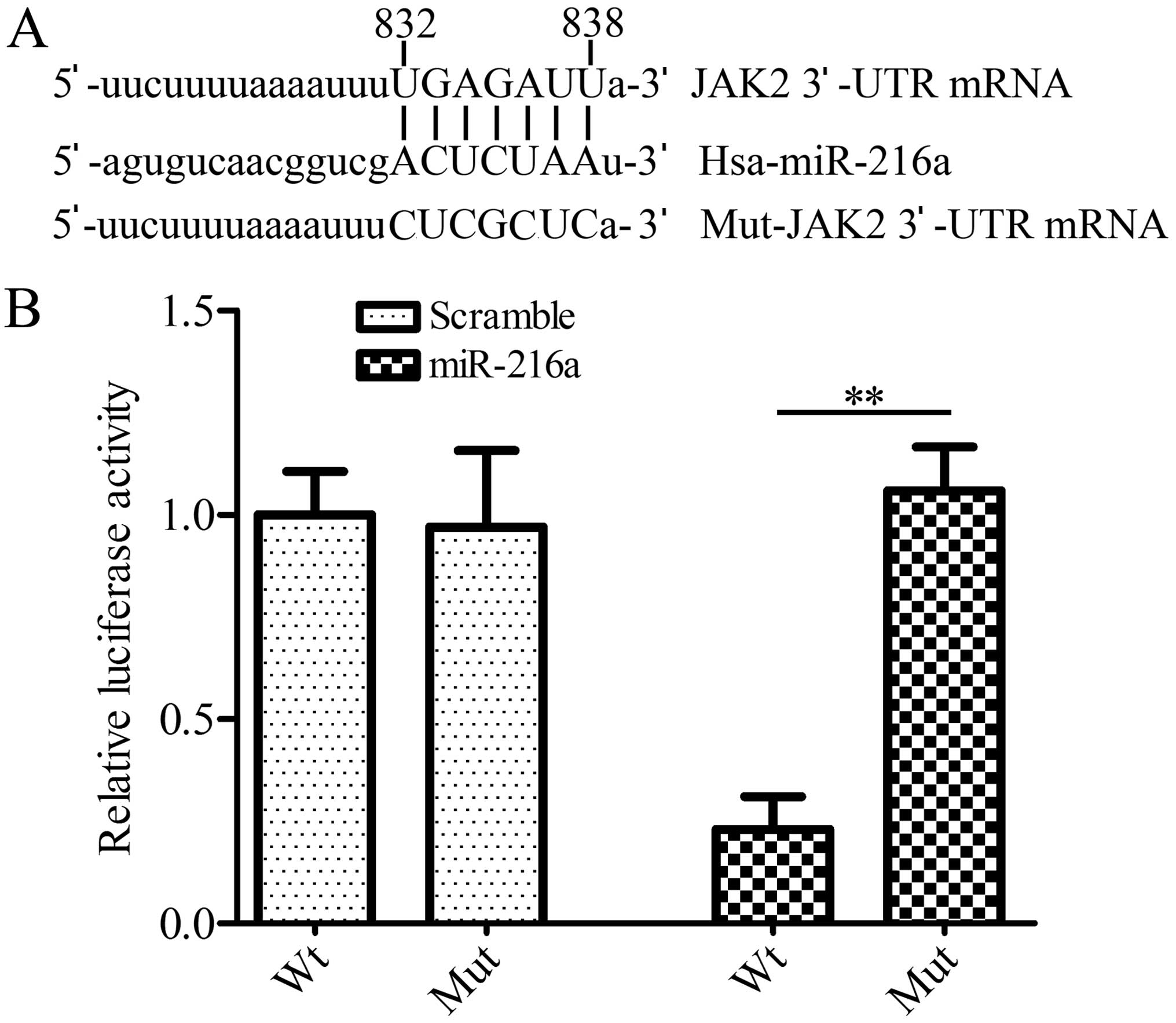
JAK2 is a target gene of miR-216a
Previous studies demonstrated that miR-216a was
significantly downregulated in pancreatic cancer (15–17).
However, the function of miR-216 in pancreatic cancer remains to be
determined. To investigate the biological role of miR-216a in
pancreatic cancer, we used bioinformatic algorithms to screen the
putative target. JAK2, which is involved in many cancer types,
including pancreatic cancer, was found to be a target gene of
miR-216a (Fig. 1A). To verify
whether miR-216a regulated JAK2 expression by directly interacting
with JAK2 mRNA, we cloned the 3′-UTR of human JKA2 downstream of
the luciferase reporter gene in pGL3 plasmid. HEK293 cells were
co-transfected with pGL3-JAK2-3′-UTR and miR216a mimics. The
results showed that miR216a mimics significantly (p<0.01)
decreased the luciferase activity compared with the control,
whereas miR216a mimics had no effect on the pGL3-mut-JAK2 3′-UTR
transfected cells (Fig. 1B). The
results suggested that miR-216a directly bound to 3′-UTR of
JAK2.
