1. Introduction
Colorectal cancer (CRC) is one of the most common
malignancies, and the third leading cause of cancer-associated
mortality worldwide (1). Although a
decrease in CRC mortality has been observed, its incidence
continues to rise. Therapeutic approaches such as conventional
surgery, chemotherapy and radiation therapy are insufficient with
regard to reversal of advanced CRC. Thus, identification of new
tumor markers for early diagnosis and therapy, is the key to
reducing the incidence of cancer, directly affecting the prevention
and treatment of CRC.
Epithelial integrity is crucial during epithelial
tumor development. Eradication of the original epithelial structure
and obtaining the peculiarity of mesenchymal cells is the first
step in tumor invasion and metastasis. This procedure is known as
epithelial-mesenchymal transformation (EMT), which plays an
important role in in situ infiltration and distant
metastasis in various types of cancer, including CRC (2).
MicroRNAs (miRNAs) are small non-coding RNA
molecules, consisting of 22–25 nucleotides, that regulate the
expression of target genes through base-pairing interactions. These
molecules are important in tumorigenesis including angiogenesis,
circulation, extravasation, intravasation, invasion and metastatic
colonization (3). Studies have
focused on tumor-associated miRNAs with regard to the expression
and function of miRNAs.
This review aimed to summarize EMT-associated miRNA
molecules in CRC to determine new types of treatment, particularly
drug-targeted therapy.
2. EMT and cancer
EMT-associated concepts
Epithelial and mesenchymal cell types are recognized
by their unique cell morphology and peculiarity in tissues.
Epithelial cells belong to a group of polarized cells that are
connected laterally via adhesion molecules and cellular junctions,
including adherent junctions, desmosomes, and tight junctions. By
contrast, mesenchymal cells enable themselves to move freely in the
extracellular matrix (ECM), without adhesion molecules or junctions
(4). In many environments, cells
change their own characteristics from epithelial-like to
mesenchymal-like and vice versa, particularly in the period of
embryonic morphogenesis processes. EMT is a biological process in
which epithelial cells lose their polarities, intercellular
junctions and epithelial-like characteristics, acquiring a
less-differentiated phenotype with motile behavior. The entire
process of EMT includes two aspects of cell morphology and genotype
changes. It involves molecular reprogramming of the cell, including
loss of cell adhesion molecules such as E-cadherin; changing shape
of cells from epithelial morphology to the spindle shape of fiber
cells and cell keratin structure; and acquiring some
characteristics of mesenchymal cells or fibroblasts, such as
overexpression of vimentin, N-cadherin, osteopontin, Snail, slug
and other interstitial proteins (5–7). Thus,
cells obtain higher abilities with regard to migration, invasion,
antiapoptosis and the degradation of extracellular matrix. For
these reasons, EMT is crucial during the early stages of invasion
and metastasis of epithelial tumors (8–9).
Molecular markers in EMT
EMT is a dynamic process comprising a number of
steps and complex regulatory mechanisms, including loss of adhesion
between cells, destruction of the basement membrane (BM) and ECM,
reconstruction of the cytoskeleton and enhancement of migration and
motility (10). Thus, the molecular
mechanisms and signaling pathways involved in EMT are mainly
associated with the above aspects.
E-cadherin/β-catenin complex
In the process of embryonic development, cellular
adhesive molecular regulation follows the principle of specific
time and space, of which cadherin is the most important. E-cadherin
is a calcium-dependent transmembrane glycoprotein on the cell
surface that mediates cell-cell adhesion, which can form the
E-/β-/α-catenin complex by binding to β-catenin inside the
cytoplasm. The complex connects directly to the actin cytoskeleton
to maintain the stability of intercellular adhesion and polarity,
maintaining epithelial cell integrity and normal function (11). Thus, loss of E-cadherin expression
or conversion between different cadherin proteins may induce EMT.
At the same time, decreasing E-cadherin expression can increase
β-catenin in the cytoplasm, which binds to transcription factors
TCF/LEF. Loss of E-cadherin expression in human tumors is most
commonly caused by methylation of its promoter (12), phosphorylation and degradation of
protein (13), and upregulation of
the transcriptional repressors Snail (SNAI1), Slug (SNAI2), Sip1
(GEMIN2) and Zeb1, targeting the E-cadherin promoter (14–15).
In the epithelial tumors, EMT cleaves cancer cells from the primary
focal, invades the blood vessels or lymph nodes and migrates. Thus,
E-cadherin is an important factor in tumor malignant
transformation, invasion and metastasis.
N-cadherin
N-cadherin is s skeleton protein present in
mesenchymal cells, which is expressed in nerve, musculoskeletal and
hematopoietic tissue, but not in normal epithelial tissue (16). Its main function involves the
induction of fibers of cell dynamic adhesion, leading to
mesenchymal cell migration. Hazan et al suggested that
conversion occurs between cadherins. Expression of E-cadherin was
reduced while that of N-cadherin was increased, an important
mechanism of EMT (17). Epithelial
tumor cells therefore cleave from the primary focal, invade
surrounding tissues, and migrate through lymphatic and blood
circulation when E-cadherin expression is reduced while N-cadherin
expression is increased (18).
Vimentin
Vimentin is an intermediate filament protein derived
from mesenchymal cells, that is hardly expressed in normal
epithelial tissue. Abnormal expression of vimentin results in the
reconstruction of cytoskeletal proteins, leading to epithelial
cells acquiring some characteristics of spindle-like fibroblasts,
which migrate more readily. Findings of previous studies have shown
the abnormal expression of vimentin in a variety of epithelial
tumors such as ovarian, breast, colonic and prostate cancer, which
is closely associated with cancer invasion and metastasis (19–22).
Fibronectin
Fibronectin is an extracellular matrix glycoprotein,
consisting of two disulfide-bound polypeptide molecular chains,
that is mainly present in mesenchymal cells of normal tissue.
Fibronectin contains a different functional and structural domain,
that binds to the cell surface. In previous studies it was shown
that, fibronectin increases migration in vivo and enhances
the effect on cell adhesion and spreading. Fibronectin has been
shown to play a role in cell morphology, cytoskeletal organization,
phagocytosis, embryonic differentiation and wound repair (23–24),
which are associated with tumor invasion and metastasis.
Transcription factors
Three core groups of transcriptional factors have
been consistently identified to be critical during various EMT
events, and are considered the core EMT regulators. The first group
is the Snail zinc-finger family, including Snail1 and Snail2, both
of which directly bind to the E-boxes of the E-cadherin promoter to
repress its transcription (25–26).
The second group is the distantly associated zinc-finger
E-box-binding homeobox family of proteins Zeb1 and Zeb2, which are
also able to suppress E-cadherin transcription via a
double-negative feedback loop controlling Zeb1/Zeb2 and miRNA-200
family expression (27–28). The third group is the basic
helix-loop-helix (bHLH) family, including Twist1 and Twist2. Twist1
can repress E-cadherin through the induction of Snail transcription
factors (29–31).
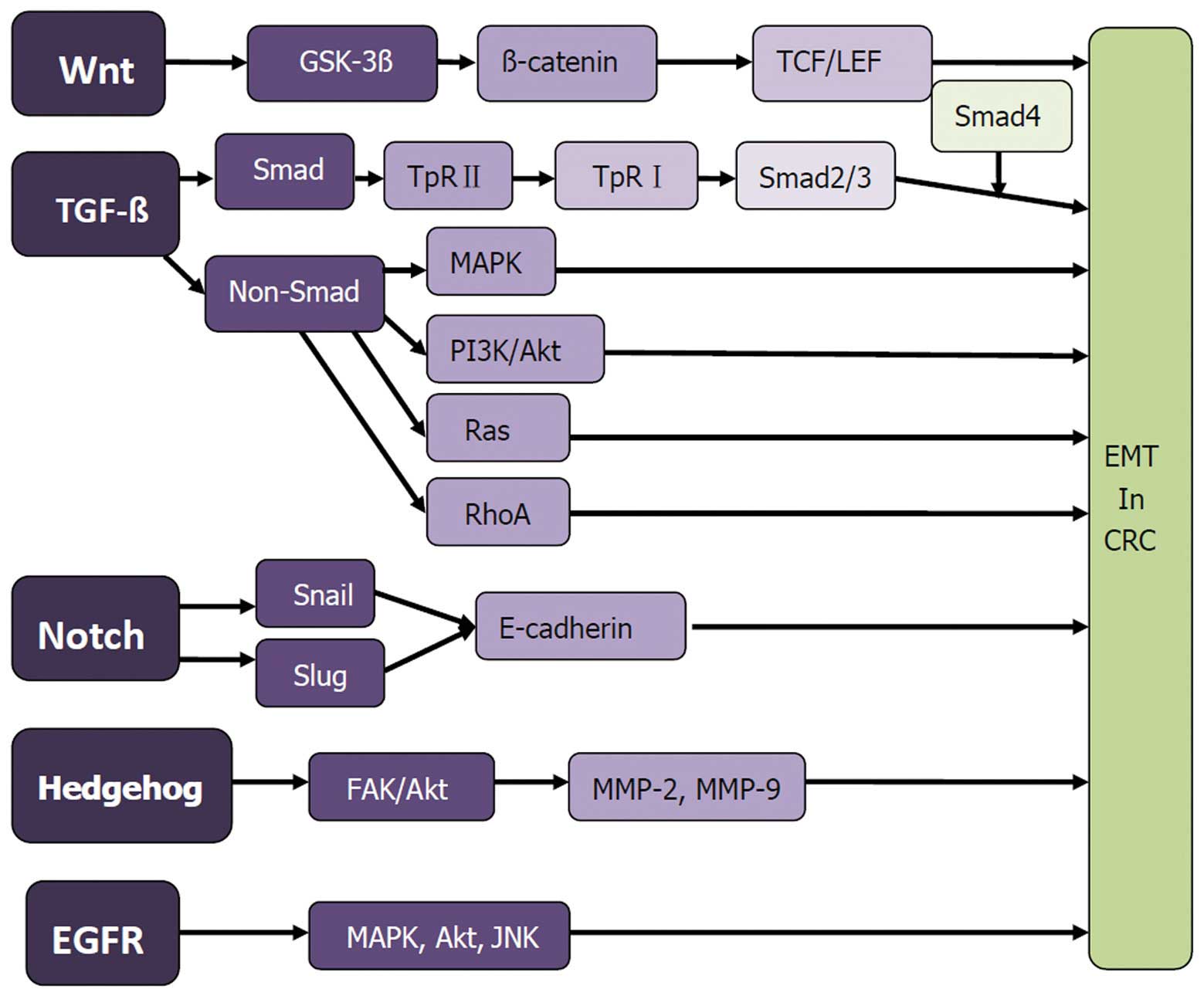
Signaling pathways and mechanisms
involved in CRC EMT
EMT is a dynamic process, involving the phenotypic
changes in protein, as an external adjustment. It is also
associated with a variety of signal transduction pathways. Signal
codes specifically bind to cell membrane surface receptors,
activating transcription factors in the nucleus through a variety
of signal transduction pathways (Fig.
