Introduction
Endometrial carcinoma (EC) is one of the most common
malignant tumors of the female genital tract and has an increasing
incidence worldwide (1).
Approximately 70–80% of sporadic endometrial carcinomas are
distinguished as type I carcinomas and are associated with
endometrial hyperplasia, hyperestrogenism and estrogen receptor
(ER) expression. The remaining 20% constitute type II carcinomas,
are generally unrelated to estrogen, and exhibit negative or low ER
expression (2). Despite a number of
studies that have identified prognostic biomarkers for EC, a
paucity of reliable markers and therapeutic targets exist to
diagnose and treat this disease.
Peroxisome proliferator-activated receptor γ (PPARγ)
is a member of the superfamily of nuclear receptors. PPARγ is
important in lipid and glucose metabolism, adipose differentiation,
inflammatory responses, macrophage differentiation and energy
homeostasis (3). In addition, the
known risk factors for EC include obesity, type II diabetes
mellitus and hypertension (4).
Thus, PPARγ ligands, which have been used for the treatment of type
II diabetes mellitus, may be important drug candidates for possible
endocrine treatment of EC. The expression of PPARγ has been
extensively studied in various human carcinomas (5–7), yet
little is known concerning PPARγ in EC. Thus, it has become very
important to obtain a better understanding of the clinical and
biological roles of PPARγ in EC tissues to improve the potential
clinical efficiency of PPARγ ligand therapy for endometrial
carcinoma patients.
EC is representative of hormone-dependent
gynecologic cancers (8). However,
attempts to treat female hormone-dependent cancers with
anti-hormonal treatments have not been effective, except in
early-stage cancers. The ERs are ligand-dependent transcription
factors and belong to a superfamily of steroid nuclear receptors
(9). To date, two ERs (ERα and
ERβ), which are encoded by different genes, have been detected
(10). It is well known that the
presence of ERα in breast and endometrial carcinoma is associated
with a less aggressive phenotype (11,12).
However, the roles of ERβ in the development and growth of these
tumors have not yet been completely elucidated (13,14).
Much of the current interest in understanding the basis of ER
actions at the molecular level is focused on the goal of
therapeutic intervention (15–17).
Despite these efforts, the exact transcriptional effects of ERα and
ERβ in EC remain obscure.
Recently, increasing physiologic significance has
been attributed to the crosstalk among nuclear receptors, which
have been observed at several levels of signal transduction
cascades (18–20). PPARγ and ERs belong to a family of
nuclear hormone receptors that have been demonstrated to affect the
transcriptional activity of each other. An area of relevance to
breast cancer is the inhibitory effect of PPARγ on ERα (ER)
promoter activation through its interaction with ER response
elements (21). PPARγ is expressed
in many types of cancer, and it is well established that activation
of the receptor inhibits cell proliferation and induces apoptosis
(22,23). Therefore, the present study aimed to
elucidate the correlation between PPARγ and ER expression in EC and
to investigate whether PPARγ activation in endometrial cancer cells
contributes to novel approaches for EC therapy.
Materials and methods
Tissue specimens
Samples of 45 endometrial adenocarcinomas and 13
normal endometrium tissues were obtained from surgical pathology
specimens at the Department of Gynecology, Qilu Hospital of
Shandong University. The samples were immediately frozen in liquid
nitrogen and stored at −70°C until analyzed. The specimens were
processed for histopathological, immunohistochemical examination
and western blot analysis. Information included body mass index
(BMI), stage and grade. BMI was calculated by dividing the weight
in kilograms by the height in meters squared. We defined obesity as
a BMI ≥25 (24). Pathological
grading was determined using histopathological analysis and the
staging process following the FIGO system. None of these patients
received preoperative chemotherapy and/or hormonal therapy or
pelvic irradiation. The present study was approved by the Research
Ethics Board of Qilu Hospital.
Immunohistochemistry
All specimens were routinely processed (10%
formalin-fixed for 24–48 h), paraffin-embedded and thin-sectioned
(4 μm). Antigen retrieval was achieved by heating the slides using
a microwave at 95°C for 15 min in citric acid buffer (2 mmol/l
citric acid, 9 mmol/l trisodium citrate dehydrate, pH 6.0). The
dilutions of antibodies used in the present study were as follows:
1:50 for PPARγ (ab19481), 1:50 for ERα (ab37438) and 1:100 for ERβ
(ab3576) (all from Abcam, Cambridge, MA, USA). A
Histostain®-Plus kit (SP-9000; ZSGB-BIO, Beijing, China)
was used to detect the immunostaining of PPARγ, ERα and ERβ.
3,3′-Diaminobenzidine tetrahydrochloride (DAB) was used to
visualize the reaction, followed by counterstaining with
hematoxylin. For each tissue section, at least three fields were
photographed under light microscopy. Two hundred cancer cells in
each field were selected, and the labeling index (LI) was
determined as the percentage of positive cells/200 cells. Cases
with an LI >10% were considered positive EC in the present
study.
Cell lines and culture conditions
The endometrial carcinoma cell lines ECC-1 and KLE
were purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA). ECC-1 cells were cultured in RPMI-1640 medium
with 10% fetal bovine serum (FBS) (Gibco, Carlsbad, CA, USA). KLE
cells were cultured in a mixture of Dulbecco’s modified Eagle’s
medium (DMEM) and Ham’s F-12 1:1 supplemented with 10% FBS in a 5%
CO2 environment at 37°C.
DNA and siRNA transient transfection
The PPARγ expression vector pGST-PPARγ plasmid was a
gift of Dr Bert Vogelstin (Johns Hopkins University, Baltimore, MD,
USA). Inhibition of PPARγ function was carried out using small
interfering RNA synthesized by GenePharma Company (Shanghai,
China). The siRNA sequences were as follows: nonsense negative
control (5′-CTG CTG ACT TTA CAG AAG AAA CA-3′) and PPARγ siRNA
(5′-AAG CCC ATT GAA GAC ATT CAA GA-3′). The cells were grown to 80%
confluency in 6-well plates for plasmid DNA transfection. In
addition, 4 μg of plasmid DNA was incubated with 8 μl of
Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA,
USA) in 0.5 ml of Opti-MEM™ I reduced serum-free medium (Gibco) for
30 min at room temperature, followed by an additional 1.5 ml of
serum-free medium. A total of 2 ml of this liposomal complex was
then added to cells. To transfect siRNA, the cells were grown to
40% confluency in 6-well plates. In each transfection reaction, 100
pmol of RNA was incubated with 5 μl of Lipofectamine 2000. Cells
were then incubated in a 5% CO2 environment at 37°C and
then switched to 2 ml of complete medium with 10% FBS after 5 h.