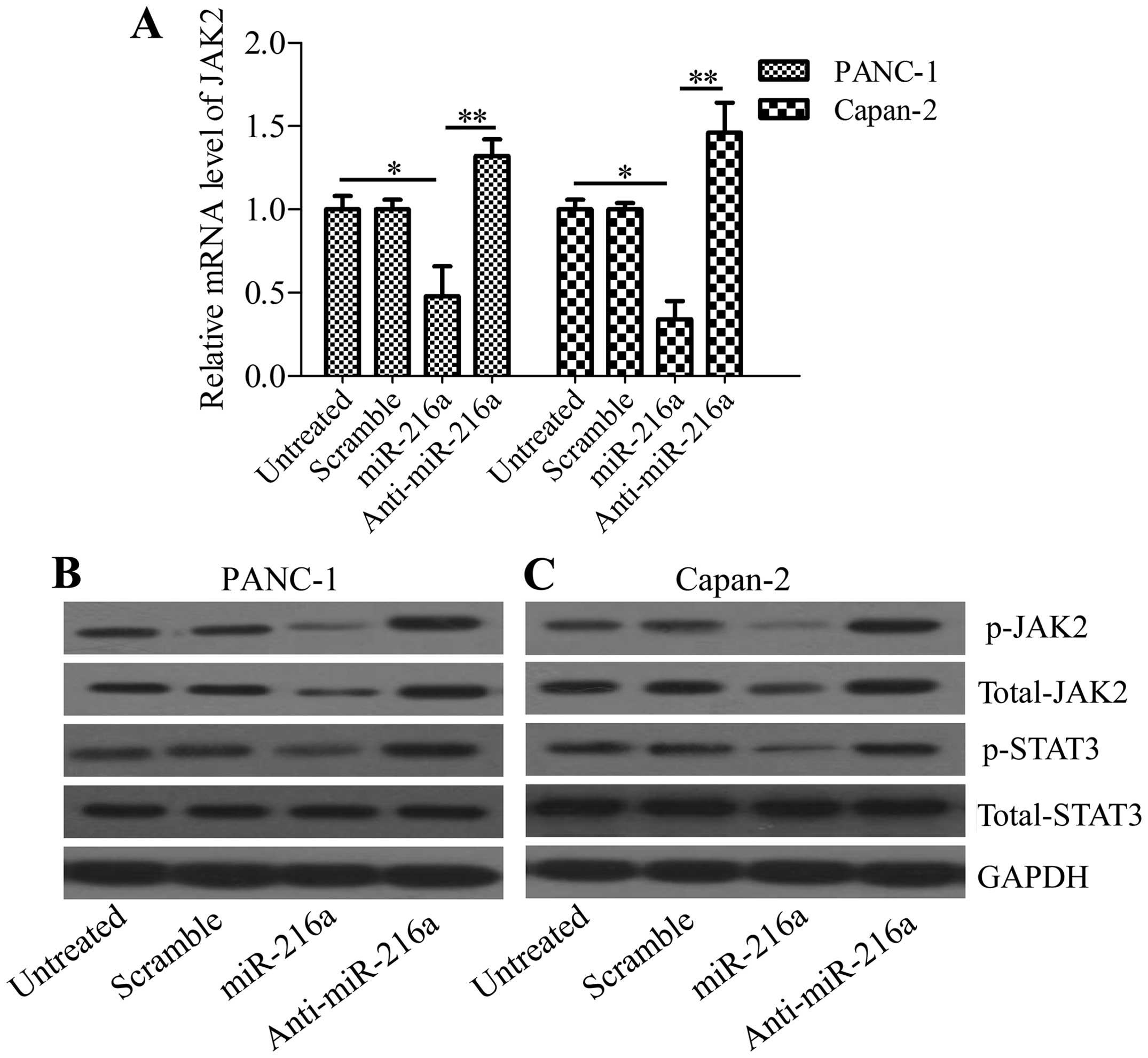
miR-216a downregulates JAK2 expression in
pancreatic cancer cells
To verify the interaction of miR-216a with JAK2,
pancreatic cancer cells were transfected with miR-216a mimics or
anti-miR-216a mimics and the expression of JAK2 was analyzed. The
results showed that miR-216a significantly downregulated JAK2 mRNA
levels, whereas anti-miR-216a markedly upregulated JAK2 mRNA levels
in PANC-1 and Capan-2 cells (Fig.