1).
Wnt/β-catenin/LEF-1 signaling
pathway
The Wnt gene was first identified and
reported in 1982 (32). This gene
forms a complex signaling pathway in tumor cells, including at
least three branches of the Wnt/β-catenin/LEF-1 pathway, Wnt/the
planar cell polarity (PCP) pathway and the Wnt/ca(2+)-mediated
pathway (32–34). The Wnt/β-catenin pathway, known as
the ‘classic Wnt pathway’, plays a role in the process of EMT in
CRC (35). The Wnt/β-catenin/LEF-1
signaling pathway is closely associated with EMT, which comprises
the Wnt protein, frizzled protein, dishevelled protein, APC
compounds, GSK-3β, axin protein, β-catenin and TCF/LEF family of
transcription factors. In tumors, due to abnormal activation, the
Wnt gene binds to the frizzled protein through the Wnt
ligands, forming a receptor complex with LRP5/6, inducing GSK-3β
inactivation. APC, an axin protein is activated in succession,
forming the GSK3β/APC/axin complex, and reducing the
phosphorylation degradation of β-catenin. Thus, large amounts of
β-catenin gather in the cytoplasm and nucleus, interacting with
TCF/LEF transcription factors, and inducing the transcription of
target genes of Wnt, such as TNC. Phosphorylation of tyrosine in
the β-catenin extremity can lead to E-cadherin/β-catenin complex
decomposition, activating the Wnt signaling pathway (36–40).
Thus, interaction between the Wnt pathway and the
E-cadherin/β-catenin complex potentially promotes tumor
development.
TGF-β signaling pathway
The transforming growth factor-β (TGF-β) signaling
pathway appears to be a primary inducer of EMT (41). Classic SMAD and other non-SMAD
pathways participate in the EMT process induced by TGF-β (41,42).
In the early stage of tumor growth, TGF-β induces growth arrest and
apoptosis as a tumor suppressor. In advanced stages of tumor
progression, TGF-β regulates transcription through
SMAD-dependent/-independent TGF-β-receptor signaling pathways,
initiating cancer growth and metastasis.
SMAD pathway
SMAD and signal-regulating proteins downstream of
TGF-β receptor complexes, are present in the cytoplasm, conducting
signals from the cell membrane to the nucleus, and regulating gene
transcription. A typical SMAD-dependent pathway involved in the EMT
process is described below. TGF-β ligand binds to the type 2 TGF-β
receptor, which recruits the type 1 TGF-β receptor. This receptor
dimerization and phosphorylation of serine/threonine residues
allows the phosphorylation of SMAD2 and SMAD3. Activated SMAD
proteins dissociate from the SMAD anchor for receptor activation
(SARA) protein, hetero-oligomerize with SMAD4, and translocate to
the nucleus, combining with the SMAD binding element located in
EMT-associated gene promoter regions. This process mediates target
gene expression or repression, such as MMP-2, SPI-1 and α-SMA. As a
tumor-suppressor gene, SMAD4 increased epithelial marker expression
of E-cadherin, strengthening the connection between cells in CRC
and SW480 (7,43–45).
Non-SMAD pathway
TGF-β signaling occurs through a number of non-SMAD
pathways, including branches of MAPK, RAS and PI3K/Akt. The main
MAPK signaling pathways involved in EMT include the JNK signaling,
p38 MAPK and Ras/Raf/MEK/ERK pathways. MAPK is mainly involved in
the regulation of cell proliferation, secretion and neuronal
differentiation. In the PI3K/Akt pathway, PI3K is activated by Ras
combining to P110 or interacting with growth-factor receptors with
phosphorylated tyrosine residues or junction protein. Akt activates
or inhibits downstream target proteins by phosphorylation,
regulating cell proliferation, differentiation, apoptosis and
migration. In addition to PI3K/Akt, RAS, and MAPK, TGF-β activates
RhoA, c-Src, m-TOR, and protein phosphatase 2A (PP2A)/p70s6K,
inducing EMT occurrence (46–51).
Notch signaling pathway
The Notch signaling pathway has been considered a
traditional pathway regulating embryogenesis and tissue formation.
However, it is involved in the occurrence of EMT. Notch can induce
tumorigenesis, causing epithelial cells to lose their polarity,
mobility and invasive ability (52). In this process, Notch regulates the
expression of E-cadherin by inducing Slug activation or regulating
Snail expression, thus enhancing the ability of tumor cell invasion
and migration (53,54). Notch signaling pathway does not
function independently, but may combine its action with other
signaling pathways to induce EMT (55).
Other signaling pathways
Studies have also shown that the Hedgehog (Hh)
signaling pathway and epidermal growth factor receptor (EGFR)
signaling cascades are involved in the EMT process. The Hh
signaling pathway is crucial in embryo nic development, formation
and maintaining of cancer stem cells (CSCs) and EMT. Binding Hh
ligands, such as Snoic, Desert, and Indian to Patched, results in
de-repression of smoothened (SMO) and its activation,
internalization and translocation to the primary cilium. The
transcription of GLI target genes is followed by the activation of
zinc-finger transcription factors GLI-1, GLI-2 and GLI-3 (56). The Hh signaling pathway induces
cancer-cell invasion by regulating FAK/AKT signaling
pathway-mediated activation, expressing MMP-2 and MMP-9 and
inducing the expression of E-cadherin, Snail (57). As mentioned above, the EMT process
is associated with ECM, otherwise, EGFR expression or activity may
lead to cancer (58). Several
auto-phosphorylation tyrosine residues in the C-terminal domain of
EGFR elicits are activated by other proteins, principally the MAPK,
Akt and JNK pathways (59). This
stimulation promotes EMT and modulates phenotypes such as cell
proliferation, migration and adhesion.
3. EMT-associated microRNA in colorectal
cancer
MicroRNAs (miRNAs) are small non-coding RNAs
regulating target mRNAs through post-transcription. Acting as
endogenous suppressors, miRNAs inhibit gene expression through the
imperfect binding of RISC subsequent to transcription. The main aim
of miRNA is the degradation or translational inhibition of target
mRNA (60). Therefore, miRNA
affects cell processes, including cell differentiation,
proliferation, metabolism and apoptosis by regulating one or two
key target genes (61). miRNAs
participate in tumor progression by acting as promoters or
suppressors. miRNAs are also important in the regulation of cancer
EMT, including CRC.
miRNA and cancer EMT
miRNA
miRNA was initially identified lin-4 in
caenorhabditis elegans (62). However, extensive studies on miRNA
were initiated following the identification of Let-7 (63). miRNAs are highly conserved, with
tissue- and sequence-specific expression (64–66).
The formation of mature miRNA binds to the imperfect complementary
site of target mRNA via base pairing. When miRNA is fully
complementary to the target site, the combination of these miRNAs
often leads to the degradation of target mRNA through a RISC
complex. When the miRNA and target mRNA sequences degree of
matching is low, the translation process is inhibited (67,68).
miRNAs have been associated with various types of cancer.
Approximately 50% of miRNAs in the genome were found to bind to
fragile sites in tumors (69),
suggesting that miRNAs play a critical role in tumorigenesis and
metastasis, including the process of EMT.
miRNAs in cancer EMT
miRNAs act as tumor suppressors in EMT. In 2007,
Hurteau et al found that the expression of miRNA-200c was
negatively associated with E-cadherin expression in breast cells
(70). miR-200 family and miR-205
were found to regulate EMT through inhibition of Zeb1 and Zeb2
E-cadherin (71,72). In another study, TGFβ2 and Zeb1 were
identified as the main targets of miR-200 family and miR-205. These
findings indicated a feed-forward loop showing Zeb1 and miR-200
members to be involved in EMT and cancer cell invasion (73). Findings of recent studies have shown
that Let-7 is associated with cancer EMT and metastasis. Let-7 was
identified as targeting HMGA2, indicating there was a relationship
between miRNA Let-7 and EMT (74).