Twenty-four hours after transfection, the cells were plated for
proliferation, migration and invasion assays. The cells were
harvested for RNA and protein analyses at 48 h after
transfection.
Quantitative real-time polymerase chain
reaction (qRT-PCR)
Total RNA was extracted using TRIzol reagent
(Invitrogen) according to the manufacturer’s instructions as
previously described (25). RNA
concentrations were quantified spectrophotometrically (Thermo
Fisher Scientific, Waltham, MA, USA). Then, 1 μg of RNA was reverse
transcribed in a total volume of 20 μl using a PrimeScript RT
reagent kit according to the manufacturer’s protocol. qRT-PCR was
performed using SYBR Premix Ex Taq (both from Takara, Tokyo,
Japan) according to the manufacturer’s protocol. The LightCycler
system (Roche Diagnostics GmbH, Basle, Switzerland) was used to
quantify the mRNA expression levels. The expression of each target
gene was normalized to the expression of β-actin. Primers were
synthesized by Takara and are listed in Table I.
 | Table ISequence of primers for qRT-PCR. |
Table I
Sequence of primers for qRT-PCR.
| Primer | Sequence (5′-3′) |
|---|
| PPARγ | F:
ATTCCATTCACAAGAACAGATCCAG
R: TTTATCTCCACAGACACGACATTCA |
| ERα | F:
CGACATGCTGCTGGCTACATC
R: AGACTTCAGGGTGCTGGACAGA |
| ERβ | F:
AGAGTCCCTGGTGTGAAGCAAGA
R: TGCAGACAGCGCAGAAGTGA |
| β-actin | F:
CTAAGGCCAACCGTGAAAAG
R: AACACAGCCTGGATGGCTAC |
Western blotting
Tissues were pulverized using a mortar and pestle,
with lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1% Triton X-100,
0.1% sodium deoxycholate, 0.1% SDS and protease inhibitor mixture).
The cells were washed twice with ice-cold PBS and then lysed using
lysis buffer. After incubation on ice for 20 min, the lysates were
cleared by centrifugation for 15 min at 12,000 rpm at 4°C. Protein
concentrations were quantified using a BCA protein assay kit
(Beyotime Biotechnology, Shanghai, China). Western blotting was
performed as previously described (26). Briefly, 30 μg of total protein was
separated by SDS-PAGE for 2 h at 80 V and then transferred onto
PVDF membranes (Millipore, Billerica, MA, USA) for 1.5 h at 200 mA.
The membranes were blocked for 2 h at room temperature in 5%
non-fat dry milk. The primary antibody was incubated overnight at
4°C, and the secondary antibody was incubated for 1 h at room
temperature. Protein expression was detected using ECL (Millipore),
and the mean intensity of the bands was quantified using ImageJ
(version 1.45). β-actin was also evaluated as an internal control.
The dilutions of the primary antibodies used In the present study
were as follows: 1:500 for PPARγ (#2453; Cell Signaling Technology,
Danvers, MA, USA), 1:100 for ERα (ab37438), 1:1,000 for ERβ
(ab3576) (both from Abcam, Cambridge, MA, USA) and 1:3,000 for
β-actin (AP0060; Bioworld, Minneapolis, MN, USA). The dilution of
the secondary goat anti-rabbit IgG-HRP antibody (BS10350; Bioworld)
was 1:20,000.
In vitro migration and invasion
assays
For the Transwell migration assays, 1×105
cells were plated in the top chamber with a non-coated membrane
(24-well insert; 8-μm pore size; Corning Costar, Tewksbury, MA,
USA). For the invasion assays, 2×105 cells were plated
in the top chamber with a Matrigel-coated membrane (BD Biosciences,
San Jose, CA, USA). In both assays, the cells were plated in medium
without serum, and medium supplemented with 10% serum was used as a
chemoattractant in the lower chamber. Following incubation for 24
h, the cells that did not migrate or invade through the pores were
removed by a cotton swab. Cells on the underside of the membrane
were fixed with methanol, stained by 0.1% crystal violet, and
photographed at ×200 magnification. The cells numbers were counted
in five randomly selected fields.
Cell proliferation assay
Cell proliferation was determined using the Cell
Counting Kit-8 (Beyotime) according to the manufacturer’s
instructions. Briefly, 24 h after transfection, the cells were
plated for the proliferation assays, with 5×103
cells/well seeded in a 96-well plate and grown at 37°C for 24 h.
After 10 μl of WST-8 dye was added to each well, the cells were
incubated at 37°C for 2 h, and the absorbance was finally
determined at 450 nm using a microplate reader (Thermo Fisher
Scientific).
Statistical analysis
All results, including transfection, were repeated
using independent experiments in triplicate. The χ2 test
was used to analyze the distribution of cases considered positive
for the biological parameters. The correlation between the
expressions was analyzed by the Pearson correlation. Statistical
analysis between small groups of subjects was performed using the
non-parametric Mann-Whiney U test. Statistical significance was
assumed at P<0.05. All calculations were performed using SPSS
17.0 software.