2A). These results were confirmed by the western blot analysis,
which revealed that phosphorylated JAK2 and total JAK2 protein
levels were decreased in miR-216a-transfected pancreatic cancer
cells. By contrast, anti-miR-216a increased phosphorylated JAK2 and
total JAK2 protein levels in anti-miR-216a-transfected PANC-1
(Fig. 2B) and Capan-2 cells
(Fig. 2C). The phosphorylation of
STAT3, the downstream gene of JAK2, was also inhibited by miR-216a
or increased by anti-miR-216a transfection, whereas the total
levels of STAT3 were unaffected (Fig.
2B and C). These data indicated that miR-216a directly targeted
JAK2 in pancreatic cancer cells.
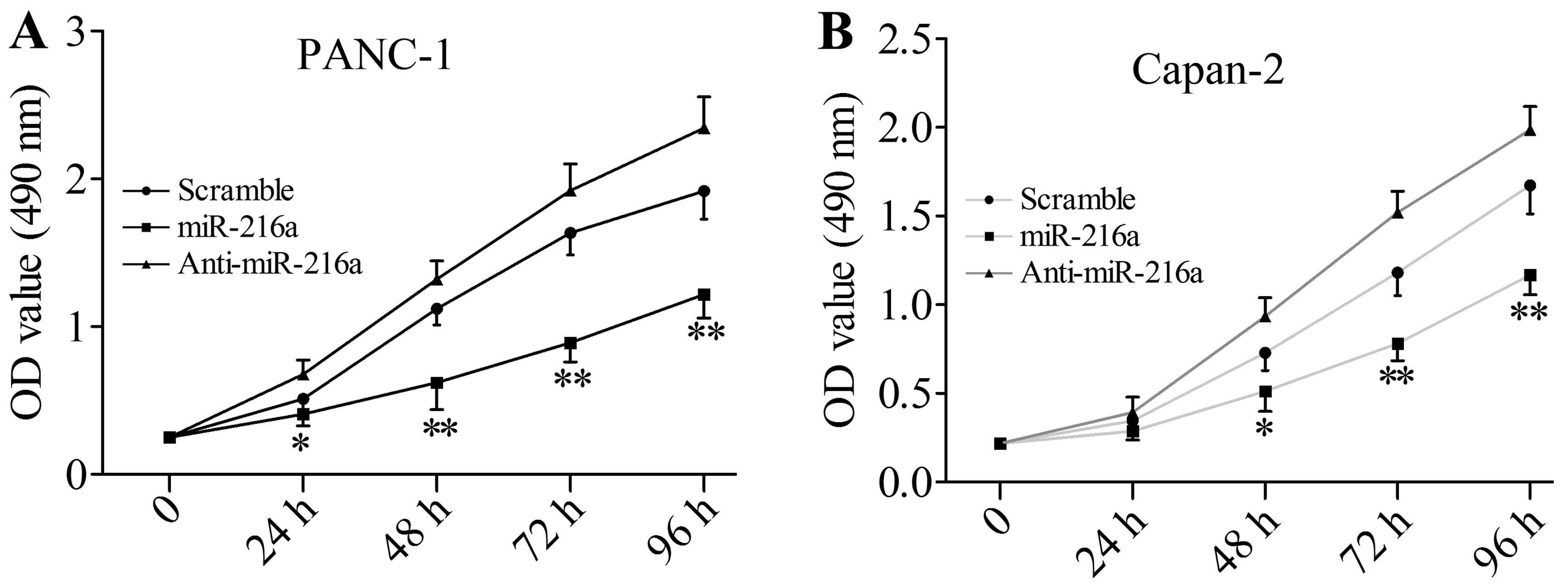
miR-216a suppresses cell growth and
viability in pancreatic cancer cells
Since JAK2 is directly targeted by miR-216a, we
investigated next whether miR-216a is able to disturb the cell
growth and viability of pancreatic cancer cells. Compared with the
scramble miRNA-transfected group, miR-216a mimics significantly
inhibited the cell growth and viability of PANC1 cells, whereas
anti-miR-216a markedly enhanced cell growth and viability (Fig. 3A). The same effect was observed in
Capan-2 cells (Fig. 3B).