Downregulation of miRNA-138 is associated with interstitial
cell-like transformation, and can enhance the metastasis and
invasion of tumor cells including EMT with a decreased expression
of E-cadherin, and enhanced expression of vimentin (75). Newly identified miRNAs, including
miR-23b, miR-29c, miR-203, miR-448 and miR-194, similarly inhibited
the EMT process (76–78).
miRNAs act as oncogenes in EMT. Overexpression of
miR-10b, upregulated by EMT transcription factor Twist, located in
the HOX gene cluster, is associated with invasiveness and
metastatic potential in breast cancer (79). Results of studies, showed that
miR-155 expression was increased in TGF-induced EMT in tumors, by
SMAD4-regulated transcription activity. Breast cancer cells with
miR-155 downregulation did not effectively induce EMT (80). In addition, miR-21 can promote the
breast cancer EMT through overexpression of the SMAD-dependent
transcription regulation matrix. By suppressing TIAM1 expression,
miR-31 significantly enhanced CRC cell migration. It also induced
cancer cell EMT process by activating transcription factors AP1 and
Zeb1, located downstream of the TGF-β signaling pathway (81–83).
miR-9, miR-661, miR-369-5p, miR-370, miR450a and miR-542-5p also
promoted EMT tumor (84–86).
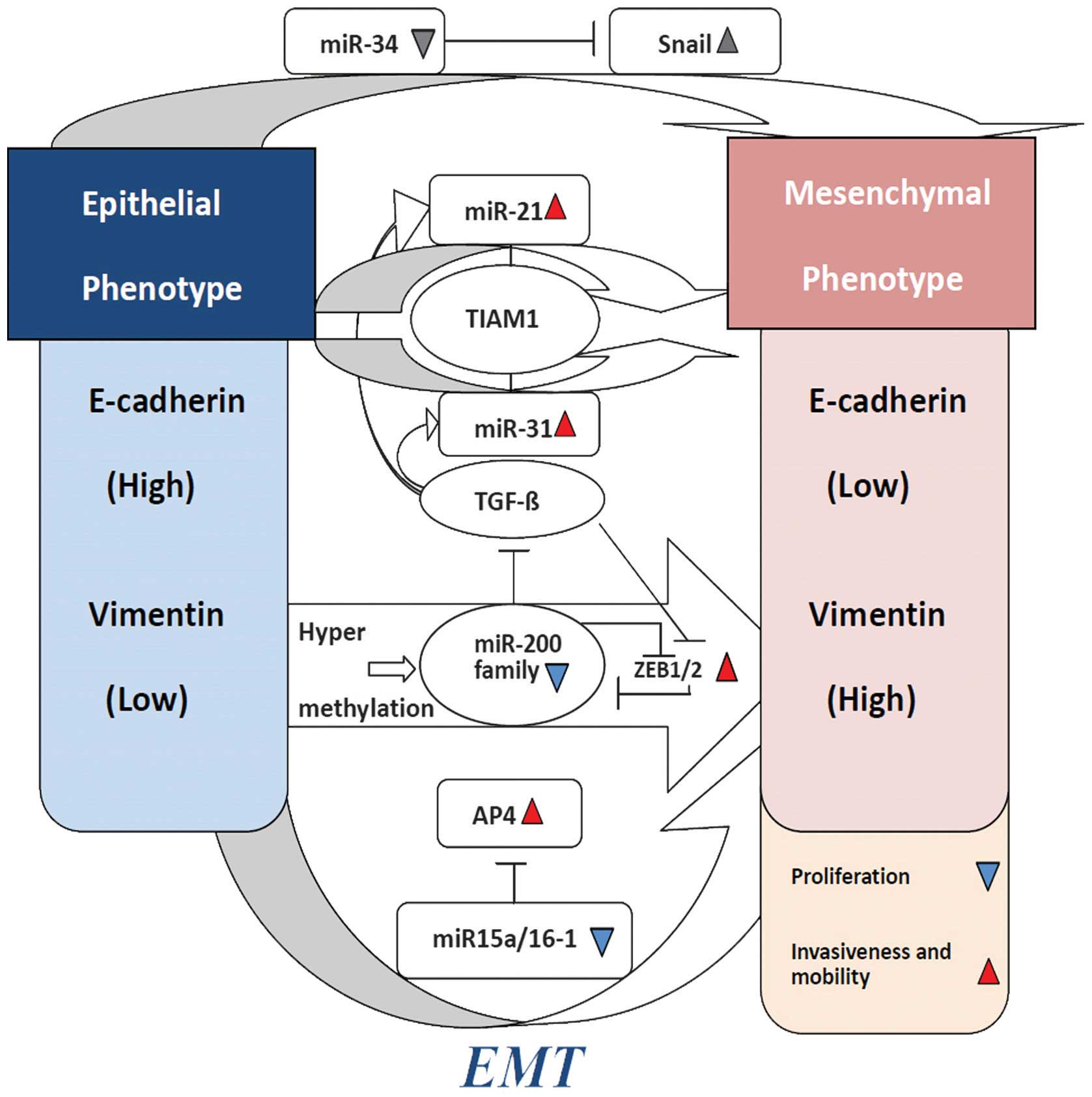
miRNA regulation of EMT in CRC
miRNAs are involved in the CRC EMT, partly by
regulating the expression of oncogenes and tumor suppressors and
partly by functioning as oncogenes or tumor suppressors themselves
(87). These important regulatory
functions of miRNA-activated upstream factors, promoted the CRC EMT
process. (Fig. 2)
miR-200 family
As mentioned earlier, the miR-200 family (miR-200a,
miR-200b, miR-200c, miR-141, and miR-429) plays a key role in
cancer EMT, including CRC. miR-200 family members were found to
enhance E-cadherin expression by targeting complementary sites in
the 3-UTR of Zeb1 and Zeb2. Loss of the basement membrane (BM), EMT
was considered a key step in induction of metastasis. Zeb1 was
found to be the key transcriptional repressor of BM components in
CRC (88,89). In addition, Zeb1 inhibits cell
polarity factors and Lgl2 expression in CRC, which is critical for
the epithelial phenotype (90).
Transforming growth factor-β (TGF-β), an EMT activator, is produced
by tumor cells, triggering the expression of Zeb1/2, an upstream
master regulator of EMT progression. Zeb1 directly suppressed the
expression of miR-141 and miR-200c (on human chromosome 12),
downregulating some essential features of EMT. After knockdown of
Zeb1 in CRC, an increased expression of miR-141 and miR-200c can
induce the epithelial phenotype, increase cell-cell adhesion,
induce E-cadherin expression, and reduce cell migration and
invasion (73). In addition, second
miRNA clusters (miR-200b, miR-200A, and miR-429) of the miR-200
family also contain highly conserved putative Zeb1 binding sites in
the upstream sequence (91).
Results of those studies showed that, Zeb1 can trigger a
miRNA-mediated feed-forward loop stabilizing the EMT and promoting
cancer-cell invasion. Otherwise, strong induction of miR-200 may
promote the differentiation and inhibition of
epithelial-mesenchymal-specific gene expression, by downregulating
Zeb1 and Zeb2 conversely. Presumably, Zeb and the miR-200 family
participate in a double-negative feedback loop stabilizing
differentiation along the EMT-MET axis (92).
miR-21 and miR-31
miR-21 is located on human chromosome 17q23.2. In
CRC, this genomic region always shows copy number gain, which is
frequently observed in metastatic tumors. This observation suggests
miR-21 is important in the CRC metastatic pathway. miR-21 encodes a
single hairpin and is regulated by its own promoter containing
binding sites for transcription factors, such as AP-1 and PU.1
(93). Upregulation of TGF-β1
increased the expression of miR-31, resulting in a higher
percentage of cells using a ‘spreading’ morphology. The levels of
miR-21 and miR-31 were markedly elevated under the synergistic
actions of TGF-β/TNF-α. The two miRNAs are likely to have a number
of different direct targets. They converge on TIAM1, a guanine
nucleotide exchange factor (GEF) for the Rac GTPase-regulating
migration and invasion of various cancer cells including CRC cells
(81). It has been identified that,
high miR-21 with low Integrin-β4 (ITGβ4) can be exclusively
expressed in polarized epithelial cells, and the level of PDCD4
expression was able to regulate EMT in CRC (94).
miR-34
The miR-34 family includes miR-34a, miR-34b and
miR-34c. The miR-34 family participates in cell cycle progression,
senescence and cell apoptosis (95–97).
The members inhibit EMT by downregulating the expression of Snail,
one of the EMT-inducing transcription factors (EMT-TF) (98,99).
It has been revealed that IL-6 induces EMT, invasiveness, and the
metastatic properties of CRC cells. By directly repressing miR-34a,
miR-34a can suppress tumor progression by inhibiting the
IL-6R/STAT3/miR-34a feedback loop. Thus it can promote and maintain
EMT, invasiveness, and metastasis in CRC (100).
miR-15a/16-1
miR-15a and miR-16-1 are located on human chromosome
13q14.3, which is highly conserved (101). Emerging evidence has shown that
they target the 3′-untranslated region (3′-UTR) of the
transcription factor AP4, which is downregulated by p53. AP4
directly represses E-cadherin via a non-canonical AP4-binding motif
and induces N-cadherin, mediating EMT in CRC. This finding suggests
that miR-15a/16-1 and AP4 are involved in a double-negative
feedback loop, stabilizing epithelial and mesenchymal states.
Respectively, they may determine metastatic prowess in CRCs
(102,103).