Results
Expression of PPARγ, ERα and ERβ in
normal endometrium and endometrial carcinomas
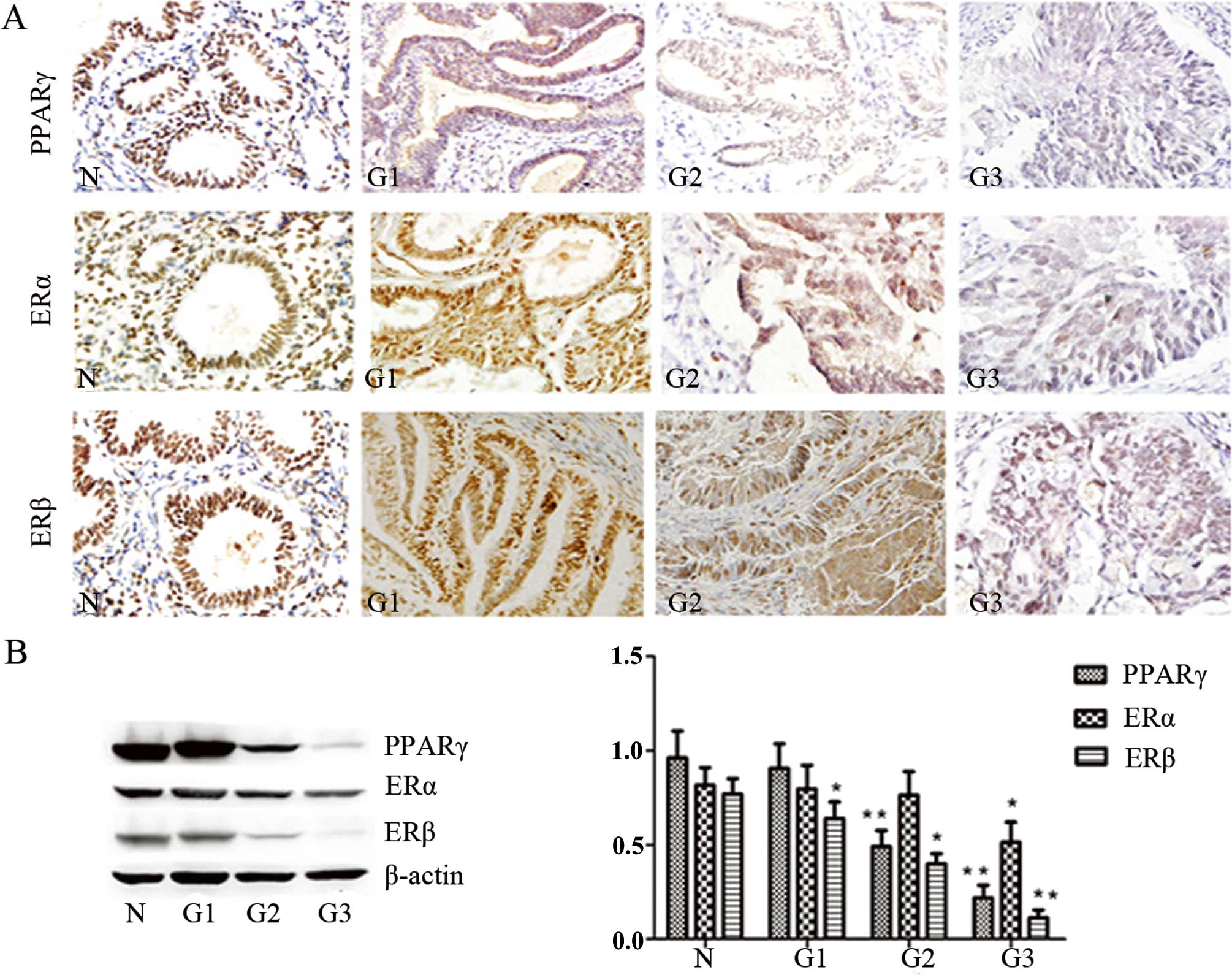
PPARγ, ERα and ERβ immunoreactivity were identified
in the cell nuclei. PPARγ immunoreactivity was significantly lower
in the EC tissues than that in the normal endometrium (P<0.05).
The statistical analysis indicated a significant correlation
between PPARγ expression and the clinicopathological variables
(P<0.05). The expression of ERα was gradually reduced in the
moderately and poorly differentiated endometrial carcinoma
(P<0.05). The expression of ERβ was only decreased in the poorly
differentiated endometrial carcinoma, and no significant
associations were detected between ERβ and the clinicopathological
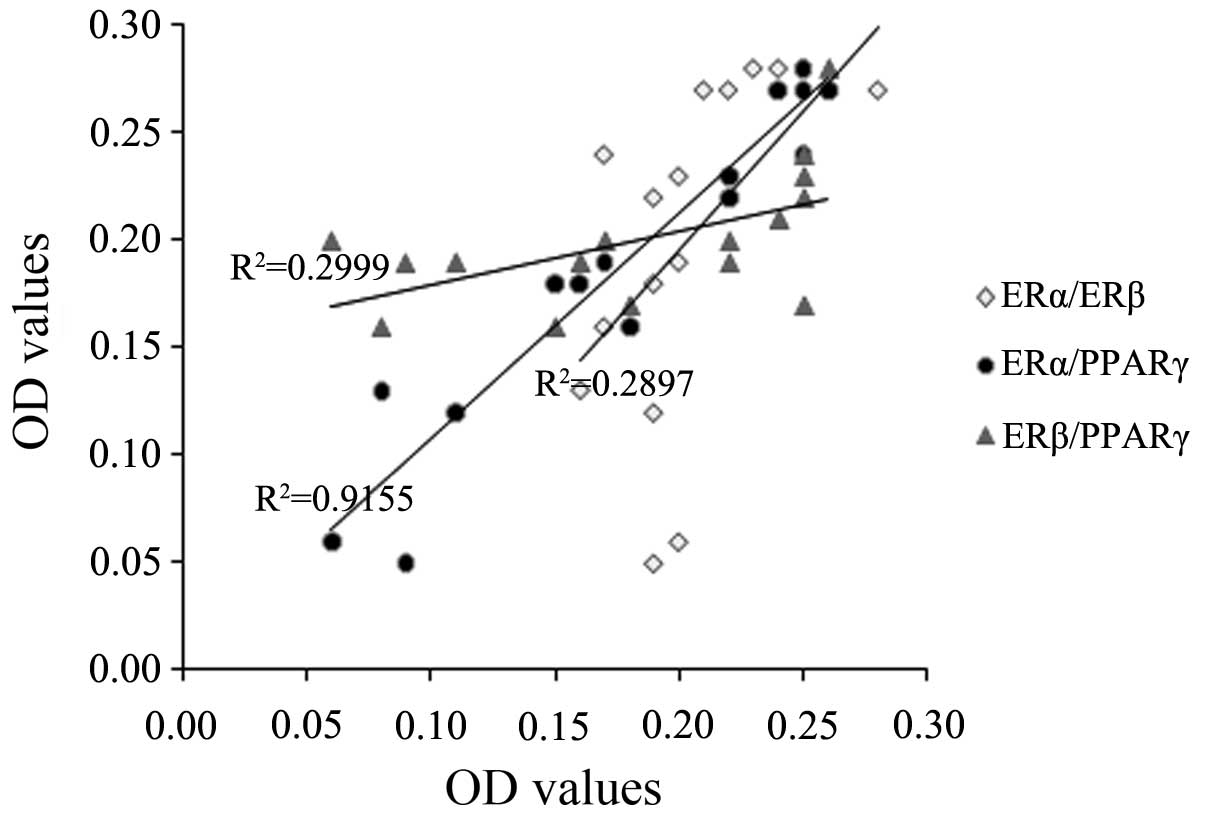
variables (Fig. 1, Table II). Furthermore, we found a
positive linear variation for PPARγ and ERα immune expression
(P<0.05) and no correlations between the expression of ERα and
ERβ, or ERβ and PPARγ (Fig. 2). The
decreased expression of PPARγ and ERα in the endometrial carcinomas
suggest that aberrant PPARγ and ERα expression may be an early
molecular event in cancer development.