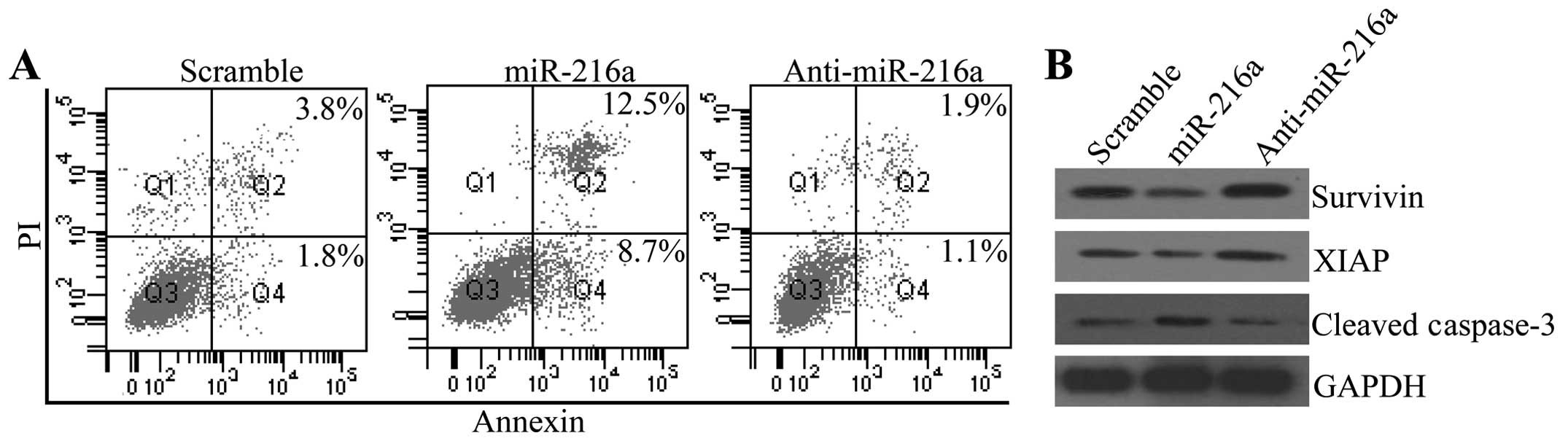
miR-216a facilitates cell apoptosis of
pancreatic cancer cells
To determine whether miR-216a was involved in the
progression of pancreatic cancer, we assessed its effect on cell
apoptosis. Fig. 4A shows that
miR-216a significantly increased apoptosis of PANC-1 cells, whereas
anti-216a inhibited cell apoptosis, compared with the control group
(Fig. 4A). To verify the inhibitory
effect of miR-216a on cell apoptosis, we measured the expression
alterations of survivin and XIAP, the downstream gene of JAK2/STAT3
that inhibited cell apoptosis. The western blot analysis
demonstrated that miR-216a transfection downregulated the protein
expression levels of survivin and XIAP, which were enhanced by
anti-miR-216a transfection. The pro-apoptotic protein cleaved
caspase-3 was also increased by miR-216a transfection and decreased
by anti-miR-216a transfection (Fig.
4B).
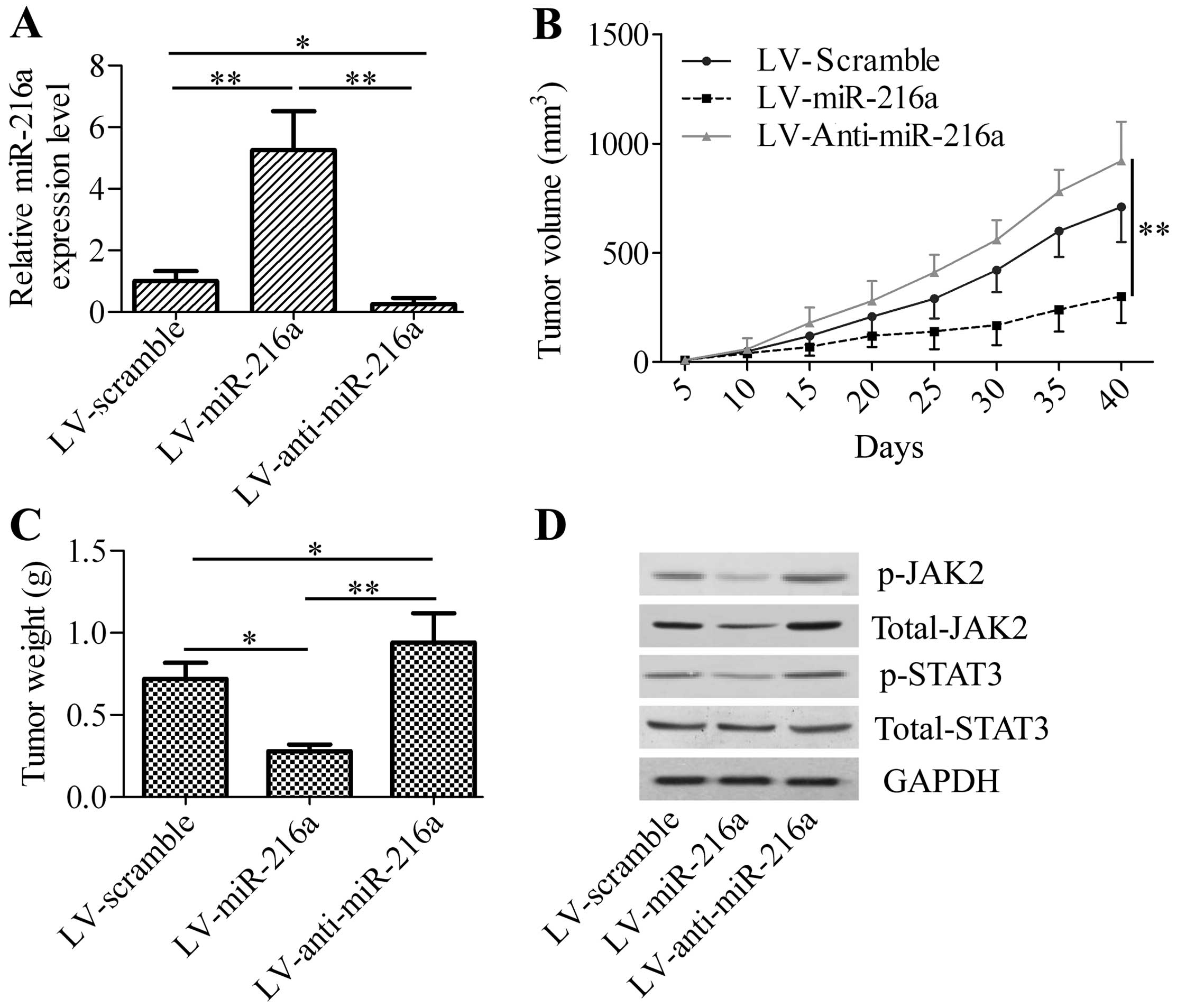
miR-216a represses xenograft tumor growth
in vivo
To gain insight into the effect of miR-216a on
pancreatic cancer, we inoculated LV-miR-216a-infected PANC-1 cells
into nude mice and measured the effect of miR-216a expression on
xenograft tumor growth. We initially detected the expression of
miR-216a in xenograft tumor tissues. The results showed that
LV-miR-216a-infected cancer cells exhibited an increase of miR-216a
levels in xenograft tumor tissues, whereas a significant decrease
of miR-216a levels was observed in the LV-anti-miR-216a-treated
group (Fig. 5A). We determined
whether miR-216a overexpression or silencing affects xenograft
tumor growth. LV-miR-216a markedly decreased xenograft tumor
growth, including tumor volume and weight (Fig. 5B and C), whereas LV-anti-miR-216a
significantly increased xenograft tumor growth compared with the
control group. Western blot analysis revealed that the
phosphorylated JAK2 and total JAK2 protein levels were decreased in
the LV-miR-216a xenograft tumor, whereas phosphorylated JAK2 and