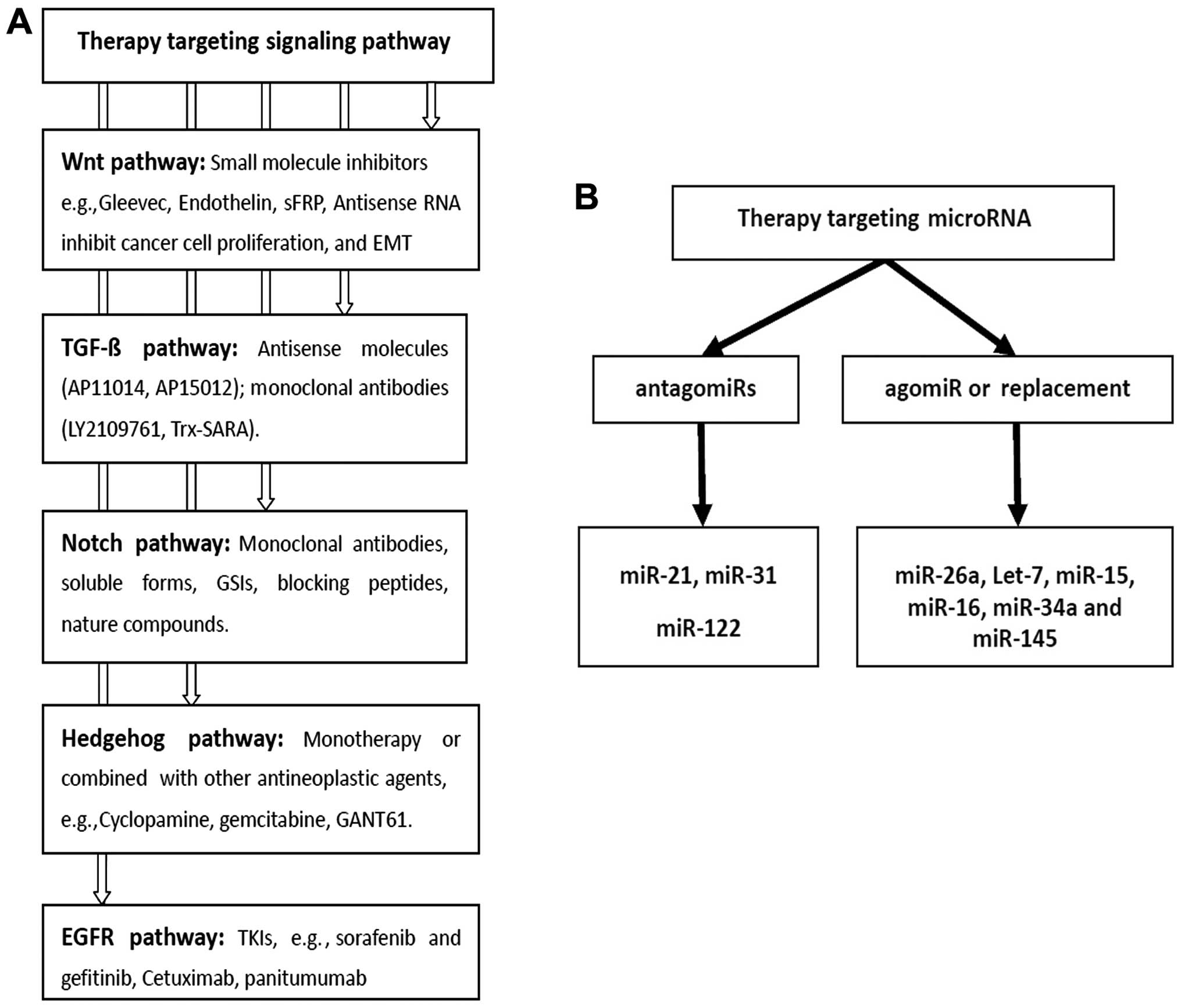
4. Strategies for therapy targeting EMT in
colorectal cancer
Improved understanding of the pathology of molecules
in cancer had led to the development of a number of target drugs,
which have demonstrated improved outcome of cancer patients with
metastasis. However, the aim of ongoing animal experiments and
clinical trials is to identify further drugs (Fig. 3).
Targeting EMT-associated molecules and
pathways in CRC
As shown in Fig. 1,
EMT-associated pathways provide therapy targets for CRC
patients.
Drugs targeting Wnt/β-catenin signaling
pathway
The Wnt signaling pathway is closely associated with
the occurrence and development of CRC. In vitro experiments
on drugs targeting the Wnt signaling pathway are currently ongoing.
Non-steroidal anti-inflammatory drugs (NSAIDs) including Aspirin
(104), Sulindac (105), Celecoxib and rofecoxib (106) downregulate the β-catenin level by
inducing β-catenin phosphorylation and degradation or inhibiting
the transcription of target genes.
Small molecule inhibitor is a research hotspot on
molecular targeting therapy. Gleevec, a type of tyrosine kinase
inhibitor, is a small molecular protein, which can significantly
inhibit CRC cell proliferation, reduce transcription activity, and
induce β-catenin migration from the cell nucleus to the membrane
(107). Endothelin (ET) can
promote the degradation of β-catenin, inhibit target-gene cyclin D1
promoter activity, and induce cell-cycle arrest, apoptosis and
tumor angiogenesis (108). The
sFRP Wnt pathway inhibitor can suppress CRC cell proliferation and
transformation ability (109).
Antisense RNA can silence β-catenin expression or inhibit CRC cell
proliferation and differentiation by eradicating the TCF/β-catenin
complex in the Wnt pathways (108). Thus, the Wnt signaling pathway is
expected to offer a new effective method for the treatment of
CRC.
Drugs targeting the TGF-β signaling
pathway
As we described earlier, the TGF-β signaling pathway
plays a key role in CRC EMT. Therapeutic strategies against TGF-β
are effective methods that are classified into the ligand,
ligand-receptor and intracellular levers (110). Powerful anti-TGF-β strategies have
been developed and tested in pre-clinical studies as well as
clinical trials. Antisense molecules can prevent TGF-β synthesis on
the ligand lever by binding to specific mRNA. AP11014 and AP15012
are antisense molecules used in pre-clinical trials for treatment
of non-small cell lung cancer, prostate carcinoma, CRC and MM,
respectively (111). By blocking
downstream signaling, monoclonal-neutralizing antibodies acting as
receptor kinase inhibitors were more efficient as compared to
ligand traps or antisense molecules (110). LY2109761 is a small molecule
inhibiting the kinase activity of TβR I and TβR II. This compound
inhibits metastatic formation in mouse models of breast cancer, CRC
and pancreatic cancer (112–114). The Trx-SMAD anchor for receptor
activation (Trx-SARA) is an example of a peptide aptamer, which
reduces the levers of TGF-β-induced SMAD-2/-3 in the complex
(115), and inhibits EMT following
TGF-β stimulation in breast cancer epithelial cells (116). Ongoing pre-clinical trials are
currently focused on other drugs targeting the TGF-β signaling
pathway. Future studies on drug development targeting EMT in CRC
remain to be conducted.
Drugs targeting Notch signaling
pathway
The Notch pathway has great potential as a new
cancer treatment target. Various Notch inhibitors, including
monoclonal antibodies against Notch receptors or ligands, soluble
forms of the extracellular domain of Notch receptors or ligands,
GSIs, blocking peptides, and nature compounds have been identified
(117–124). GSIs are the most widely studied
Notch receptors, having potential for bio-distribution and
pan-Notch inhibition (125). GSIs
are utilized in Phase I clinical trials as a monotherapy or in
combination with other drugs (126). Therefore Notch is a potential
antitumor therapeutic target and its implementation should occur
subsequent to in-depth study with a focus on the Notch signaling
pathway.
Drugs targeting Hedgehog signaling
pathway
Currently, all of the small-molecules Hedgehog (Hh)
pathway therapeutics in clinical trials are designed to target
Smoothened (SMO). The reagents act as monotherapy or are combined
with other antineoplastic agents for the treatement of various
tumors including colon cancer. The Hh inhibitor cyclopamine is an
antagonist of the Hh signaling pathway component SMO. Studies have
shown that cyclopamine can eliminate pancreatic cancer metastasis
(127). Cyclopamine inhibits the
invasion of pancreatic cancer by suppressing EMT, upregulating
E-cadherin and downregulating Snail. In an orthotopic xenograft
model, the combination of cyclopamine and gemcitabine can restore
metastases (128). GLI was found
to be activated by other signaling pathways, such as Ras, TGF-β and
Wnt signaling pathways. In human colon carcinoma cells, specific
targeting of GLI, which is downstream of SMO, can induce cancer
cell death (129). GLI has a
broader role, blocking the downstream gene transcription and
activation (130–131).
Drugs targeting the EGFR signaling
pathway
The epidermal growth factor receptor (EGFR) is known
to be overexpressed in CRC EMT, while anti-EGFR therapy is
considered effective. Tyrosine kinase inhibitors (TKIs), such as
sorafenib and gefitinib, were designed to block the cascade of
reactions which are crucial to tumor development and survival
(132). Sorafenib is currently
being tested in phase II trials of CRC. A recent study has
indicated that the ability of TKIs strongly suppress P13K signaling
in CRC, suggesting that such treatments have great therapeutic
value (133). Cetuximab and
panitumumab, monoclonal antibodies to EGFR, were approved by the
FDA for the treatment of CRC in 2004 and 2006, respectively. The
two antibodies prevent EGFR auto-phosphorylation by binding to the
extracellular domain. They also inhibit activation of the MAPK and
P13K downstream cell signaling pathways (132). In addition, the two drugs
exhibited significant antitumor activity, although limitations and
side effects were observed (134–137).