| Table IICorrelation between PPARγ, ERα, ERβ
expression immunoreactivity and clinical parameters in EC. |
Table II
Correlation between PPARγ, ERα, ERβ
expression immunoreactivity and clinical parameters in EC.
| | PPARγ-positive | ERα-positive | ERβ-positive |
|---|
| |
|
|
|
|---|
| Parameter | No. of cases | No. (%) | P-value | No. (%) | P-value | No. (%) | P-value |
|---|
| All cases | 58 | 37 (62.07) | | 39 (67.24) | | 48 (82.76) | |
| BMI | | | | | | | |
| ≥25 | 23 | 11 (47.83) | | 12 (52.17) | | 18 (78.26) | |
| <25 | 35 | 26 (74.29) | 0.040 | 27 (77.14) | 0.047 | 30 (85.71) | 0.462 |
| Diabetes mellitus
(type II) | | | | | | | |
| Yes | 15 | 6 (40.00) | | 7 (46.67) | | 11 (73.33) | |
| No | 43 | 31 (72.09) | 0.026 | 32 (74.42) | 0.049 | 37 (86.05) | 0.262 |
| Grade | | | | | | | |
| G1 | 14 | 12 (85.71) | | 12 (85.71) | | 13 (92.86) | |
| G2–G3 | 31 | 17 (54.84) | 0.045 | 16 (51.61) | 0.029 | 23 (74.19) | 0.147 |
| FIGO stage | | | | | | | |
| I–II | 35 | 22 (62.86) | | 23 (65.71) | | 28 (80.00) | |
| III–IV | 10 | 2 (20.00) | 0.017 | 3 (30.00) | 0.044 | 7 (70.00) | 0.502 |
Stimulation of PPARγ downregulates
expression of the ERs in endometrial carcinoma cell lines
The PPARγ immunoreactivity results demonstrated a
strong association between PPARγ and ERα in EC (P<0.05),
suggesting a possible interaction of these two nuclear receptors in
human EC cells. Thus, we used two EC cell lines in the following
experiments in vitro. The expression of ERs in each cell
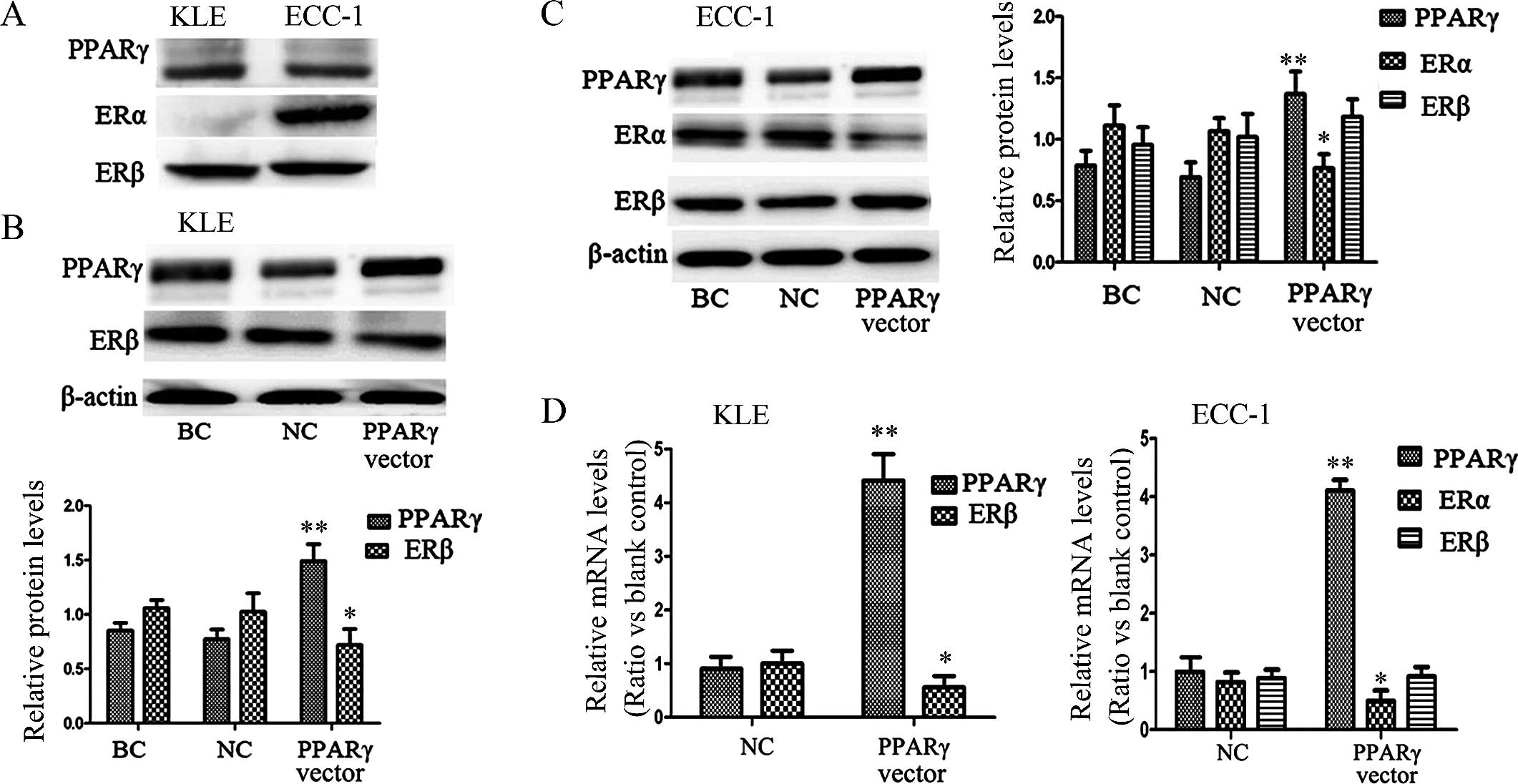
line was evaluated using western blotting. Only ERβ was expressed
in the KLE cells, whereas ERα and ERβ were expressed in the ECC-1
cells, and PPARγ was expressed in both cell lines (Fig. 3A).
When KLE cells were transfected with the expression
vector pGST-PPARγ for 48 h, the expression of PPARγ protein
increased 49.04%, and the expression of ERβ decreased 28.15%
significantly (P<0.01), when compared with the negative control
cells (Fig. 3B). In the transfected
ECC-1 cells under the same conditions, we found increased
expression of PPARγ protein of 36.96% and decreased expression of
ERα of 23.41% (P<0.01); however, we did not observe the
noticeable depression of ERβ protein in this cell line (Fig. 3C). According to previous studies,
PPARγ inhibits ER transcriptional activity through its interaction
with ER response elements (ERE) (27,28).