total JAK2 protein levels were increased in LV-anti-miR-216a
xenograft tumor. The phosphorylation of STAT3, the downstream gene
of JAK2, was also significantly downregulated in the LV-miR-216a
xenograft tumor and upregulated in the LV-anti-miR-216a xenograft
tumor. However, LV-miR-216a or LV-anti-miR-216a exhibited no effect
of total STAT3 (Fig. 5D). These
results confirmed the tumor-suppressor activity of miR-216a on
pancreatic cancer cell growth by targeting and regulating JAK2.
Discussion
Previous studies have demonstrated that the
expression of miR-216a is significantly decreased in pancreatic
cancer patients (15) or a murine
model of pancreatic cancer (26).
Link et al suggested that miR-216a downregulation in feces
may be used as fecal miRNA biomarkers for pancreatic cancer
(16). Decreased miR-216a
expression is also verified in pancreatic intraepithelial neoplasms
(17). The abovementioned studies
suggest that miR-216a may be used as a diagnostic marker and as a
target for pancreatic cancer. However, the potential precise
mechanism of miR-216a in regulating pancreatic cancer remains to be
determined. To the best of our knowledge, the present study is the
first to provide evidence that miR-216a regulates pancreatic cancer
by directly targeting 3′-UTR of JAK2.
miR-216a is a member of the miR-216 family, which is
widely conserved in many species (27). The function of miR-216a is not well
delineated. miR-216a together with miR-217 is induced by
transforming growth factor-β target phosphatase and tensin
homologue, leading to Akt activation in glomerular mesangial cells
in kidney disorders (28). miR-216a
targets YB-1, which mediates Tsc-22 post-transcriptional
regulation, having an important function in the pathogenesis of
diabetic nephropathy (29).
Jeyapalan et al found that miR-216 suppresses tumorigenesis
and angiogenesis by targeting 3′-UTR of CD44 (27). In early hepatocarcinogenesis,
miR-216a, elevated by the androgen pathway, inhibits the tumor
suppressor in lung cancer-1 (30).