Targeting miRNA in CRC EMT
miRNAs play a key role in proliferation, cell cycle,
evasion of apoptosis, invasion, migration and EMT. One of the most
appealing properties of miRNA as therapeutic drugs in comparison
with targeting single genes is their ability to target multiple
molecules in the network. Abnormal expression levels of miRNAs show
that, miRNAs can be used as drugs in CRC. Aiming at an abnormally
high expression of onco-miR in tumor, small molecule drugs known as
‘antagomiRs’ have been designed, to knock out abnormal miRNAs or
suppress expression of miRNAs. Targeting low or no expression of
tumor-supressor miRNAs, corresponding exogenous miRNAs are known as
‘agomiRs’. AgomiRs can be guided into the patient’s body, restoring
or improving the function of miRNAs in tissues (138).
Strategies for antagomiRs
Anti-miRNA oligonucleotides
Anti-miRNA oligonucleotides (AMOs) are
single-stranded oligonucleotides consisting of 22–25 nucleotides,
inhibiting miRNAs (139,140). Because of not being chemically
decorated, antisense oligomeric nucleotide chain is not stable,
almost invalid in the body. AMOs are often modified chemically,
such as 2′-O-methylation (141),
2′-O-methoxyl ethylation and locked nucleic acids (LNAs) (142,143). The modification of AMOs can
suppress miRNAs effectively. Experimental results showed that
injecting inhibitors in mice specifically silenced their target
miR-122 or miR-16 in liver and other organs, such as kidney, lung,
heart and colon etc. (144).
LNA-modified anti-miRNA oligonucleotides have been shown to inhibit
miR-21 in breast cancer and in glioblastoma cells, resulting in the
inhibition of tumor cells in vitro and in vivo. In
xenograft tumor models, 2′-O-methyl-modified oligonucleotides
(anti-miR-21) were injected into MCF-7 cells as instantaneous
transfection. The results showed that the transfected tumor cells
were reduced by ~50%. The study proved that 2′-O-methyl-modified
oligonucleotides were a potential cancer treatment candidate
(145,146). Of note, inhibition of miR-21
expression in extrahepatic cholangiocarcinoma cells can increase
the drug sensitivity of gemcitabine (147). Studies showed that the miRNA-based
antitumor treatment combined with chemotherapy can improve
treatment efficiency. However, the mechanism of downregulating the
miRNA activity through AMOs inhibitors remains to be elucidated.
The mechanism may occur by blocking miRNA biosynthesis via
interaction with prior pre-miRNAs, or binding to mature miRNA to
exert its role in the RISC compounds.
Other novel inhibitions
miRNAs sponges, miRNA-masking antisense
oligodeoxynucleotides and small molecule inhibitors also suppress
the overexpression of miRNA. Synthetized chemically, miRNAs sponges
is a single-stranded RNA (148),
blocking interactions between endogenous miRNA and its target mRNA
(149). The sponge-mediated
inhibition of the anti-metastatic miR-31 has been successfully
applied in an orthotopic breast-cancer model. Results of that study
demonstrated that a significant induction of lung metastases
appeared, even with use of non-aggressive breast cancer cells
(138–150). miRNA-masking antisense
oligodeoxynucleotides bind perfectly to sites of complementary
sequence, forming dimers with higher affinity, closing the sites of
miRNAs through prior combining with target mRNA, and inhibiting the
combination of miRNA and target mRNA (151). Other miRNA inhibitors, such as
azobenzene, can block the transcription of miR-21, providing
evidence that small organic molecules can be used in the miRNA
context as well (152).
Strategies for agomiR or
replacement
Treatment strategies, such as targeting low
expression of miRNAs in CRC EMT, replacement therapy or agomiRs to
restore physiological miRNA levels, can be applied. Current
replacement therapy includes gene therapy and transfection of miRNA
mimics. The approach of gene therapy involves generation of the
pre-miRNA hairpin structure, transfecting and ensuring long-term
and stable expression of mature miRNA. In addition, miRNA mimics,
small double-stranded RNAs synthetized chemically, are widely used.
It can stimulate active endogenous miRNA molecular and
target-specific mRNA through transfection in vitro or in
systemic administration in vivo by liposomes or
nanoparticles polymer. Through virus-mediated miRNA re-introduction
of miR-26a in liver cancer mouse, investigators observed the
inhibition of cancer cell proliferation and induction of
tumor-specific apoptosis (153).
In vitro, Let-7 inhibits cancer cell growth in lung cancer
cells (154). In an autochthonous
NSCLC mouse model, the overexpression of Let-7 through lentiviral
vector can lead to a significant growth reduction (155). Another example of miRNAs
replacement therapy is miR-15 and miR-16, targeting apoptotic
inhibition gene Bcl-2. The re-introduction of miR-15a/miR-16
into prostate carcinoma xenografts by lentiviruses can also reduce
tumor growth (156). In addition,
using lipid-based delivery reagents, miR-34 led to the inhibition
of non-small lung cancer and reduction of metastatic tumor load in
the lung, basing on survivin inhibition (157,158). The expression lever of miR-145,
and downregulation in tumor tissues of CRC patients were increased
in vitro. Thus, cell proliferation was reduced while
sensitivity to radiotherapy was increased (159).
miRNAs and antisense miRNA drugs are currently at
the pre-clinical stage. Owing to poor stability, low efficiency of
transmission and toxic reaction, the clinical application of
targeting miRNA requires additional investigation.
5. Conclusion and perspective
In the present review, we attempted to summarize the
molecules, mechanisms and corresponding targeted therapies involved
in the EMT process. In the past few years, it has become clear that
EMT can reprogram normal epithelial cells. The process occurs
systematically at the transcriptional, post-transcriptional,
translational and post-translational levels. An improved
understanding of the EMT process in CRC may lead to the
identification of novel drug-targeted therapies and approaches.
Acknowledgements
This study was supported by a grant from the
Shanghai Science and Technology Department of Medicine guiding
biomedical projects (no. 134119a1400) and a grant from the Research
Fund for the Doctoral Program of Higher Education (New Teacher)
(no. 20130071120048).
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
Statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thiery JP: Epithelial-mesenchymal
transitions in development and pathologies. Curr Opin Cell Biol.
15:740–746. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Croce CM and Iorio MV: MicroRNA
dysregulation in cancer: diagnostics, monitoring and therapeutics.
EMBO Mol Med. 4:143–159. 2012. View Article : Google Scholar
|
|
4
|
Jeff JH and Yang J: Epithelial-mesenchymal
plasticity in carcinoma metastasis. Genes Dev. 27:2192–2206. 2013.
View Article : Google Scholar
|
|
5
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal-transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jang J and Weinberg RA:
Epithelial-mesenchymal transition: at the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar
|
|
7
|
Pino MS, Kikuchi H, Zeng M, et al:
Epithelial to mesenchymal transition is impaired in colon cancer
cells with microsatellite instability. Gastroenterology.
138:1406–1417. 2010. View Article : Google Scholar :
|
|
8
|
Savagner P: The epithelial-mesenchymal
transition phenomenon. Ann Oncol. 21:89–92. 2010. View Article : Google Scholar
|
|
9
|
Bonnomet A, Brysse A, Tachsidis A, et al:
Epithelial-tomesenchymal transitions and circulating tumor cells. J
Mammary Gland Biol Neoplasia. 15:261–273. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lun H and Shourong S: EMT phenomenon and
related microRNAs in the malignant progression of tumor. Chin
Oncol. 21:725–730. 2011.
|
|
11
|
Cavallaro U, Schaffhauser B and
Christofori G: Cadherins and the tumour progression: is it all in a
switch? Cancer Lett. 176:123–128. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Saito Y, Takazawa H, Uzawa K, et al:
Reduced expression of E-cadherin in oral squamous cell carcinoma:
Relationship with DNA methylation of 5′ CpG island. Int J Oncol.
12:293–298. 1998.PubMed/NCBI
|
|
13
|
Lester RD, Jo M, Montel V, et al: uPAR
induces epithelial-mesenchymal transition in hypoxic breast cancer
cells. J Cell Biol. 178:425–436. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Berx G and Van Roy F: The
E-cadherin/catenin complex: an important gatekeeper in breast
cancer tumorigenesis and malignant progression. Breast Cancer Res.
3:289–293. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Canel M, Serrels A, Frame MC, et al:
E-cadherin-integrin crosstalk in cancer invasion and metastasis. J
Cell Sci. 126:393–401. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Derycke LD and Bracke ME: N-cadherin in
the spotlight of cell-cell adhesion, differentiation,
embryogenesis, invasion and signaling. Int J Dev Biol. 48:463–476.
2004. View Article : Google Scholar
|
|
17
|
Hazan RB, Qiao R, Keren R, et al: Cadherin
switch in tumor progression. Ann NY Acad Sci. 1014:155–163. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rosivatz E, Becker I, Bamba M, et al:
Neoexpression of N-cadherin in E-cadherin positive colon cancers.