To provide further insight into the effects of PPARγ on the
activity of ERs, we investigated mRNA levels using qRT-PCR. As
expected, the downstream ER activity was confirmed as a decrease in
ER gene expression after transfection with the PPARγ expression
vector (Fig. 3D). As shown by
qRT-PCR and western blot analysis, stimulation of PPARγ did not
significantly inhibit transactivation of ERβ in the ECC-1 cells,
indicating that in the ECC-1 cell line, the PPARγ effect occurred
predominantly through ERα.
Inhibition of PPARγ upregulates ERα
expression in the endometrial carcinoma cell lne ECC-1
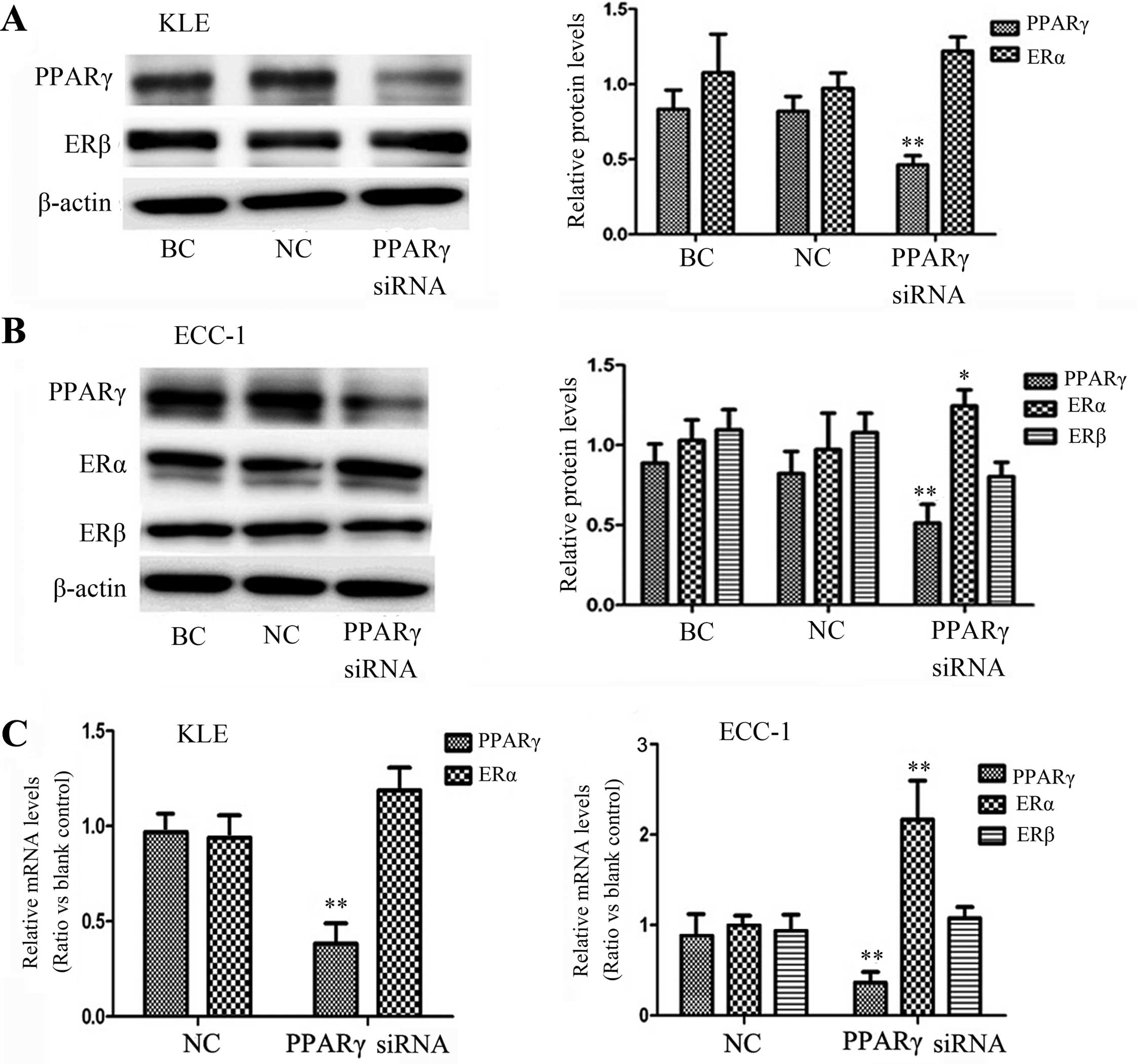
To obtain a better understanding of whether
suppression of PPARγ expression is associated with ER expression,
we transfected PPARγ siRNA and then analyzed both the protein and
mRNA levels of ERs. As shown in Fig. 4A
and B, after PPARγ siRNA transfection, the PPARγ protein levels
in the KLE and ECC-1 cells were decreased by 53.64 and 48.71%,
respectively. We also noted increased ERα expression in the ECC-1
cell line, yet not ERβ, in both cell lines. The mRNA levels were
confirmed by qRT-PCR analysis (Fig.
4C). These data demonstrate a negative crosstalk between the
PPARγ and ER signaling pathways (P<0.05). ECC-1 expressed both
the ERα and ERβ receptors, and KLE only expressed the ERβ receptor.
Although we did not evaluate whether or not PPARγ regulated ER
expression via ERα only, ERα may be more closely related with the
mechanism than ERβ.
