Introduction
Hepatocellular carcinoma (HCC) is a major health
concern worldwide and is the main cause of death among cirrhotic
patients (1). It is one of the most
lethal malignancies in the world with more than half a million new
cases diagnosed annually (2).
Hepatocarcinogenesis is a complex and multi-step process, which is
associated with many risk factors (3). Epidemiological studies have
demonstrated that hepatitis B virus (HBV) infection is one of the
key risk factors for HCC (4),
particularly in eastern Asia.
Hepatitis B virus X (HBx) protein, a 17-kDa protein
encoded by the HBV genome, plays a critical role in the progression
of HBV-related HCC (5,6). In addition, HBx protein can interfere
with several signaling pathways that are associated with cell
proliferation and apoptosis. However, the exact role of HBx protein
in the progression of HCC remains to be determined. Investigation
of the mechanism is important to clarify the tumorigenic pathway
and is helpful for developing new biomarkers for early detection
and accurate diagnosis.
MicroRNAs (miRNAs) are a class of ~21–25 nucleotide
non-coding RNAs, which mediate the post-transcriptional regulation
of mRNAs by interfering with mRNA stability or protein translation
(7). They have been identified as
being involved in biological processes, including cell
proliferation, apoptosis as well as cell differentiation (8,9).
Deregulation of miRNAs has been found to be essential in cancer
development and progression (10).
Growing evidence reveals that many miRNAs are involved in HCC
tumorigenesis, including miR-221 (11,12).
miR-221 upregulation indicates the activation of stellate cells and
the progression of liver fibrosis (13).
Previous data indicate that estrogen receptor-α
(ERα) has protective effects on HCC (14). The relationship between miR-221 and
ERα was studied by Zhao et al (15). They found that miR-221 negatively
regulates ERα expression by interaction with the 3′-untranslated
region (3′UTR) of ERα in breast cancer. To date, however, the role
of miR-221 in HBV-related hepatocarcinogenesis and the molecular
mechanisms by which miR-221 modulates malignant phenotypes of HCC
cells remain largely unknown.
In the present study, we found that upregulation of
miR-221 was unregulated by HBx protein in HCC. The overexpression
of miR-221 was found to induce cell proliferation. Finally, an
inverse correlation between the expression of miR-221 and ERα was
found in HCC. Our findings will help to determine the oncogenic
role of miR-221 in hepatocarcinogenesis and the molecular mechanism
of HBx protein in the progression of HCC.
Materials and methods
Tissue specimens and cell culture
HepG2 and MCF-7 cells were purchased from the
American Type Culture Collection. The HepG2.215 cell line, which
persistently produces HBV due to the integrated HBV genome, was
kindly provided by Professor Ailong Huang (Key Laboratory of
Molecular Biology on Infection Diseases of the Ministry of
Education, Chongqing Medical University). The cells were cultured
in minimum essential medium (MEM; HyClone) supplemented with 10%
(v/v) FCS, 2 mmol/l glutamine, 1 mM sodium pyruvate, 100 U/ml
penicillin-streptomycin solution in a 5% CO2 incubator
at 37°C.
Fourteen formalin-fixed and paraffin-embedded (FFPE)
hepatic tissue specimens were obtained from patients chronically
infected with HBV at the First Affiliated Hospital of Chongqing
Medical University and were confirmed as HCC by histopathological
evaluation. All of the tissue specimens were obtained after consent
was obtained by the patients.
Construction of the Ad-HBx adenovirus and
the infection of HepG2 cells
Both the Ad-HBx and the control Ad-EGFP adenoviruses
were constructed with the AdEasy system. HepG2 cells were plated
into a 6-well plate in 2 ml of complete medium at a density of
1.5×105 cells/well and incubated overnight until 30–50%
confluency. The Ad-HBx and Ad-EGFP adenoviruses were respectively
infected into HepG2 cells at an MOI of 50. The infection efficiency
was evaluated by observation of EGFP expression under a
fluorescence microscope after 48 h.
Semi-quantitative reverse-transcription
PCR
After incubation for 48 h, cells infected with the
adenovirus were harvested. Total RNA was then extracted using
TRIzol reagent (Invitrogen) and quantified by spectrophotometry at
OD260. First-strand cDNA was synthesized using
oligo(dT)12-18 primers and SuperScript II RNase
H-Reverse Transcriptase (both from Invitrogen) according to the
manufacturer’s recommendations. The target cDNA was amplified using
Platinum Taq DNA polymerase (Invitrogen) for 28–30 cycles.
PCR was performed with the following primers: P1 and P2 for HBx, P3
and P4 for GAPDH: P1, 5′-ACCGACCTTGAGGCCTACTT-3′; P2, 5′-GC
TTGGCAGAGGTGAAAAAG-3′; P3, 5′-GAGTCAACGGAT TTGGTCGT-3′; and P4,
5′-TTGATTTTGGAGGGATCT CG-3′. Each sample was then analyzed by
electrophoresis, and the resulting gel was photographed using a gel
documentation system (Bio-Rad).
miRNA isolation and quantitative reverse
transcription-polymerase chain reaction
Total RNA was isolated from HepG2 cells using the
mirVana PARIS kit (Applied Biosystems, Inc.) according to the
manufacturer’s instructions. Quantification of miR-221 was
performed using the TaqMan miRNA assay kit (Applied Biosystems,
Inc.) according to the manufacturer’s instructions. U6 small
nuclear RNA was used as an internal control for determining the
relative miRNA expression level, followed by detection with the
Applied Biosystems 7500HT Sequence Detection System (P/N: 4329002)
(both from Applied Biosystems). In relation to the expression of
small nuclear U6 RNA, the expression level of miR-221 for each RNA
sample was calculated, reflected by the value of ΔCt (Ct of miR-221
− Ct of U6).