Increased miR-216a level is considered a biomarker of pancreatitis
(31,32). Thus, miR-216a apparently has a wide
biological effect on different conditions.
In the present study, we identified that miR-216a
interacted with JAK2 to negatively regulate pancreatic cancer.
Forced expression of miR-216a significantly inhibited cell growth
and promoted the cell apoptosis of pancreatic cancer cells. The
JAK2/STAT3 signaling pathway was markedly disturbed by miR-216a.
The aberrant activation of the JAK2/STAT3 signaling pathway is
considered to be extensively involved in tumorigenesis (33). In recent years, the JAK2V617F
mutation has been found to be involved in myeloproliferative
disorders, in which the 617th amino acid mutation leads to the
sustained activation of JAK2 and STAT3 (34). Disruption of JAK2/STAT3 leads to the
inhibition of gastric carcinogenesis (35). The JAK2/STAT3 signaling pathway is
also involved in pancreatic cancer. STAT3 is a critical factor in
Kras-induced pancreatic cancers (36). Disruption of STAT3 in the transgenic
pancreas profoundly suppresses the pancreatic metaplasia (37). NADPH oxidase promotes pancreatic
cancer by maintaining the sustained phosphorylation of JAK2 by
tyrosine phosphatases (38).
Inhibition of JAK2 and Notch together has a better effect than
single inhibitions on pancreatic cell progression (39). Activated STAT3 induces the elevation
of anti-apoptotic genes (40,41).
In the present study, we found that miR-216a inhibited the
phosphorylation of STAT3, and the downstream anti-apoptotic genes
survivin and XIAP were downregulated, leading to the apoptosis of
pancreatic cancer cells. Thus, targeting JAK2/STAT3 has potential
for the treatment of pancreatic cancer.
Application of miRNAs in the treatment of cancer has
achieved convincing antitumor effects in animal models (42). An increasing number of studies have
been performed to develop potential miRNAs in the treatment of
pancreatic cancer. miR-219-1-3p is a negative regulator of the
mucin MUC4 expression and inhibits the progression of pancreatic
cancer (43). miRNA-141 exerts an
inhibitory effect on cell proliferation and invasion by directly
targeting MAP4K4 (44). Miao et
al revealed that miR-203 suppresses cancer cell migration and
invasion by targeting caveolin-1 in pancreatic cancer (45). More recently, miR-196a has been
found to bind nuclear factor κ-B-inhibitor α to facilitate
pancreatic cancer progression (46). miR let-7 blocks STAT3 activation by
JAK2 by increasing SOCS3 expression in pancreatic cancer cells
(47). miR-375 significantly
downregulates in pancreatic cancer (48) targeting JAK2/STAT3 to suppress
gastric carcinogenesis (35),
suggesting an inhibitory role of miR-375 on JAK2 in pancreatic
cancer cells. In the present study, we provided evidence that
miR-216a, which is frequently down-regulated in pancreatic cancer,
directly targeted JAK2 to inhibit pancreatic cancer growth in
vitro and in vivo. A recent study has demonstrated that
miR-216a targeting beclin 1, an autophagy-related gene, inhibits
cell autophagy in endothelial cells, suggesting a potential
function in the pathogenesis in cardiovascular disorders (49). More investigations are required to
determine whether miR-216a is associated with autophagy in
pancreatic cancer cells due to the important function of autophagy
in pancreatic cancer development (50).
In summary, to the best of our knowledge, this is
the first study to demonstrate the direct interaction between
miR-216a and JAK2, and the regulation effect of miR-216a on JAK2 in
pancreatic cancer cells. Considering the tumorigenic function of
JAK2 in pancreatic cancer, targeting JAK2/STAT3 signaling pathway
by miR-126a provides novel insight for the development of useful
therapeutic strategy against pancreatic cancer.
Abbreviations:
|
miR-216a
|
microRNA-216a
|
|
JAK2
|
Janus kinase 2
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
XIAP
|
X-linked inhibitor of apoptosis
protein
|
|
UTR
|
untranslated region
|
References
|
1
|
Vincent A, Herman J, Schulick R, Hruban RH
and Goggins M: Pancreatic cancer. Lancet. 378:607–620. 2011.