Stimulation of PPARγ inhibits the
migratory and invasive abilities of endometrial carcinoma cell
lines
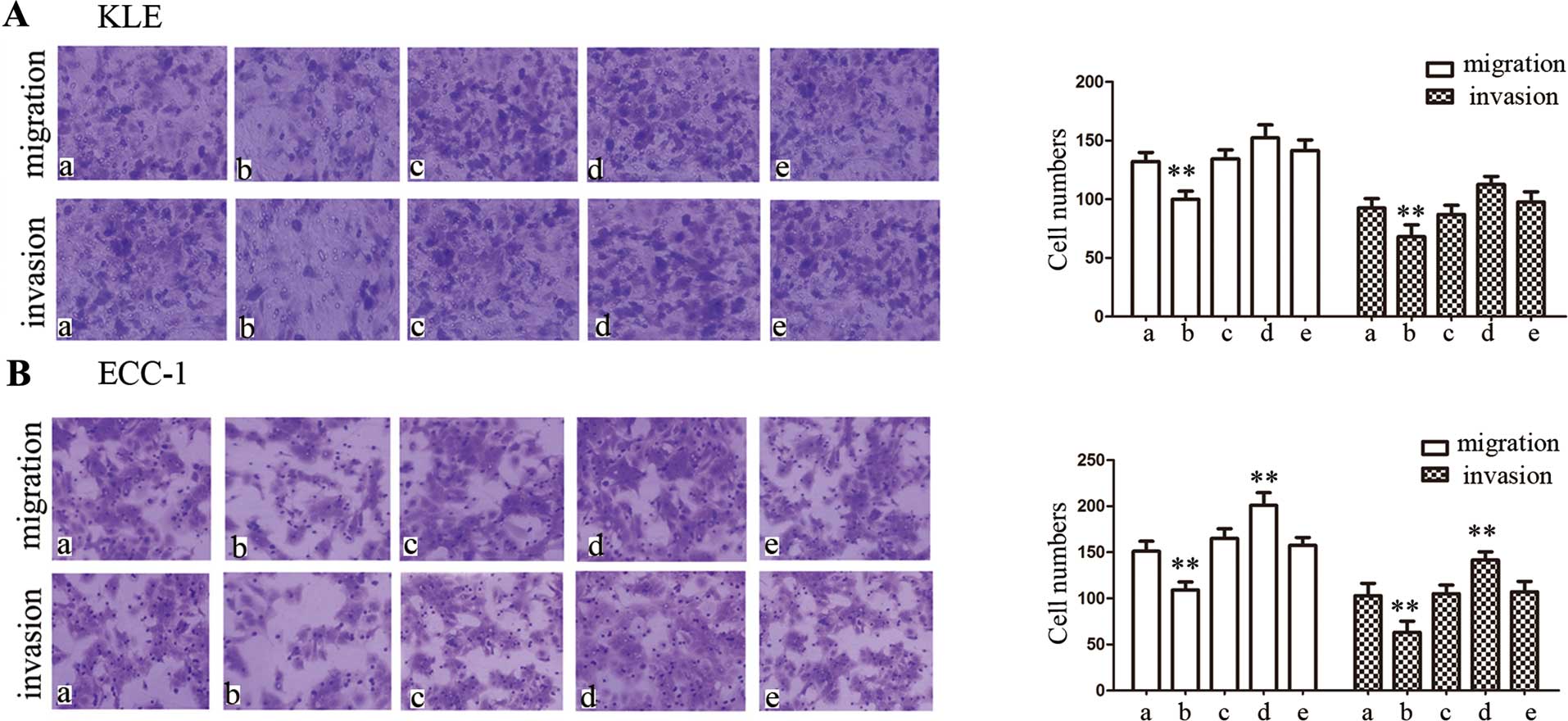
Having analyzed the interactions of protein and gene
expression between PPARγ and ERs, we next evaluated the biological
effects of upregulating or downregulating the PPARγ expression in
EC cells. In the present study, we investigated cell migration and
invasion using Transwell migration and invasion assays. After 24 h
of transfection with the PPARγ expression vector, the migratory or
invasive cell numbers were significantly decreased compared with
the controls in both cell lines (P<0.01) (Fig. 5). However, after 24 h of
transfection with PPARγ siRNA in KLE cells, there were no
differences in migration or invasive cell numbers compared with the
controls (Fig. 5A). On the
contrary, downregulation of PPARγ expression enhanced the migratory
and invasive abilities in the ECC-1 cells (P<0.01) (Fig 5B).
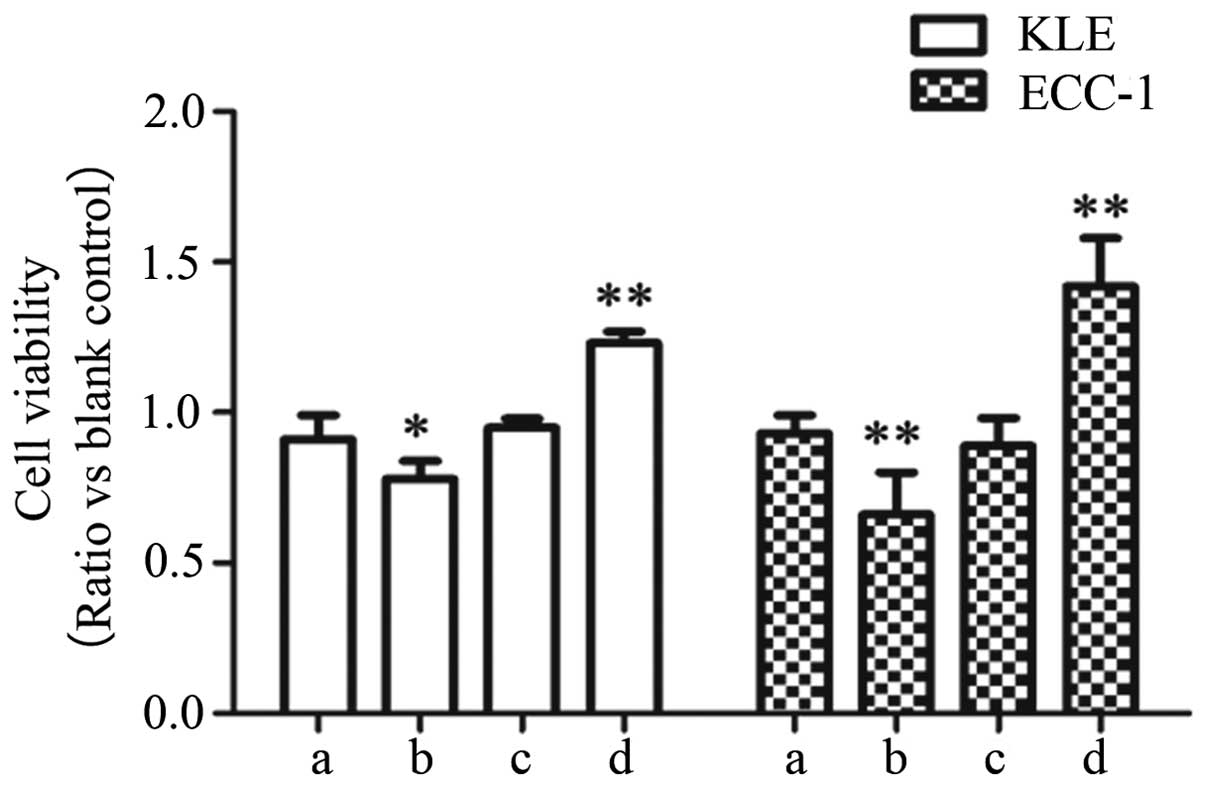
Enhanced PPARγ expression inhibits cell
proliferation in endometrial carcinoma cell lines
The stimulation of PPARγ has demonstrated growth
inhibitory effects on different tumor cell types, including colon
(5), lung (6) and breast cancer (7). Therefore, PPARγ has been considered as
a molecular target for cancer chemoprevention (29). Thus, we would expect that PPARγ
activation results in decreased cell proliferation in endometrial
carcinoma cells. Here, to investigate whether the aberrant
expression of PPARγ influences EC cell viability, we performed a
CCK-8 assay in each cell line. As expected, the number of KLE and
ECC-1 cells was significantly decreased after transfection with the
PPARγ expression vector (P<0.05) (Fig. 6). Moreover, our data revealed that
the proliferation of the two cell lines was significantly promoted
after transfection with PPARγ siRNA (P<0.05) (Fig. 6).