Western blot analysis
M-PER mammalian protein extraction reagent (Cell
Signaling) was employed to lyse the cells. Proteins were resolved
on 10% SDS-PAGE gels and transferred to PVDF membranes. The
membranes were blocked with 5% BSA, and incubated with antibodies
against ERα (R&D Systems) and GAPDH according to the standard
western blot protocol. Blots were developed using SuperSignal West
Pico chemiluminescent substrate (Pierce), imaged and analyzed using
the Bio-Rad Gel Imaging System (Bio-Rad).
Cell transfection
The miR-221 mimic or the miR-221 inhibitor (both
from Qiagen, Valencia, CA, USA) was transfected (4.5 μM) with
HiPerFect Transfection Reagent (Qiagen) according to the
manufacturer’s procedures. Negative control premiR (Qiagen)was also
transfected into the HepG2 cells as the control.
Cell growth assay
HepG2 cells (4×103/well) were plated in
96-well plates, and then the cells were transfected (final miRNA
concentration of 3 nM), or infected. Cell proliferation was
documented every 24 h for 3 days using a cell counting kit
(Dojindo), and absorbance at 450 and 610 nm was evaluated using a
SpectraMax 190 microplate reader (Molecular Devices, Sunnyvale, CA,
USA).
Flow cytometry for cell cycle and
apoptosis analysis
For cell cycle analysis, the cells from each well
were harvested 48 h after transfection, and then the transfected
cells were fixed with 70% cold ethanol for at least 1 h after
collecting and washing. Cells were stained with propidium iodide,
and cell cycle distribution was examined by a
fluorescence-activated cell sorting (FACS) flow cytometer, and DNA
histograms were analyzed with modified software.
Luciferase reporter assay
The 3′UTR of the candidate gene was cloned and
inserted into the downstream of the luciferase gene in the
pGL3/luciferase vector. The mutant 3′UTR of the candidate gene was
cloned using the wild-type 3′UTR as a template and inserted into
pGL3/luciferase as described for the wild-type 3′UTR. The cells
were co-transfected with miRNA mimics and the wild-type or mutant
3′UTR of the candidate gene. Luciferase activity was determined 48
h after transfection using the Dual-Luciferase Reporter Assay
system (Promega).
Statistical analysis
All data are expressed as mean ± standard deviation
(SD) from at least 3 separate experiments. Statistical significance
was determined with the paired Student’s t-test, except as noted
for analyses of microarray data, which were examined with Fisher’s
exact tests. P-value <0.05 was considered to indicate a
statistically significant result. All statistical analyses were
performed with SPSS software (version 17.0).
Results
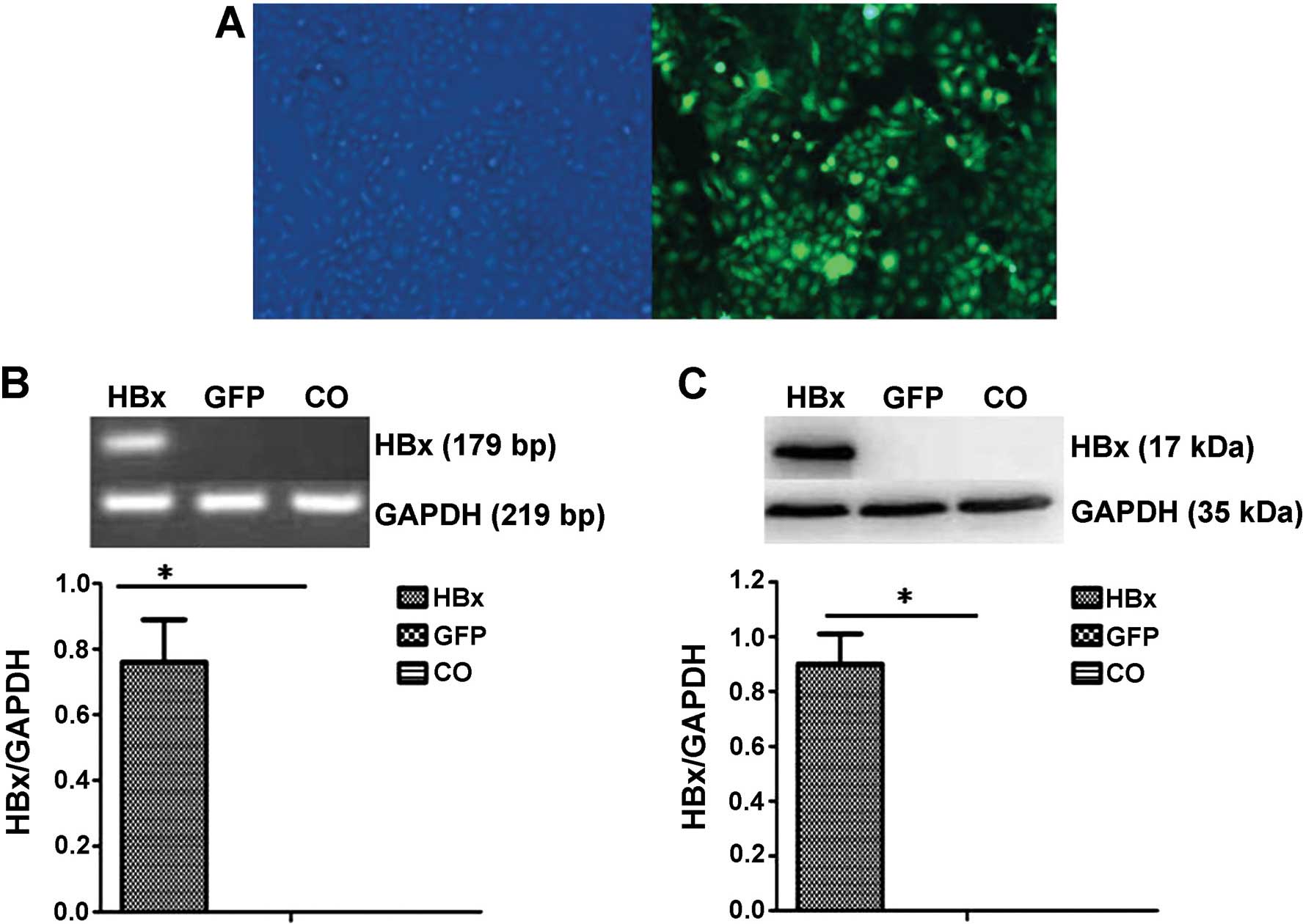
Ad-HBx-transfected HCC cells
HepG2 cells were infected with Ad-HBx core or Ad-GFP
at an MOI of 50, and the infection efficiency was evaluated by
observation of EGFP expression under a fluorescence microscope
(Fig. 1A). We used
semi-quantitative reverse transcription-PCR to confirm the
infection efficacy of HBx (Fig.