View Article : Google Scholar
|
|
2
|
Kleeff J, Michalski CW, Friess H and
Büchler MW: Surgical treatment of pancreatic cancer: the role of
adjuvant and multimodal therapies. Eur J Surg Oncol. 33:817–823.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Burris HA III, Moore MJ, Andersen J, et
al: Improvements in survival and clinical benefit with gemcitabine
as first-line therapy for patients with advanced pancreas cancer: a
randomized trial. J Clin Oncol. 15:2403–2413. 1997.PubMed/NCBI
|
|
4
|
Poplin E, Feng Y, Berlin J, et al: Phase
III, randomized study of gemcitabine and oxaliplatin versus
gemcitabine (fixed-dose rate infusion) compared with gemcitabine
(30-minute infusion) in patients with pancreatic carcinoma E6201: a
trial of the Eastern Cooperative Oncology Group. J Clin Oncol.
27:3778–3785. 2009. View Article : Google Scholar
|
|
5
|
Heinemann V, Quietzsch D, Gieseler F, et
al: Randomized phase III trial of gemcitabine plus cisplatin
compared with gemcitabine alone in advanced pancreatic cancer. J
Clin Oncol. 24:3946–3952. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Conroy T, Desseigne F, Ychou M, et al:
FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N
Engl J Med. 364:1817–1825. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schneider G and Schmid RM: Genetic
alterations in pancreatic carcinoma. Mol Cancer. 2:152003.
View Article : Google Scholar
|
|
8
|
Hezel AF, Kimmelman AC, Stanger BZ,
Bardeesy N and Depinho RA: Genetics and biology of pancreatic
ductal adenocarcinoma. Genes Dev. 20:1218–1249. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chiu J and Yau T: Metastatic pancreatic
cancer: are we making progress in treatment? Gastroenterol Res
Pract. 2012:8989312012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mendell JT and Olson EN: MicroRNAs in
stress signaling and human disease. Cell. 148:1172–1187. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Winter J, Jung S, Keller S, Gregory RI and
Diederichs S: Many roads to maturity: microRNA biogenesis pathways
and their regulation. Nat Cell Biol. 11:228–234. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lujambio A and Lowe SW: The microcosmos of
cancer. Nature. 482:347–355. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Drakaki A and Iliopoulos D: MicroRNA-gene
signaling pathways in pancreatic cancer. Biomed J. 36:200–208.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hou B, Jian Z, Chen S, et al: Expression
of miR-216a in pancreatic cancer and its clinical significance. Nan
Fang Yi Ke Da Xue Xue Bao. 32:1628–1631. 2012.(In Chinese).
|
|
16
|
Link A, Becker V, Goel A, Wex T and
Malfertheiner P: Feasibility of fecal microRNAs as novel biomarkers
for pancreatic cancer. PLoS One. 7:e429332012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu J, Li A, Hong SM, Hruban RH and Goggins
M: MicroRNA alterations of pancreatic intraepithelial neoplasias.
Clin Cancer Res. 18:981–992. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Scholz A, Heinze S, Detjen KM, et al:
Activated signal transducer and activator of transcription 3
(STAT3) supports the malignant phenotype of human pancreatic
cancer. Gastroenterology. 125:891–905. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhong Z, Wen Z and Darnell JE Jr: Stat3: a
STAT family member activated by tyrosine phosphorylation in
response to epidermal growth factor and interleukin-6. Science.
264:95–98. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shuai K, Horvath CM, Huang LH, et al:
Interferon activation of the transcription factor Stat91 involves
dimerization through SH2-phosphotyrosyl peptide interactions. Cell.
76:821–828. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chan KS, Sano S, Kiguchi K, et al:
Disruption of Stat3 reveals a critical role in both the initiation
and the promotion stages of epithelial carcinogenesis. J Clin
Invest. 114:720–728. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang T, Niu G, Kortylewski M, et al:
Regulation of the innate and adaptive immune responses by Stat-3
signaling in tumor cells. Nat Med. 10:48–54. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Frank DA: STAT3 as a central mediator of
neoplastic cellular transformation. Cancer Lett. 251:199–210. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kumar C, Purandare AV, Lee FY and Lorenzi
MV: Kinase drug discovery approaches in chronic myeloproliferative
disorders. Oncogene. 28:2305–2313. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ali S, Banerjee S, Logna F, et al:
Inactivation of Ink4a/Arf leads to deregulated expression of miRNAs
in K-Ras transgenic mouse model of pancreatic cancer. J Cell
Physiol. 227:3373–3380. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jeyapalan Z, Deng Z, Shatseva T, et al:
Expression of CD44 3′-untranslated region regulates endogenous
microRNA functions in tumorigenesis and angiogenesis. Nucleic Acids
Res. 39:3026–3041. 2011.
|
|
28
|
Kato M, Putta S, Wang M, et al: TGF-β
activates Akt kinase through a microRNA-dependent amplifying
circuit targeting PTEN. Nat Cell Biol. 11:881–889. 2009.