Discussion
Although PPARγ was first isolated in 1990 by
Issemann and Green (30), its
function has not yet been clearly elucidated. Our finding of a
decreased level of PPARγ in endometrial carcinoma is consistent
with previous observations (24,29)
and provides strong evidence supporting the biological significance
and clinical relevance in EC (Fig.
1, Table II). Our data also
demonstrated that the expression levels of ERα decreased with the
degree of differentiation and the stage of the tumor, with
significant correlation in this respect (P<0.05), whereas the
expression of ERβ was only decreased in poorly differentiated EC,
and no significant associations were detected between ERβ and the
clinicopathological variables. Other studies have disputed the
importance of ERα and ERβ expression and have failed to demonstrate
direct correlations with the tumor grade or the stage of the
differentiation. Saegusa and Okayasu examined ERα and ERβ
expression in normal and malignant endometrium and found a stepwise
decrease in ERβ with increasing grade, whereas ERβ levels remained
unchanged. They concluded that ERα expression and a shift in the
ratio of the two subtypes play an important role during endometrial
tumorigenesis (31). However, among
the few published reports that investigated ERβ expression in EC,
the decreased expression of ERβ was observed in EC compared with
normal endometrium, and there was a significant association with
tumor clinicopathological variables. Thus, they indicated that ERβ
alterations may be more important in EC (32). Therefore, it would be of value to
reexamine the expression levels of ERα and ERβ in human EC tissues
using a larger sample size.
In the present study, we also demonstrated that ERα
expression was regulated by PPARγ, and the evidence was obtained
with an ERα-positive EC cell line (ECC-1). To date, studies on the
crosstalk of ER and PPARγ in target tissues have mostly been
concerned with breast cancer, and little is known about their
involvement in EC. Our data confirmed that stimulating PPARγ
expression suppressed ERα expression both at the mRNA and protein
levels in ECC-1 cells, yet no noticeable suppression of ERβ was
detected. In addition, after inhibiting the expression of PPARγ in
ECC-1 cells, we found that ERα was significantly increased but not
ERβ. For PPARγ, the heterodimers formed with RXR are able to bind
to diverse hormone responsive elements such as ERE (27,28),
and negatively interfere with ER transcription. Although the exact
mechanism remains to be clarified, our investigation indicated that
the molecular mechanism occurred predominantly through ERα in
EC.
Recent studies have mainly focused on the physical
association between the crosstalk of ERα and PPARγ, and until now,
there have been few reports on the biological effects on
endometrial carcinoma cells after upregulating or downregulating
PPARγ expression. On the basis of our findings, PPARγ activation
inhibited the migratory and invasive abilities and the growth of EC
cells, and ECC-1 cells were more sensitive to this inhibition. In
our investigation into the effects of downregulating PPARγ
expression, we found enhanced migratory and invasive abilities in
the ECC-1 but not in the KLE cell line. These findings indicate
that in EC cells, the PPARγ effect occurred predominantly through
ERα. Previous studies have reported the ability to inhibit growth
and differentiation induced by PPARγ activation, and nuclear
expression of PPARγ has been reported to be associated with a lower
risk of recurrence of female breast cancer (33). Therefore, PPARγ has been considered
an important molecular target for cancer chemoprevention. Notably,
the effects of PPARγ activation on EC are largely unknown, and our
investigation may open a new direction for the development of novel
therapeutic methods targeting PPARγ in endometrial carcinoma
treatment, particularly ERα-positive carcinomas.
Taken together, our results indicate that PPARγ and
ERα play a role in the onset and progression of EC. It is likely
that the activation of PPARγ mediates ER transactivation mainly
through ERα. Therefore, PPARγ activation may potentiate
anti-estrogen therapy in ERα-positive endometrial carcinoma.
Further research needs to investigate whether targeting PPARγ has
potential as a clinical treatment for ERα-positive endometrial
tumors.
Acknowledgements
We thank Dr Bert Vogelstin (John Hopkins University,
USA) for providing the PPARγ expression vector pGST-PPARγ plasmid.
The present study was supported by the Natural Scientific
Foundation of Shandong Province (ZR2010HM102).
References
|
1
|
Boll D, Karim-Kos HE, Verhoeven RH, et al:
Increased incidence and improved survival in endometrioid
endometrial cancer diagnosed since 1989 in The Netherlands: a
population based study. Eur J Obstet Gynecol Reprod Biol.
166:209–214. 2013. View Article : Google Scholar
|
|
2
|
Gadducci A, Cosio S and Genazzani AR: Old
and new perspectives in the pharmacological treatment of advanced
or recurrent endometrial cancer: hormonal therapy, chemotherapy and
molecularly targeted therapies. Crit Rev Oncol Hematol. 58:242–256.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tavares V, Hirata MH and Hirata RD:
Peroxisome proliferator-activated receptor γ (PPAR γ): molecular
study in glucose homeostasis, lipid metabolism and therapeutic
approach. Arq Bras Endocrinol Metabol. 51:526–533. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Berstein LM, Kvatchevskaya JO, Poroshina
TE, Kovalenko IG, et al: Insulin resistance, its consequences for
the clinical course of the disease, and possibilities of correction
in endometrial cancer. J Cancer Res Clin Oncol. 130:687–693. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tsukahara T and Haniu H: Peroxisome
proliferator-activated receptor gamma overexpression suppresses
proliferation of human colon cancer cells. Biochem Biophys Res
Commun. 424:524–529. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Reka AK, Goswami MT, Krishnapuram R,
Standiford TJ and Keshamouni VG: Molecular cross-regulation between
PPAR-γ and other signaling pathways: implications for lung cancer
therapy. Lung Cancer. 72:154–159. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jiang Y, Huang Y, Cheng C, et al:
Combination of thiazolidinedione and hydralazine suppresses
proliferation and induces apoptosis by PPARγ up-expression in
MDA-MB-231 cells. Exp Mol Pathol. 91:768–774. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ito K, Utsunomiya H, Niikura H, Yaegashi N
and Sasano H: Inhibition of estrogen actions in human gynecological
malignancies: new aspects of endocrine therapy for endometrial
cancer and ovarian cancer. Mol Cell Endocrinol. 340:161–167. 2011.