1B). As shown in Fig. 1C, the
efficient transient expression of the HBx protein was noted in the
HepG2 cells after Ad-HBx infection at 48 h.
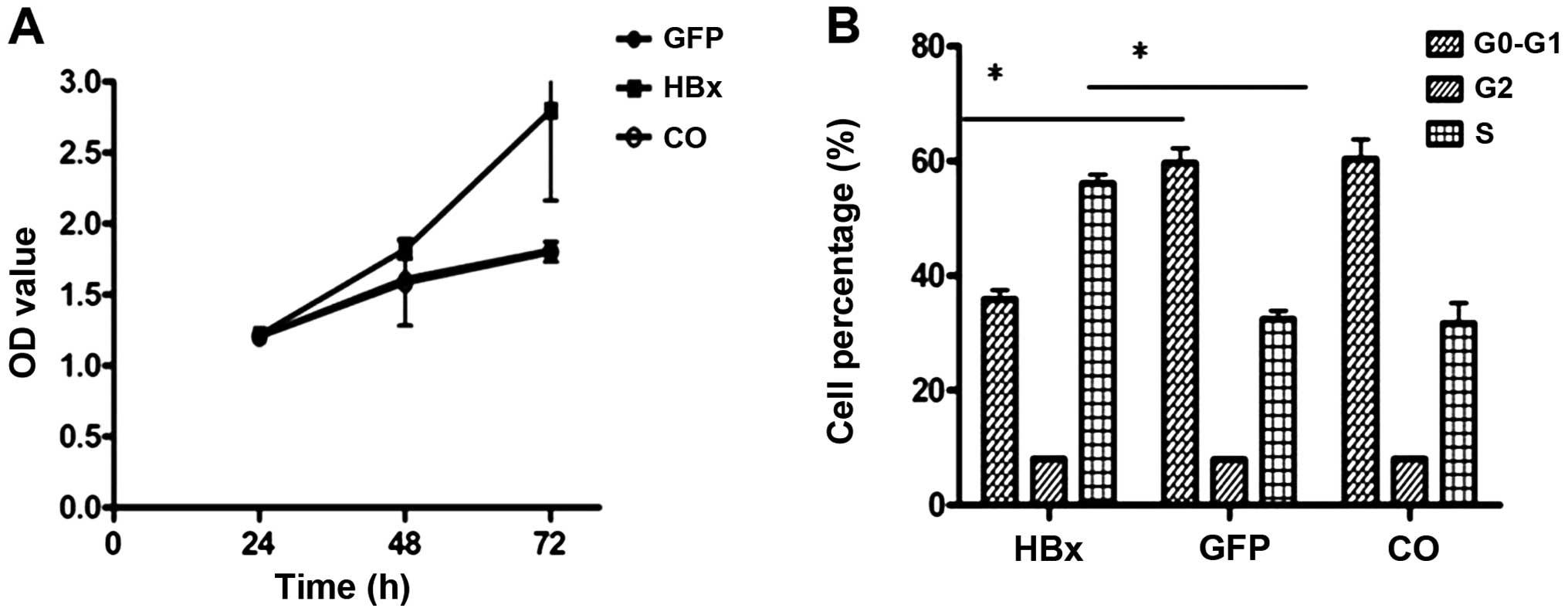
HBx promotes HCC cell proliferation
After HepG2 cells were infected with Ad-HBx and
Ad-GFP for 48 h, cell proliferation assays indicated significant
growth promotion in the Ad-HBx-infected cells (Fig. 2A). To further investigate the effect
of HBx overexpression on cell cycle distribution, cells were
analyzed for cell cycle status using propidium iodide staining and
flow cytometry. An obvious accumulation of cells in the S phase was
detected after HBx overexpression compared with the control
(Fig. 2B).
HBx downregulates ERα expression in
HBV-related HCC
To determine whether HBx protein alters ERα
expression, we detected the ERα protein level after the transient
infection of Ad-HBx into HepG2 cells. Our results confirmed that
ERα expression was inhibited by HBx (Fig. 3A and B).