|
|
29
|
Kato M, Wang L, Putta S, et al:
Post-transcriptional up-regulation of Tsc-22 by Ybx1, a target of
miR-216a, mediates TGF-β-induced collagen expression in kidney
cells. J Biol Chem. 285:34004–34015. 2010.PubMed/NCBI
|
|
30
|
Chen PJ, Yeh SH, Liu WH, et al: Androgen
pathway stimulates microRNA-216a transcription to suppress the
tumor suppressor in lung cancer-1 gene in early
hepatocarcinogenesis. Hepatology. 56:632–643. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Usborne AL, Smith AT, Engle SK, et al:
Biomarkers of exocrine pancreatic injury in 2 rat acute
pancreatitis models. Toxicol Pathol. 42:195–203. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Blenkiron C, Askelund KJ, Shanbhag ST, et
al: MicroRNAs in mesenteric lymph and plasma during acute
pancreatitis. Ann Surg. Feb 6–2014.(Epub ahead of print).
|
|
33
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: a leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gabler K, Behrmann I and Haan C: JAK2
mutants (e.g, JAK2V617F) and their importance as drug targets in
myeloproliferative neoplasms. JAKSTAT. 2:e250252013.PubMed/NCBI
|
|
35
|
Miao L, Liu K, Xie M, Xing Y and Xi T:
miR-375 inhibits Helicobacter pylori-induced gastric
carcinogenesis by blocking JAK2-STAT3 signaling. Cancer Immunol
Immunother. 63:699–711. 2014.
|
|
36
|
Corcoran RB, Contino G, Deshpande V, et
al: STAT3 plays a critical role in KRAS-induced
pancreatic tumorigenesis. Cancer Res. 71:5020–5029. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Miyatsuka T, Kaneto H, Shiraiwa T, et al:
Persistent expression of PDX-1 in the pancreas causes
acinar-to-ductal metaplasia through Stat3 activation. Genes Dev.
20:1435–1440. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee JK, Edderkaoui M, Truong P, et al:
NADPH oxidase promotes pancreatic cancer cell survival via
inhibiting JAK2 dephosphorylation by tyrosine phosphatases.
Gastroenterology. 133:1637–1648. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Palagani V, Bozko P, El Khatib M, et al:
Combined inhibition of Notch and JAK/STAT is superior to
monotherapies and impairs pancreatic cancer progression.
Carcinogenesis. 35:859–866. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Butturini E, Carcereri de Prati A,
Chiavegato G, et al: Mild oxidative stress induces
S-glutathionylation of STAT3 and enhances chemosensitivity of
tumoural cells to chemotherapeutic drugs. Free Radic Biol Med.
65:1322–1330. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim JK, Kim JY, Kim HJ, et al: Scoparone
exerts anti-tumor activity against DU145 prostate cancer cells via
inhibition of STAT3 activity. PLoS One. 8:e803912013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ørom UA, Kauppinen S and Lund AH:
LNA-modified oligo-nucleotides mediate specific inhibition of
microRNA function. Gene. 372:137–141. 2006.PubMed/NCBI
|
|
43
|
Lahdaoui F, Delpu Y, Vincent A, et al:
miR-219-1-3p is a negative regulator of the mucin MUC4 expression
and is a tumor suppressor in pancreatic cancer. Oncogene. 0:1–9.
2014.PubMed/NCBI
|
|
44
|
Zhao G, Wang B, Liu Y, et al: miRNA-141,
downregulated in pancreatic cancer, inhibits cell proliferation and
invasion by directly targeting MAP4K4. Mol Cancer Ther.
12:2569–2580. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Miao L, Xiong X, Lin Y, et al: miR-203
inhibits tumor cell migration and invasion via caveolin-1 in
pancreatic cancer cells. Oncol Lett. 7:658–662. 2014.PubMed/NCBI
|
|
46
|
Huang F, Tang J, Zhuang X, et al: MiR-196a
promotes pancreatic cancer progression by targeting nuclear factor
kappa-B-inhibitor alpha. PLoS One. 9:e878972014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Patel K, Kollory A, Takashima A, et al:
MicroRNA let-7 down-regulates STAT3 phosphorylation in pancreatic
cancer cells by increasing SOCS3 expression. Cancer Lett.
347:54–64. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Song S, Zhou J, He S, et al: Expression
levels of microRNA-375 in pancreatic cancer. Biomed Rep. 1:393–398.
2013.PubMed/NCBI
|
|
49
|
Menghini R, Casagrande V, Marino A, et al:
MiR-216a: a link between endothelial dysfunction and autophagy.
Cell Death Dis. 5:e10292014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yang S, Wang X, Contino G, et al:
Pancreatic cancers require autophagy for tumor growth. Genes Dev.
25:717–729. 2011. View Article : Google Scholar : PubMed/NCBI
|