Int J Cancer. 111:711–719. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Colomiere M, Findlay J, Ackland L and
Ahmed N: Epidermal growth factor-induced ovarian carcinoma cell
migration is associated with JAK2/STAT3 signals and changes in the
abundance and localization of alpha6betal integrin. Int J Biochem
Cell Biol. 41:1034–1045. 2009. View Article : Google Scholar
|
|
20
|
Vora HH, Patel NA, Rajvik KN, et al:
Cytokerain and vimentin expression in breast cancer. Int J Biol
Markers. 24:38–46. 2009.PubMed/NCBI
|
|
21
|
Shirahata A, Sakata M, Sakuraba K, et al:
Vimentin methylation as a marker or advanced colorectal carcinoma.
Anticancer Res. 29:279–281. 2009.PubMed/NCBI
|
|
22
|
Wei J, Xu G, Wu M, et al: Overexpression
of vimentin contributes to prostate cancer invasion and metastasis
via src regulation. Anticancer Res. 28:327–334. 2008.PubMed/NCBI
|
|
23
|
Pankov R and Yamada KM: Fibronectin at a
glance. J Cell Science. 115:3861–3863. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Birchler MT, Milisavlijevic D, Pfaltz M,
et al: Expression of the extra domain B of fibronectin, a marker of
angiogenesis, in head and neck tumors. Laryngoscope. 113:1231–1237.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cano A, Perez-Moreno MA, Rodrigo I, et al:
The transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
12:76–83. 2000. View Article : Google Scholar
|
|
26
|
Hajra KM, Chen DY and Fearon ER: The SLUG
zinc-finger protein represses E-cadherin in breast cancer. Cancer
Res. 62:1613–1618. 2002.PubMed/NCBI
|
|
27
|
Eger A, Aigner K, Sonderegger S, et al:
DeltaEF1 is a transcriptional repressor of E-cadherin and regulates
epithelial plasticity in breast cancer cells. Oncogene.
24:2375–2385. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim T, Veronese A, Pichiorri F, et al: p53
regulates epithelial-mesenchymal transition through microRNAs
targeting ZEB1 and ZEB2. J Exp Med. 208:875–883. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang J, Mani SA, Donaher JL, et al: Twist,
a master regulator of morphogenesis, plays an essential role in
tumor metastasis. Cell. 117:927–939. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fang X, Cai Y, Liu J, et al: Twist2
contributes to breast cancer progression by promoting an
epithelial-mesenchymal transition and cancer stem-like cell
self-renewal. Oncogene. 30:4707–4720. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Casas E, Kim J, Bendesky A, et al: Snail2
is an essential mediator of Twist1-induced epithelial mesenchymal
transition and metastasis. Cancer Res. 71:245–254. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nusse R and Varmus HE: Many tumors induced
by the mouse mammary tumor virus contain a provirus integrated in
the same region of the host genome. Cell. 31:99–109. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kurayoshi M, Oue N, Yamamoto H, et al:
Expression of Wnt-5a is correlated with aggressiveness of gastric
cancer by stimulating cell migration and invasion. Cancer Res.
66:10439–10448. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dissanayake SK, Wade M, Johnson CE, et al:
The Wnt5A/protein kinase C pathway mediates motility in melanoma
cells via the inhibition of metastasis suppressors and initiation
of an epithelial to mesenchymal transition. J Biol Chem.
282:17259–17271. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huelsken J and Behrens J: The Wnt
signaling pathway. J Cell Sci. 115:3977–3978. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
King TD, Zhang W, Suto MJ, et al:
Frizzled7 as an emerging target for cancer therapy. Cell Signal.
24:846–851. 2012. View Article : Google Scholar :
|
|
37
|
MacDonald BT, Tamai K and He X:
Wnt/beta-catenin signaling: components, mechanisms, and diseases.
Dev Cell. 17:9–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Deka J, Wiedemann N, Anderle P, et al:
Bcl9/Bcl9l are critical for Wnt-mediated regulation of stem cell
traits in colon epithelium and adenocarcinomas. Cancer Res.
70:6619–6628. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hlubek F, Spaderna S, Schmalhofer O, et
al: Wnt/FZD signaling and colorectal cancer morphogenesis. Front
Biosci. 12:458–470. 2007. View
Article : Google Scholar
|
|
40
|
Beiter K, Hiendlmeyer E, Brabletz T, et
al: Beta-catenin regulates the expression of tenascin-C in human
colorectal tumors. Oncogene. 24:8200–8204. 2005.PubMed/NCBI
|
|
41
|
Katsuno Y, Lamouille S and Derynck R:
TGF-β signaling and epithelial-mesenchymal transition in cancer
progression. Curr Opin Oncol. 25:76–84. 2013. View Article : Google Scholar
|
|
42
|
Watanabe Y, Itoh S, Goto T, et al: TMEPAI,
a transmembrane TGF-beta-inducible protein, sequesters Smad
proteins from active participation in TGF-beta signaling. Mol Cell.
37:123–134. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shi Y and Massague J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tsukazaki T, Chiang TA, Davison AF, et al:
SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta
receptor. Cell. 95:779–791. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Müller N, Reinacher-Schick A, Baldus S, et
al: SMAD4 induces the tumor suppressor E-cadherin and P-cadherin in
colon carcinoma cells. Oncogene. 21:6049–6058. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mu Y, Gudey SK and Landström M: Non-SMAD
signaling pathways. Cell Tissue Res. 347:11–20. 2012. View Article : Google Scholar
|
|
47
|
Hong M, Wilkes MC, Penheiter SG, et al:
Non-Smad transforming growth factor-β signaling regulated by focal
adhesion kinase binding the p85 subunit of phosphatidylinositol
3-kinase. J Biol Chem. 286:17841–17850. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kang JS, Liu C and Derynck R: New
regulatory mechanisms of TGF-beta receptor function. Trends Cell
Biol. 19:385–394. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hartsough MT and Mulder KM: Transforming
growth factor beta activation of p44mapk in proliferating cultures
of epithelial cells. J Biol Chem. 270:7117–7124. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Park SS, Eom YW, Kim EH, et al:
Involvement of c-Src kinase in the regulation of TGF-beta1-induced
apoptosis. Oncogene. 23:6272–6281. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yu L, Hebert MC and Zhang YE: TGF-beta
receptor-activated p38 MAP kinase mediates Smad-independent
TGF-beta responses. EMBO J. 21:3749–3759. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Timmerman LA, Grego Bessa J, Raya A, et
al: Notch promotes epithelial-mesenchymal transition during cardiac
development and oncogenic transformation. Genes Dev. 18:99–115.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Leong KG, Niessen K, Kulic I, et al:
Jagged l-mediated Notch activation induces
epithelial-to-mesenchymal transition through Slug-induced
repression of E-cadherin. J Exp Med. 204:2935–2948. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sahlgren C, Gustafsson MV, Jin S, et al:
Notch signaling mediates hypoxia induced tumor cell migration and
invasion. Proc Natl Acad Sci USA. 105:6392–6397. 2008. View Article : Google Scholar
|
|
55
|
Veenendaal LM, Kranenburg O, Smakman N, et
al: Differential Notch and TGFbeta signaling in primary colorectal
tumors and their corresponding metastases. Cell Oncol. 30:1–11.
2008.PubMed/NCBI
|
|
56
|
Murone M, Rosenthal A and de Sauvage FJ:
Sonic hedgehog signaling by the patched-smoothened receptor
complex. Curr Biol. 9:76–84. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chen JS, Huang XH, Wang Q, et al: Sonic
hedgehog signaling pathway induces cell migration and invasion
through focal adhesion kinase/AKT signaling-mediated activation of
matrix metalloproteinase (MMP)-2 and MMP-9 in liver cancer.
Carcinogenesis. 34:10–19. 2013. View Article : Google Scholar
|
|
58
|
Zhang H, Berezov A, Wang Q, et al: ErbB
receptors: from oncogenes to targeted cancer therapies. J Clin
Invest. 117:2051–2058. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Oda K, Matsuoka Y, Funahashi A, et al: A
comprehensive pathway map of epidermal growth factor receptor
signaling. Mol Syst Biol. 1:2005.00102005. View Article : Google Scholar
|
|
60
|
Wu L, Fan J and Belasco JG: MicroRNAs
direct rapid deadenylation of mRNA. Proc Natl Acad Sci USA.
103:4034–4039. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wienholds E, Koudijs MJ, van Eeden FJ, et
al: The microRNA-producing enzyme Dicer1 is essencial for zebrafish
development. Nat Genet. 35:217–218. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lee RC, Feinbaum RL and Ambros V: The C.
elegans heterochronic gene lin-4 encodes small RNAs with antisense
complementarity to lin-14. Cell. 75:843–854. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Pasquinelli AE, Reinhart BJ, Slack F, et
al: Conservation of the sequence and temporal expression of let-7
heterochronic regulatory RNA. Nature. 408:86–89. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Fang Y, Shi C, Manduchi E, et al:
MicroRNA-10a regulation of proinflammatory phenotype in
athero-susceptible endothelium in vivo and in vitro. Proc Natl Acad
Sci USA. 107:13450–13455. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Tsai KW, Wu CW, Hu LY, et al: Epigenetic
regulation of miR-34b and miR-129 expression in gastric cancer. Int
J Cancer. 129:2600–2610. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yan W, Song G-X and Li Q: Advances in
understanding the relationship between microRNAs and colorectal
cancer. World Chin J Digestol. 19:3426–3431. 2011.
|
|
67
|
Ng EK, Chong WW, Jin H, et al:
Differential expression of microRNAs in plasma of colorectal cancer
patients: a potential marker for colorectal cancer screening. Gut.