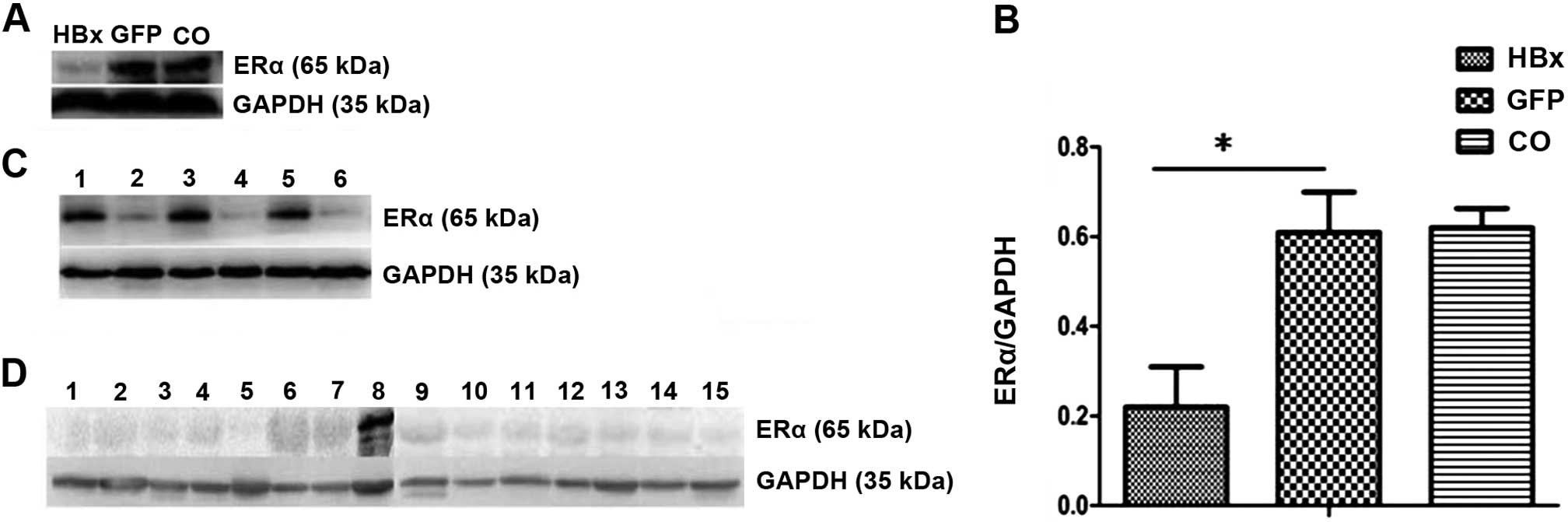 | Figure 3HBx downregulates ERα expression in
HBV-related HCC. (A and B) Expression of ERα was determined by
western blotting in HepG2 cells infected with the Ad-HBx or Ad-GFP
adenovirus. GAPDH was used as an internal control. (C) Expression
of ERα was determined by western blotting in HepG2 and HepG2.215
cells. GAPDH was used as an internal control. Lanes 1, 3 and 5,
HepG2 cells; lanes 2, 4 and 6, HepG2.215 cells. (D) Expression of
ERα was determined by western blotting in 14 HBV-related HCC tumor
specimens. MCF-7 cells were used as a positive control. GAPDH was
used as an internal control. Lanes 1–7, tumor specimens; lane 8,
MCF-7 cells; lanes 9–15, tumor specimens. *P<0.05.
CO, control; HBx, hepatitis B virus X protein; ERα, estrogen
receptor-α. |
HepG2.215 is a cell line that is derived from HepG2.
The only difference between them is that HepG2.215 cells are able
to produce infectious viruses through the HBV genome integrated in
the cell chromosome (16–18). The results suggest that the
expression of ERα was obviously higher in the HepG2 cells than that
in the HepG2.215 cells (Fig.
3C).
Additionally, in accordance with the results
obtained in the cancer cells, ERα was validated to be downregulated
in HBV-related HCC tumor tissues. MCF-7 breast cancer cells were
used as a positive control (Fig.
3D).