View Article : Google Scholar
|
|
9
|
Shabani N, Kuhn C, Kunze S, et al:
Prognostic significance of oestrogen receptor alpha (ERα) and beta
(ERβ), progesterone receptor A (PR-A) and B (PR-B) in endometrial
carcinomas. Eur J Cancer. 43:2434–2444. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ito K, Utsunomiya H, Yaegashi N and Sasano
H: Biological roles of estrogen and progesterone in human
endometrial carcinoma - new developments in potential endocrine
therapy for endometrial cancer. Endocr J. 54:667–679. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pearce ST and Jordan VC: The biological
role of estrogen receptors α and β in cancer. Crit Rev Oncol
Hematol. 50:3–22. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kurebayashi J, Otsuki T, Kunisue H, Tanaka
K, Yamamoto S and Sonoo H: Expression levels of estrogen
receptor-α, estrogen receptor-β, coactivators, and corepressors in
breast cancer. Clin Cancer Res. 6:512–518. 2000.PubMed/NCBI
|
|
13
|
Mann S, Laucirica R, Carlson N, et al:
Estrogen receptor beta expression in invasive breast cancer. Hum
Pathol. 32:113–118. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin CY, Ström A, Li Kong S, et al:
Inhibitory effects of estrogen receptor beta on specific
hormone-responsive gene expression and association with disease
outcome in primary breast cancer. Breast Cancer Res. 9:R252007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jensen EV and Jordan VC: The estrogen
receptor: a model for molecular medicine. Clin Cancer Res.
9:1980–1989. 2003.PubMed/NCBI
|
|
16
|
Yuan H, Kopelovich L, Yin Y, Lu J and
Glazer RI: Drug-targeted inhibition of peroxisome
proliferator-activated receptor-gamma enhances the chemopreventive
effect of anti-estrogen therapy. Oncotarget. 3:345–356.
2012.PubMed/NCBI
|
|
17
|
Tateishi Y, Kawabe Y, Chiba T, et al:
Ligand-dependent switching of ubiquitin-proteasome pathways for
estrogen receptor. EMBO J. 23:4813–4823. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kato S: Estrogen receptor-mediated
cross-talk with growth factor signaling pathways. Breast Cancer.
8:3–9. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Z, Zhou D, Lai Y, et al: Estrogen
induces endometrial cancer cell proliferation and invasion by
regulating the fat mass and obesity-associated gene via PI3K/AKT
and MAPK signaling pathways. Cancer Lett. 319:89–97. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Glass CK, Holloway JM, Devary OV and
Rosenfeld MG: The thyroid hormone receptor binds with opposite
transcriptional effects to a common sequence motif in thyroid
hormone and estrogen response elements. Cell. 54:313–323. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang X and Kilgore MW: Signal cross-talk
between estrogen receptor alpha and beta and the peroxisome
proliferator-activated receptor gamma1 in MDA-MB-231 and MCF-7
breast cancer cells. Mol Cell Endocrinol. 194:123–133. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nickkho-Amiry M, McVey R and Holland C:
Peroxisome proliferator-activated receptors modulate proliferation
and angiogenesis in human endometrial carcinoma. Mol Cancer Res.
10:441–453. 2012. View Article : Google Scholar
|
|
23
|
Mansure JJ, Nassim R and Kassouf W:
Peroxisome proliferator-activated receptor γ in bladder cancer: a
promising therapeutic target. Cancer Biol Ther. 8:6–15. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ota K, Ito K, Suzuki T, et al: Peroxisome
proliferator-activated receptor γ and growth inhibition by its
ligands in uterine endometrial carcinoma. Clin Cancer Res.
12:4200–4208. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Brody F, Hill S, Celenski S, et al:
Expression of ectonucleotide pyrophosphate phosphodiesterase and
peroxisome proliferator activated receptor γ in morbidly obese
patients. Surg Endosc. 21:941–944. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Houston KD, Copland JA, Broaddus RR,
Gottardis MM, Fischer SM and Walker CL: Inhibition of proliferation
and estrogen receptor signaling by peroxisome
proliferator-activated receptor γ ligands in uterine leiomyoma.
Cancer Res. 63:1221–1227. 2003.PubMed/NCBI
|
|
27
|
Keller H, Givel F, Perroud M and Wahli W:
Signaling cross-talk between peroxisome proliferator-activated
receptor/retinoid X receptor and estrogen receptor through estrogen
response elements. Mol Endocrinol. 9:794–804. 1995.PubMed/NCBI
|
|
28
|
Kliewer SA, Umesono K, Noonan DJ, Heyman
RA and Evans RM: Convergence of 9-cis retinoic acid and peroxisome
proliferator signalling pathways through heterodimer formation of
their receptors. Nature. 358:771–774. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Knapp P, Chabowski A, Blachnio-Zabielska
A, Jarzabek K and Wołczyński S: Altered peroxisome-proliferator
activated receptors expression in human endometrial cancer. PPAR
Res. 2012:4715242012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Issemann I and Green S: Activation of a
member of the steroid hormone receptor superfamily by peroxisome
proliferators. Nature. 347:645–650. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Saegusa M and Okayasu I: Changes in
expression of estrogen receptors alpha and beta in relation to
progesterone receptor and pS2 status in normal and malignant
endometrium. Jpn J Cancer Res. 91:510–518. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chakravarty D, Srinivasan R, Ghosh S,
Rajwanshi A and Gopalan S: Estrogen receptor beta (ERbeta) in
endometrial simple hyperplasia and endometrioid carcinoma. Appl
Immunohistochem Mol Morphol. 16:535–542. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jiang Y, Zou L, Zhang C, et al: PPARγ and
Wnt/β-catenin pathway in human breast cancer: expression pattern,
molecular interaction and clinical/prognostic correlations. J
Cancer Res Clin Oncol. 135:1551–1559. 2009. View Article : Google Scholar : PubMed/NCBI
|