58:1375–1381. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lu J, Getz G, Miska EA, et al: MicroRNA
expression profiles classify human cancers. Nature. 435:834–838.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Calin GA, Sevignani C, Dumitru CD, et al:
Human microRNA genes are frequently located at fragile sites and
genomic regions involved in cancers. Proc Natl Acad Sci USA.
101:2999–3004. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Hurteau GJ, Carlson JA, Spivack SD, et al:
Overexpression of the microRNA has-miR-200c leads to reduced
expression of transcription factor 8 and increased expression of
E-cadherin. Cancer Res. 67:7972–7976. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Gregory PA, Bert AG, Paterson EL, et al:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Park SM, Gaur AB, Lengyel E, et al: The
miR-200 family determines the epithelial phenotype of cancer cells
by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev.
22:894–907. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Burk U, Schubert J, Wellner U, et al: A
reciprocal repression between ZEB1 and members of the miR-200
family promotes EMT and invasion in cancer cells. EMBO Rep.
9:582–589. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Shell S, Park SM, Radjabi AR, et al: Let-7
expression defines two differentiation stages of cancer. Proc Natl
Acad Sci USA. 104:11400–11405. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Liu X, Wang C, Chen Z, et al: MicroRNA-138
suppresses epithelial-mesenchymal transition in squamous cell
carcinoma lines. Biochem J. 440:23–31. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Castilla MA, Moreno-Bueno G, Romero-Perez
L, et al: Micro-RNA signature of the epithelial-mesenchymal
transition in endometrial carcinosarcoma. J Pathol. 233:72–80.
2011. View Article : Google Scholar
|
|
77
|
Li QQ, Chen ZQ, Cao XX, et al: Involvement
of NF-κB/miR-448 regulatory feedback loop in chemotherapy-induced
epithelial-mesenchymal transition of breast cancer cells. Cell
Death Differ. 18:16–25. 2011. View Article : Google Scholar :
|
|
78
|
Meng Z, Fu X, Chen X, et al: miR-194 is a
marker of hepatic epithelial cells and suppresses metastasis of
liver cancer cells in mice. Hepatology. 52:2148–2157. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ma L, Teruya-Feldstein J and Weinberg RA:
Tumor invasion and metastasis initiated by microRNA-10b in breast
cancer. Nature. 449:682–688. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Kong W, Yang H, He L, et al: MicroRNA155
is regulated by the transforming growth factor beta/Smad pathway
and contributes to epithelial cell plasticity by targeting RhoA.
Mol Cell Biol. 28:6773–6784. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Cottonham CL, Kaneko S and Xu L: miR-21
and miR-31 converge on TIAM1 to regulate migration and invasion of
colon carcinoma cells. J Biol Chem. 285:35293–35302. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Asangani IA, Rasheed SA, Nikolova DA, et
al: MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor
suppressor Pdcd4 and stimulates invasion, intravasation and
metastasis in colorectal cancer. Oncogene. 27:2128–2136. 2008.
View Article : Google Scholar
|
|
83
|
Wang P, Zho F, Zheng X, et al: microRNA-21
negatively regulates Cdc25A and cell cycle progression in colon
cancer cells. Cancer Res. 69:8157–8165. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Ma L, Young J, Prabhala H, et al: miR-9, a
MYC/MYCN-activated microRNA, regulates E-cadherin and cancer
metastasis. Nat Cell Biol. 12:247–256. 2010.PubMed/NCBI
|
|
85
|
Vetter G, Saumet A, Moes M, et al: miR-661
expression in SNAI1-induced epithelial to mesenchymal transition
contributes to breast cancer cell invasion by targeting Nectin-1
and StarD10 messengers. Oncogene. 29:4436–4448. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Castilla MA, Moreno-Bueno G, Romero-Pérez
L, et al: Micro-RNA signature of the epithelial-mesenchymal
transition in endometrial carcinosarcoma. J Pathol. 223:72–80.
2011. View Article : Google Scholar
|
|
87
|
De Krijger I, Mekenkamp LJ, Punt CJ and
Nagtegaal ID: MicroRNAs in colorectal cancer metastasis. J Pathol.
224:438–447. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Liu M and Chen H: The role of microRNAs in
colorectal cancer. J Genet Genomics. 37:347–358. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Spaderna S, Schmalhofer O, Hlubek F, et
al: A transient, EMT-linked loss of basement membranes indicates
metastasis and poor survival in colorectal cancer.
Gastroenterology. 131:830–840. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Spaderna S, Schmalhofer O, Wahlbuhl M, et
al: The transcriptional repressor ZEB1 promotes metastasis and loss
of cell polarity in cancer. Cancer Res. 68:537–544. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Korpal M, Lee ES, Hu G and Kang Y: The
miR-200 family inhibits epithelial-mesenchymal transition and
cancer cell migration by direct targeting of E-cadherin
transcriptional repressors ZEB1 and ZEB2. J Biol Chem.
283:14910–14914. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Brabletz S and Brabletz T: The ZEB/miR-200
feedback loop - a motor of cellular plasticity in development and
cancer? EMBO Rep. 11:670–677. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Fujita S, Ito T, Mizutani T, et al: miR-21
gene expression triggered by AP-1 is sustained through a
double-negative feedback mechanism. J Mol Biol. 378:492–504. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Ferraro A, Kontos CK, Boni T, et al:
Epigenetic regulation of miR-21 in colorectal cancer: ITGB4 as a
novel miR-21 target and a three-gene network (miR-21-ITGB4-PDCD4)
as predictor of metastatic tumor potential. Epigenetics. 9:129–141.
2014. View Article : Google Scholar :
|
|
95
|
Bommer GT, Gerin I, Feng Y, et al:
p53-mediated activation of miRNA34 candidate tumor-suppressor
genes. Curr Biol. 17:1298–1307. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Chang TC, Wentzel EA, Kent OA, et al:
Transactivation of miR-34a by p53 broadly influences gene
expression and promotes apoptosis. Mol Cell. 26:745–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Yamakuchi M, Ferlito M and Lowenstein CJ:
miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci
USA. 105:13421–13426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Siemens H, Jackstadt R, Hünten S, et al:
miR-34 and SNAIL form a double-negative feedback loop to regulate
epithelial-mesenchymal transitions. Cell Cycle. 10:4256–4271. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Kim NH, Kim HS, Li XY, et al: A
p53/miRNA-34 axis regulates Snail1-dependent cancer cell
epithelial-mesenchymal transition. J Cell Biol. 195:417–433. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Rokavec M, Oner MG, Li H, et al:
IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal
cancer invasion and metastasis. J Clin Invest. 124:1853–1867. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Bandi N, Zbinden S, Gugger M, et al:
miR-15a and miR16 are implicated in cell cycle regulation a
Rb-dependent manner and are frequently deleted or down-regulated in
non-small cell lung cancer. Cancer Res. 69:5553–5559. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Jackstadt R, Röh S, Neumann J, et al: AP4
is a mediator of epithelial-mesenchymal transition and metastasis
in colorectal cancer. J Exp Med. 210:1331–1350. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Shi L, Jackstadt R, Siemens H, et al:
p53-induced miR-15a/16-1 and AP4 form a double-negative feedback
loop to regulate epithelial-mesenchymal transition and metastasis
in colorectal cancer. Cancer Res. 74:532–542. 2014. View Article : Google Scholar
|
|
104
|
Dihlmann S, Siermann A and von Knebel
Doeberitz M: The nonsteroidal anti-inflammatory drugs aspirin and
indomethacin attenuate beta-catenin/TCF-4 signaling. Oncogene.
20:645–653. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Boon EM, Keller JJ, Wormhoudt TA, et al:
Sulindac targets nuclear beta-catenin accumulation and Wnt
signaling in adenomas of patients with familial adenomatous
polyposis and in human colorectal cancer cell lines. Br J Cancer.
90:224–229. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Linna L and Shoujun Y: Wnt/β-catenin
signaling pathway and strategy of originating and treatment in
colorectal cancer. World Chin J Digestol. 14:201–206. 2006.
|
|
107
|
Zhou L, An N, Haydon RC, et al: Tyrosine
kinase inhibitor STI-571/Gleevec down-regulatates the beta-catenin
signaling activity. Cancer Lett. 193:161–170. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Green DW, Roh H, Pippin JA and Drebin JA:
Beta-catenin antisense treatment decreases beta-catenin expression
and tumor growth rate in colon carcinoma xenografts. J Surg Res.
101:16–20. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Suzuki H, Watkins DN, Jair KW, et al:
Epigenetic inactivation of SFRP genes allows constitutive WNT
signaling in colorectal cancer. Nat Genet. 36:417–422. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Katz LH, Li Y, Chen JS, et al: Targeting
TGF-β signaling in cancer. Expert Opin Ther Targets. 17:743–760.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Lampropoulos P, Zizi-Sermpetzoglou A,
Rizos S, et al: TGF-beta signaling in colon carcinogenesis. Cancer
Lett. 314:1–7. 2012. View Article : Google Scholar
|
|
112
|
Melisi D, Ishiyama S, Sclabas GM, et al:
LY2109761, a novel transforming growth factor beta receptor type I
and type II dual inhibitor, as a therapeutic approach to
suppressing pancteatic cancer metastasis. Mol Cancer Ther.
7:829–840. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Korpal M, Yan J, Lu X, et al: Imaging
transforming growth factor-beta signaling dynamics and therapeutic
response in breast cancer bone metastasis. Nat Med. 15:960–966.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Zhang B, Halder SK, Zhang S and Datta PK:
Targeting transforming growth factor-beta signaling in liver
metastasis of colon cancer. Cancer Lett. 277:114–120. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Connolly EC, Freimuth J and Akhurst RJ:
Comlexities of TGF-β targeted cancer therapy. Int J Biol Sci.