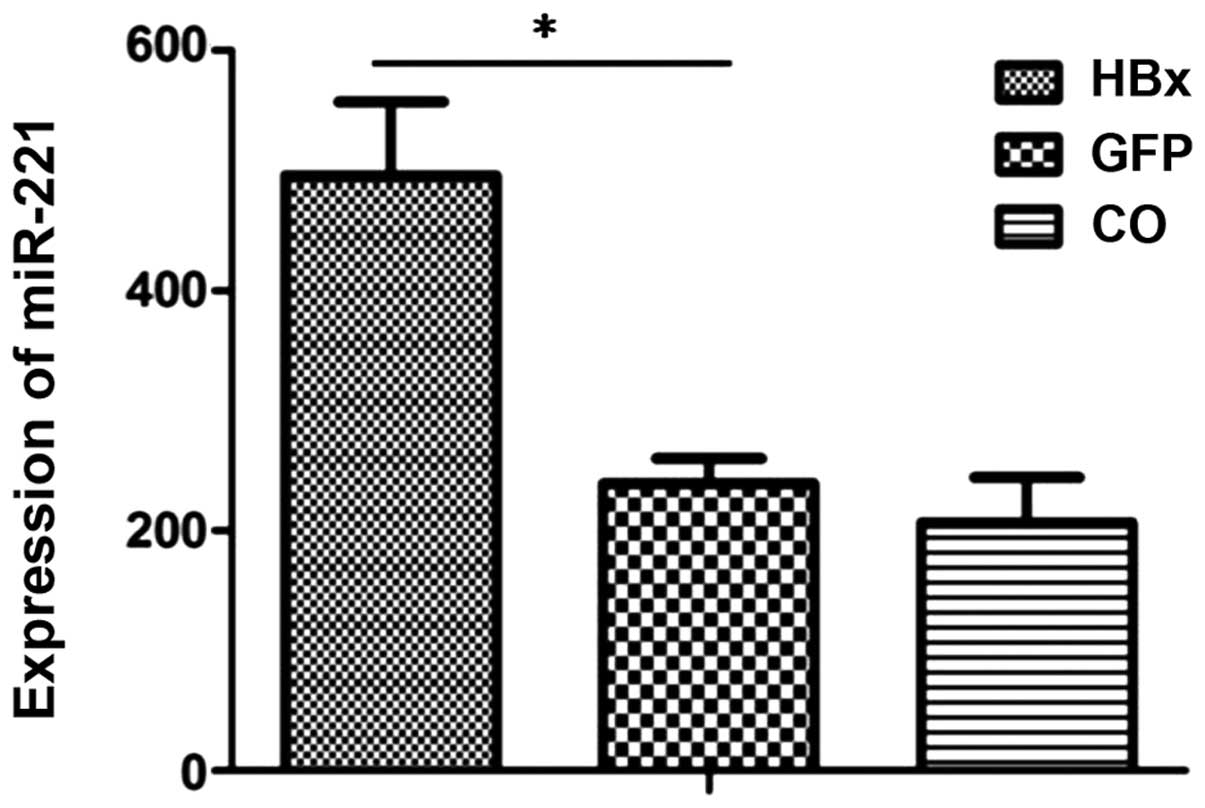
HBx unregulates miR-221 in HepG2
cells
We aimed to ascertain whether the miR-221 level is
affected by HBx overexpression. As shown in Fig. 4, the level of miR-221 was
significantly higher in the HepG2-HBx cells than that in the
HepG2-GFP control group or HepG2-mock control group.
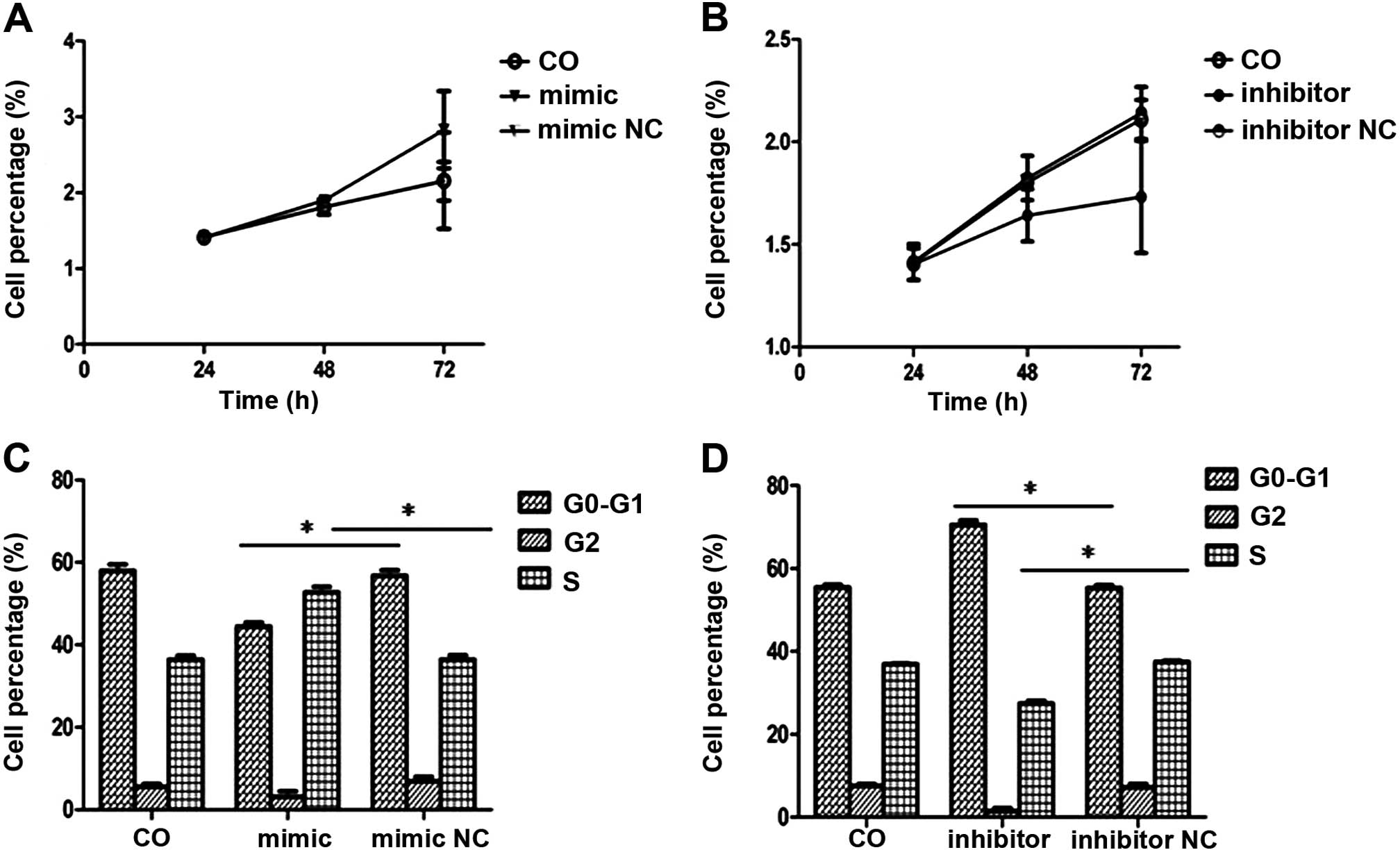
HBx promotes cell growth and G1-S
transition via upregulation of miR-221 in HepG2 cells
To test the hypothesis that the HBx protein
influences the proliferation of HepG2 cells via its upregulation of
miR-221, we evaluated the effects of transient transfection of the
miR-221 inhibitor and miR-221 mimics on cell growth using MTT and
flow cytometry. Upon the construction of cell growth curves, we
found that the miR-221 mimic promoted the growth of HepG2 cells
compared to the control cells (Fig.
5A). However, the miR-221 inhibitor significantly inhibited
cell growth (Fig. 5B). To test the
hypothesis that the HBx protein promotes G1/S transition via its
upregulation of miRA-221, we performed gain-of-function and
loss-of-function studies of miR-221, and evaluated the effects on
the cell cycle profile. We observed profound promotion of G1/S
transition by the miR-221 mimics (Fig.
5C), while the miR-221 inhibitor significantly inhibited G1/S
progression (Fig. 5D). Overall,
these data suggest that miR-221 promotes HCC cell
proliferation.
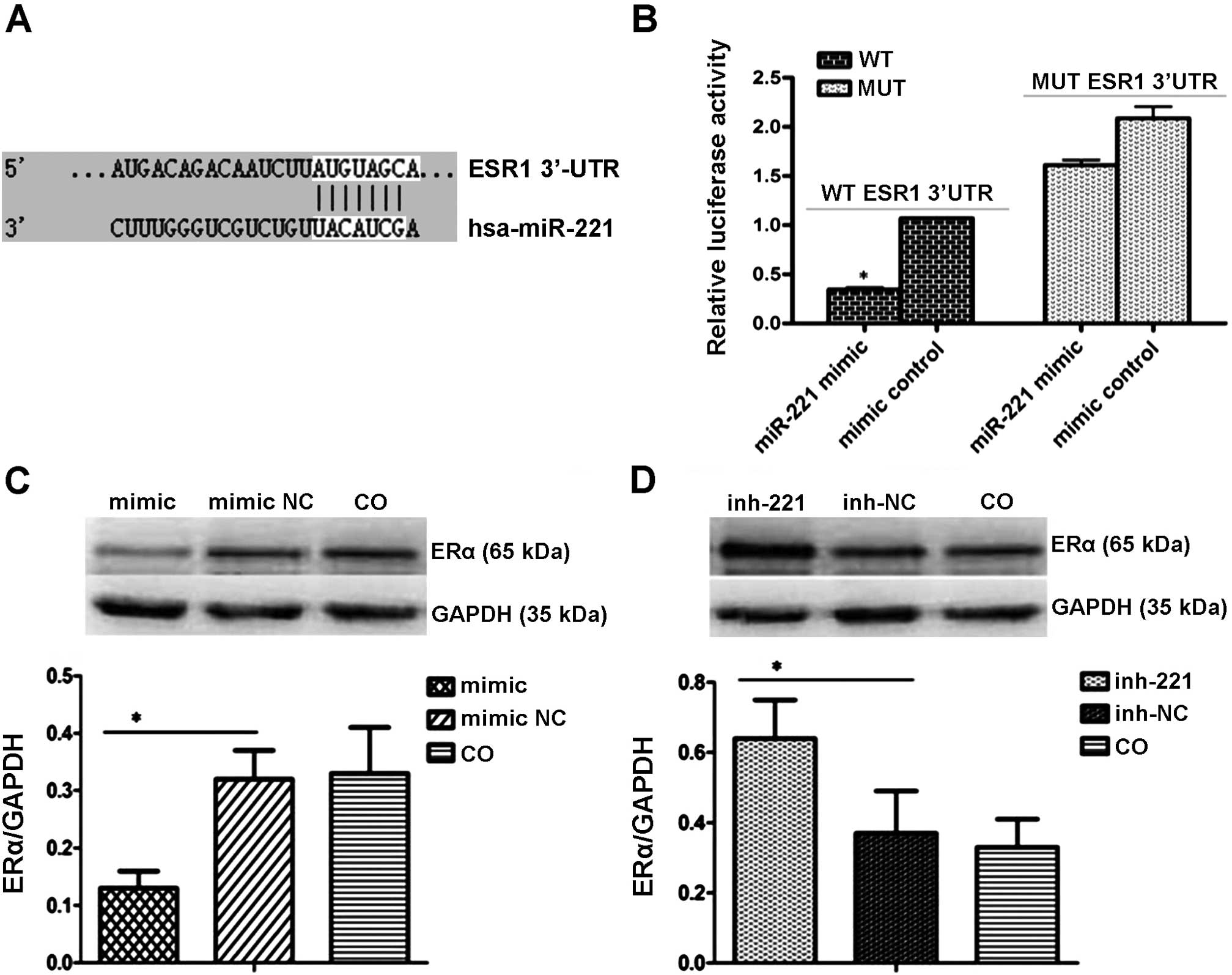
ERα is a direct target of miR-221
Online bioinformatic analyses showed that the 3′UTR
of ERα-encoded mRNA contains a sequence that is partially
complementary to miR-221 (Fig. 6A).