8:964–978. 2012. View Article : Google Scholar
|
|
116
|
Zhao BM and Hoffmann FM: Inhibition of
transforming growth factor-beta1-induced signaling and
epithelial-to-mesenchymal transition by the Smad-binding peptide
aptamer Trx-SARA. Mol Biol Cell. 17:3819–3831. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Espinoza I, Pochampally R, Xing F, et al:
Notch signaling: targeting cancer stem cells and
epithelial-to-mesenchymal transition. Onco Targets Ther.
6:1249–1259. 2013.PubMed/NCBI
|
|
118
|
Yan M and Plowman GD: Delta-like 4/Notch
signaling and its therapeutic implications. Clin Cancer Res.
13:7243–7246. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Hayashi I, Takatori S, Urano Y, et al:
Neutralization of the γ-secretase activity by monoclonal antibody
against extracellular domain of nicastrin. Oncogene. 31:787–798.
2012. View Article : Google Scholar
|
|
120
|
Funahashi Y, Hernandez SL, Das I, et al: A
notch1 ectodomain construct inhibits endothelial notch signaling,
tumor growth, and angiogenesis. Cancer Res. 68:4727–4735. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Tammam J, Ware C, Efferson C, et al:
Down-regulation of the Notch pathway mediated by a gamma-secretase
inhibitor induces anti-tumor effects in mouse models of T-cell
leukaemia. Br J Pharmacol. 158:1183–1195. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Fouladi M, Stewart CF, Olson J, et al:
Phase I trial of MK_0752 in children with refractory CNS
malignancies: a pediatric brain tumor consortium study. J Clin
Oncol. 29:3529–3534. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Moellering RE, Cornejo M, Davis TN, et al:
Direct inhibition of the NOTCH transcription factor complex.
Nature. 462:182–188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Zhou W, Kallifatidis G, Baumann B, et al:
Dietary polyphenol quercetin targets pancreatic cancer stem cells.
Int J Oncol. 37:551–561. 2010.PubMed/NCBI
|
|
125
|
Kallifatidis G, Labsch S, Rausch V, et al:
Sulforaphane increases drug-mediated cytotoxicity toward cancer
stem-like cells of pancreas and prostate. Mol Ther. 19:188–195.
2011. View Article : Google Scholar :
|
|
126
|
Espinozal I and Miele L: Notch inhibitors
for cancer treatment. Pharmacol Ther. 139:95–110. 2013. View Article : Google Scholar
|
|
127
|
Kelleher FC: Hedgehog signaling and
therapeutics in pancreatic cancer. Carcinogenesis. 32:445–451.
2011. View Article : Google Scholar
|
|
128
|
Feldmann G, Dhara S, Fendrich V, et al:
Blockade of Hedgehog signaling inhibits pancreatic cancer invasion
and metastases: a new paradigm for combination therapy in solid
cancers. Cancer Res. 67:2187–2196. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Agyeman A, Mazumdar T and Houghton JA:
Regulation of DNA damage following termination of Hedgehog (HH)
survival signaling at the level of the GLI genes in human colon
cancer. Oncotarget. 3:854–868. 2012.PubMed/NCBI
|
|
130
|
Stecca B, Ruiz I and Altaba A:
Context-dependent regulation of the GLI code in cancer by Hedgehog
and non-Hedgehog signals. J Mol Cell Biol. 2:84–95. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Lauth M and Toftqard R: Non-canonical
activation of GLI transcription factors: implications for targeted
anti-cancer therapy. Cell Cycle. 6:2458–2463. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Hegan S, Orr MC and Doyle B: Targeted
therapies in colorectal cancer - an integrative view by PPPM. EPMA
J. 4:32013. View Article : Google Scholar
|
|
133
|
Ebi H, Corcoran RB, Singh A, et al:
Receptor tyrosine kinases exert dominant control over PI3K
signaling in human KRAS mutant colorectal cancers. J Clin Invest.
121:4311–4321. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Van Cutsem E, Peeters M, Siena S, et al:
Open-label phase III trial of panitumumab plus best supportive care
compared with best supportive care alone in patients with
chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol.
25:1658–1664. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Jonker DJ, O’Callaghan CJ, Karapetis CS,
et al: Cetuximab for the treatment of colorectal cancer. N Engl J
Med. 357:2040–2048. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Van Cutsem E, Köhne CH, Lang I, et al:
Cetuximab plus irinotecan, fluorouracil, and leucovorin as
first-line treatment for metastatic colorectal cancer: updated
analysis of overall survival according to tumor KRAS and BRAF
mutation status. J Clin Oncol. 29:2011–2019. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Heinemann V, von Weikersthal LF, Decker T,
et al: Randomized comparison of FOLFIRI plus cetuximab versus
FOLFIRI plus bevacizumab as first-line treatment of KRAS wild-type
metastatic colorectal cancer: German AIO study KRK-0306 (FIRE-3). J
Clin Oncol. 31:LBA3506 ASCO Annual Meeting. 2013.
|
|
138
|
Dassow H and Aigner A: MicroRNAs (miRNAs)
in colorectal cancer: from aberrant expression towards therapy.
Curr Pharm Des. 19:1242–1252. 2013.PubMed/NCBI
|
|
139
|
Weiler J, Hunziker J and Hall J:
Anti-miRNA oligonucleotides (AMOs): ammunition to target miRNAs
implicated in human disease. Gene Ther. 13:496–502. 2006.
View Article : Google Scholar
|
|
140
|
Garzon R, Marcucci G and Croce CM:
Targeting microRNAs in cancer: rationale, strategies and
challenges. Nat Rev Drug Discov. 9:775–789. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Yang Z, Vilkaitis G, Yu B, et al:
Approaches for studying MicroRNA and small interfering RNA
methylation in vitro and in vivo. Methods Enzymol. 427:139–154.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Ørom UA, Kauppinen S and Lund AH:
LNA-modified oligonucleotides mediate specific inhibition of
microRNA function. Gene. 10:137–141. 2006. View Article : Google Scholar
|
|
143
|
Vester B and Wengel J: LNA (locked nucleic
acid): high-affinity targeting of complementary RNA and DNA.
Biochemistry. 43:13233–13241. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Krützfeldt J, Rajewsky N, Braich R, et al:
Silencing of microRNAs in vivo with ‘antagomirs’. Nature.
438:685–689. 2005. View Article : Google Scholar
|
|
145
|
Si ML, Zhu S, Wu H, et al: miR-21-mediated
tumor growth. Oncogene. 26:2799–2803. 2007. View Article : Google Scholar
|
|
146
|
Corsten MF, Miranda R, Kasmieh R, et al:
MicroRNA-21 knockdown disrupts glioma growth in vivo and displays
synergistic cytotoxicity with neural precursor cell delivered
S-TRAIL in human gliomas. Cancer Res. 67:8994–9000. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Meng F, Henson R, Lang M, et al:
Involvement of human microRNA in growth and response to
chemotherapy in human cholangiocarcinoma cell lines.
Gastroenterology. 130:2113–2129. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Ebert MS, Neilson JR and Sharp PA:
MicroRNA sponges: competitive inhibitors of small RNAs in mammalian
cells. Nat Methods. 4:721–726. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Ebert MS and Sharp PA: MicroRNA sponges:
progress and possibilities. RNA. 16:2043–2050. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Valastyan S, Reinhardt F, Benaich N, et
al: A pleiotropically acting microRNA, miR-31, inhibits breast
cancer metastasis. Cell. 137:1032–1046. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Xiao J, Yang B, Lin H, et al: Novel
approaches for gene-specific interference via manipulating actions
of microRNAs: examination on the pacemaker channel genes HCN2 and
HCN4. J Cell Physiol. 212:285–292. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Gumireddy K, Young DD, Xiong X, et al:
Small-molecule inhibitors of microrna miR-21 function. Angew Chem
Int Ed Engl. 47:7482–7484. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Lennox KA and Behlke MA: Chemical
modification and design of anti-miRNA oligonucleotides. Gene Ther.
18:1111–1120. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Yang N, Kaur S, Volinia S, et al: MicroRNA
microarray identifles Let-7i as a novel biomarker and therapeutic
target in human epithelial ovarian cancer. Cancer Res.
68:10307–10314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Kumar MS, Erkeland SJ, Pester RE, et al:
Suppression of non-small cell lung tumor development by the let-7
microRNA family. Proc Natl Acad Sci USA. 105:3903–3908. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Bonci D, Coppola V, Musumeci M, et al: The
miR-15a-miR-16–1 cluster controls prostate cancer by targeting
multiple oncogenic activities. Nat Med. 14:1271–1277. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Wiggins JF, Ruffino L, Kelnar K, et al:
Development of a lung cancer therapeutic based on the tumor
suppressor microRNA-34. Cancer Res. 70:5923–5930. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Chen Y, Zhu X, Zhang X, et al:
Nanoparticles modified with tumor-targeting scFv deliver siRNA and
miRNA for cancer therapy. Mol Ther. 18:1650–1656. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
La Rocca G, Badin M, Shi B, et al:
Mechanism of growth inhibition by MicroRNA 145: the role of the
IGF-1 receptor signaling pathway. J Cell Physiol. 220:485–491.
2009. View Article : Google Scholar : PubMed/NCBI
|