Cells were transiently transfected with miR-221 mimics or the
inhibitor. The luciferase reporter assay indicated that miR-221 led
to inhibition of the wild-type 3′UTR by 47.01%, yet it had no
effect on the luciferase intensity controlled by the mutant 3′UTR
(Fig. 6B). To further determine
whether miR-221 affects ERα expression in the HCC intracellular
environment, we analyzed changes in ERα expression in HepG2 cells
after transient transfection of miR-221 mimics or the inhibitor.
Western blot analysis showed that the miR-221 inhibitor upregulated
ERα expression while the miR-221 mimics markedly inhibited its
expression (Fig. 6C and D). Taken
together, these results indicate that miR-221 negatively regulates
ERα by directly targeting its 3′UTR.
Discussion
miRNAs have been shown to exhibit regulatory
functions in numerous cellular processes, including cell growth,
differentiation and apoptosis. Moreover miRNAs were demonstrated to
induce hepatocyte apoptosis (16),
liver fibrosis (17) and
hepatocarcinogenesis (18).
Previous reports suggest that miR-221 is upregulated
in various types of cancers (19),
including HCC (20). Meanwhile,
overexpression of miR-221 was shown to promote cancer cell
proliferation by inhibiting the expression of cell cycle
controllers CDKN1B/p27 (21) and
CDKN1C/p57 (22). In addition,
miR-221 silencing was found to inhibit HCC cell proliferation and
promote survival (23).
In the present study, we investigated the role and
the functional target of miR-221 in HBV-related HCC.
Our data indicated that HBx overexpression promoted
cell proliferation and upregulated miR-221. Suppression of miR-221
significantly inhibited HCC cell growth. All of these results were
supported by data from other reports. For example, miR-221 was
found to promote cell proliferation of breast cancer (24). MiR-221 was also found to be
essential for the EMT phenotype, migration and growth of pancreatic
cancer cells (25). Moreover, the
level of miR-221 consistently reflects the stage of acute myeloid
leukemia, serving as a tumor promoter (26).
As for the mechanism of miRNA regulation in cancer,
it is crucial to validate target genes. Our data demonstrated that
HBx protein stimulated miR-221 but inhibited ERα expression.
Further studies showed that miR-221 directly targeted the 3′UTR of
ERα. MiR-221 was also shown to reduce the protein levels of ERα,
resulting in increased proliferation activity in hepatoma cell
lines. These results suggest a new mechanism by which to suppress
the estrogen signaling pathway, which has been long known to
protect against HCC development (27–29).
Previous report found that ERα was significantly
downregulated in HCCs, both by immunohistochemical staining or by
receptor binding assay (30–32).
Obviously downregulation of ERα was found in HCC cancer tissues
compared with their non-tumor counterparts. Thus, ERα suppression
promotes the proliferation of HCC cells (33).
We demonstrated that ERα expression was
downregulated in both HBV-related HCC cell lines and tumor
specimens, which is consistent with the result from a recent
publication by Liu et al (33). Studies have demonstrated that the
methylation of a CpG island at the promoter of ESR1 is one of the
main contributors to ERα downregulation in HCC (34).
Estrogens are well accepted as cancer-promoting
agents in both the breast and the uterus (35). How they function in a contradictory
manner to protect individuals against HCC is intriguing. We presume
that estrogens through tissue-specific selective ER modulators
(36), may exert opposite effects
on HCC and breast cancer. This can be addressed in future studies
by overexpressing miR-221 specifically in the liver or in the
breast of transgenic mice.
In conclusion, the present study indicates that HBx
increases miR-221 expression, and miR-221 promotes cell
proliferation through the suppression of ERα, functioning as a
tumor promoter. Our data imply that the overexpression of miR-221
may serve as a novel biomarker for HBV-related HCC and may also be
a potential target for an miRNA-based molecular therapeutic
strategy.
Acknowledgements
This study was supported by a grant from the
National Natural Science Foundation of China (no. 81101826), and
the National Key Clinical Specialities Construction Program of
China.
References
|
1
|
Sangiovanni A, Del Ninno E, Fasani P, et
al: Increased survival of cirrhotic patients with a hepatocellular
carcinoma detected during surveillance. Gastroenterology.
126:1005–1014. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Han Z: Recent progress in genomic
[corrected] research of liver cancer. Sci China C Life Sci.
52:24–30. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lavanchy D: Hepatitis B virus
epidemiology, disease burden, treatment, and current and emerging
prevention and control measures. J Viral Hepat. 11:97–107. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Neuveut C, Wei Y and Buendia MA:
Mechanisms of HBV-related hepatocarcinogenesis. J Hepatol.
52:594–604. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ng SA and Lee C: Hepatitis B virus X gene
and hepatocarcinogenesis. J Gastroenterol. 46:974–990. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brenneck J, Hipfner DR, Stark A, et al:
bantam encodes a developmentally regulated microRNA that controls
cell proliferation and regulates the proapoptotic gene hid in
Drosophila. Cell. 113:25–36. 2003. View Article : Google Scholar
|
|
9
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shi XB, Tepper CG and deVere White RW:
Cancerous miRNAs and their regulation. Cell Cycle. 7:1529–1538.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Santhekadur PK, Das SK, Gredler R, et al:
Multifunction protein staphylococcal nuclease domain containing 1
(SND1) promotes tumor angiogenesis in human hepatocellular
carcinoma through novel pathway that involves nuclear factor κB and
miR-221. J Biol Chem. 287:13952–13958. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park JK, Koqure T, Nuovo GJ, et al:
miR-221 silencing blocks hepatocellular carcinoma and promotes
survival. Cancer Res. 71:7608–7616. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ogawa T, Enomoto M, Fujii H, et al:
MicroRNA-221/222 upregulation indicates the activation of stellate
cells and the progression of liver fibrosis. Gut. 61:1600–1609.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Naugler WE, Sakurai T, Kim S, et al:
Gender disparity in liver cancer due to sex differences in
MyD88-dependent IL-6 production. Science. 317:121–124. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao JJ, Lin J, Yang H, et al:
MicroRNA-221/222 negatively regulates estrogen receptor α and is
associated with tamoxifen resistance in breast cancer. J Biol Chem.
283:31079–31086. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Acs G, Sells MA, Purcell RH, et al:
Hepatitis B virus produced by transfected HepG2 cells causes
hepatitis in chimpanzees. Proc Natl Acad Sci USA. 84:4641–4644.
1987. View Article : Google Scholar
|
|
17
|
Sells MA, Chen ML and Acs G: Production of
hepatitis B virus particles in HepG2 cells transfected with cloned
hepatitis B virus DNA. Proc Natl Acad Sci USA. 84:1005–1009. 1987.
View Article : Google Scholar
|
|
18
|
Mott JL, Kobayashi S, Bronk SF and Gores
GJ: miR-29 regulates Mcl-1 protein expression and apoptosis.
Oncogene. 26:6133–6140. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roderburq C, Urban GW, Bettermann K, et
al: Micro-RNA profiling reveals a role of miR-29 in human and
murine liver fibrosis. Hepatology. 53:209–218. 2011. View Article : Google Scholar
|
|
20
|
Pineau P, Volinia S, McJunkin K, et al:
miR-221 overexpression contributes to liver tumorigenesis. Proc
Natl Acad Sci USA. 107:264–269. 2010. View Article : Google Scholar :
|
|
21
|
Gramantieri L, Ferracin M, Formari F, et
al: Cyclin G1 is a target of miR-122a, a microRNA frequently
down-regulated in human hepatocellular carcinoma. Cancer Res.
67:6092–6099. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
le Sage C, Nagel R, Egan DA, et al:
Regulation of the p27Kip1 tumor suppressor by miR-221
and miR-222 promotes cancer cell proliferation. EMBO J.
26:3699–3708. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Medina R, Zaidi SK, Liu CG, et al:
MicroRNAs 221 and 222 bypass quiescence and compromise cell
suvival. Cancer Res. 68:2773–2780. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rehman SK, Li SH, Wyszomierski SL, et al:
14-3-3ζ orchestrates mammary tumor onset and progression via
miR-221-mediated cell proliferation. Cancer Res. 74:363–373. 2014.
View Article : Google Scholar :
|
|
25
|
Su A, He S, Tian B, et al: MicroRNA-221
mediates the effects of PDGF-BB on migration, proliferation, and
the epithelial-mesenchymal transition in pancreatic cancer cells.
PLoS One. 8:e713092013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rommer A, Steinleitner K, Hackl H, et al:
Overexpression of primary microRNA 221/222 in acute myeloid
leukemia. BMC Cancer. 13:3642013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu MW, Chang HC, Chang SC, et al: Role of
reproductive factors in hepatocellular carcinoma: impact on
hepatitis B- and C-related risk. Hepatology. 38:1393–1400.
2003.PubMed/NCBI
|
|
28
|
Vesselinovitch SD, Itze L, Mihailovich N
and Rao KV: Modifying role of partial hepatectomy and gonadectomy
in ethylnitrosourea-induced hepatocarcinogenesis. Cancer Res.
40:1538–1542. 1980.PubMed/NCBI
|
|
29
|
Nakatani T, Roy G, Fujimoto N, et al: Sex
hormone dependency of diethylnitrosamine-induced liver tumors in
mice and chemoprevention by leuprorelin. Jpn J Cancer Res.
92:249–256. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ng IO, Ng M and Fan ST: Better survival in
woman with respected hepatocellular carcinoma is not related to
tumor proliferation or expression of hormone receptors. Am J
Gastroenterol. 92:1355–1358. 1997.PubMed/NCBI
|
|
31
|
Jonas S, Bechstein WO, Heinze T, et al:
Female sex hormone receptor status in advanced hepatocellular
carcinoma and outcome after surgical resection. Surgery.
121:456–461. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nagasue N, Kohno H, Chang YC, et al:
Clinicopathologic comparisons between estrogen receptor-positive
and -negative hepatocellular carcinomas. Ann Surg. 212:150–154.
2009. View Article : Google Scholar
|
|
33
|
Liu WH, Yeh SH, Lu CC, et al: MicroRNA-18a
prevents estrogen receptor-α expression, promoting proliferation of
hepatocellular carcinoma cells. Gastroenterology. 136:683–693.
2009. View Article : Google Scholar
|
|
34
|
Shen L, Ahuja N, Shen Y, et al: DNA
methylation and environmental exposures in human hepatocellular
carcinoma. J Natl Cancer Inst. 94:755–761. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pike MC and Spicer DV: Hormonal
contraception and chemoprevention of female cancers. Endocr Relat
Cancer. 7:73–83. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dutertre M and Smith CL: Molecular
mechanisms of selective estrogen receptor modulator (SERM) action.
J Pharmacol Exp Ther. 295:431–437. 2000.PubMed/NCBI
|