Introduction
Cervical cancer is the second most common cancer
among women worldwide and it is a leading cause of cancer-related
deaths (1). It is widely accepted
that cervical cancer is caused by HPV infections and expression of
oncogenes, such as c-MYC, Ha-RAS and ERB-2 (2,3).
Cervical cancer is generally curable when detected early, but
treatment of this cancer is poorly effective and is usually
accompanied by adverse effects. Gene therapy is a promising
therapeutic approach for chemoresistant cervical cancers (4–6).
Hence, therapeutic interventions targeting the key factors
contributing to the initiation and progression of cervical cancer
may be a more effective treatment strategy.
The protein tyrosine phosphatase receptor J (PTPRJ)
is a 220-kDa transmembrane protein that belongs to the family of
receptor-type protein tyrosine phosphatases which are involved in
multiple signaling pathways (7).
PTPRJ is expressed in many cancer cell lines, including breast,
colorectal, lung cancers and mammary and pancreatic tumors.
Previous studies demonstrated that PTPRJ is a candidate tumor
suppressor and its overexpression in different carcinoma cell lines
negatively regulates cell proliferation and transformation
(7–10). Mechanistically, the
anti-proliferative effect of PTPRJ may account for its inhibition
of growth factor signaling through the dephosphorylation of various
receptor tyrosine kinases, such as FLT3, PDGFR, VEGFR2, MET, and
ERK1/2 (11–15). However, the roles and function
mechanism of PTPRJ in human cervical carcinoma remain largely
unknown.
Here, we analyzed the expression of PTPRJ in human
cervical tumor and non-tumor tissues and observed a striking
downregulation of PTPRJ in cancer tissues. We further showed that
PTPRJ functioned as a negative regulator of cell proliferation and
migration in the human cervical cancer cell line C33A. In addition,
sustained inhibition of PTPRJ increased the cell resistance to
5-fluorouracil (5-FU)-induced apoptosis while high levels of PTPRJ
accelerated cell death. Mechanistically, PTPRJ decreased the
phosphorylation levels of Janus kinase 1 (JAK1) and signal
transducer and activator of signal transducer and activator of
transcription 3 (stat3) and modulated the expression of the
downstream factors of the JAK1/stat3 signaling pathway. The present
study provided a clinical guide to the treatment of human cervical
cancer.
Materials and methods
Tumor samples
For the analysis of mRNA and protein levels of
PTPRJ, 8-paired human cervical tumor and non-tumor samples were
obtained from patients with stage I–III adenocarcinoma and
immediately frozen in liquid nitrogen and stored at the Department
of Pathology, Gansu Provincial Hospital in Lanzhou, China.
Immunochemistry methods were used to characterize these
tissues.
Cells and reagents
Human cervical carcinoma cell line, C33A, was
purchased from the American Type Culture Collection (Rockville, MD,
USA) and maintained in RPMI-1640 (Invitrogen Corp., Carlsbad, CA,
USA) supplemented with 10% fetal bovine serum (Hyclone, South
Logan, UT, USA), 100 U/ml penicillin and 100 mg/ml streptomycin at
37°C in a humidified incubator with 5% CO2. 5-FU was
obtained from Sigma Chemical.
Plasmid construction
PTPRJ cDNA was PCR amplified from the whole genome
of HEK293T using primers for PTPRJ: 5′-ATGAAGCCGGCGGCGCGGGA-3′
(sense) and 5′-TTAGGCGATGTAACCATTGG-3′ (antisense). The cDNAs were
ligated into pSicoR after being digested by EcoRI and
XhoI to create pSicoR-PTPRJ. RNA interference vectors used
in the present study were constructed in pLKO.1 puro. Target
sequences are as follows: NTC (AATTCTCCGAACGTGTCACGT), shPTPRJ-1
(ACGAGTCGTCATCTAACTATA), shPTPRJ-2 (CCGATACAATGCCACCGTTTA).
Stable cell line establishment and
treatment
The indicated constructs were transfected into the
HEK293T cells in combination with the lentiviral packaging vectors
pRSV-Rev, pMD2.G, and pCMV-VSV-G using Lipofectamine 2000
(Invitrogen). After 60 h, the media containing the viruses were
collected using a 0.22-μm filter (Millipore, Bedford, MA, USA).
Viral supernatant from a 6-well plate was used to infect
~103 C33A cells for 48 h. After a 24-h infection, the
C33A cells were selected for 48 h in 2.5 μg/ml puromycin and two
colonies were chosen to expand and detect the protein levels. For
5-FU treatment, the indicated stable cell lines were plated in
6-well plates and cultured for 24 h. Then the culture media were
changed with fresh DMEM containing 20 μg/ml 5-FU or DMSO.
Western blotting
The cells were washed with PBS and lysed in ice-cold
lysis buffer consisting of 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1%
NP-40, 0.5% sodium deoxycholate, 0.1% SDS along with phosphatase
and protease inhibitors. After sonication on ice, protein lysates
were obtained by centrifugation at 12,000 × g at 4°C for 10 min.
The protein concentration was determined using the BCA assay. Fifty
micrograms of the total protein was mixed with 4X loading buffer
[360 mM TrisHCl (pH 6.8), 30% glycerol and 10% SDS]. The protein
samples were then heated at 95°C for 5 min and electroseparated
using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) and transferred onto a PVDF membrane (Immobilon-P;
Millipore). After being blocked with 0.5% skim milk, the membrane
was probed with a monoclonal antibody for PTPRJ (R&D, MAB1934),
or PARP (#9532), pro-caspase-3 (#9665), cleaved-caspase-3 (#9661),
phospho-STAT3 (Tyr705) (#9145), phospho-JAK1 (Tyr1022/1023)
(#3331), STAT3 (#9132), JAK1 (#1013) and GAPDH (#3683), which were
purchased from Cell Signaling Technology. The membrane was then
incubated with a horseradish peroxidase-conjugated secondary
antibody and developed with the SuperSignal chemiluminescence kit
(Immobilon Western Chemiluminescent HRP Substrate; EMD
Millipore).
Cell viability and proliferation
assays
C33A cells were cultured into 96-well plates at
~5,000 cells/well. After 24 h at 37°C, the cells were transfected
with the indicated plasmids and allowed another 48-h culture. Cell
viability was assessed using the Dojindo Cell Counting Kit-8
(CCK-8; Dojindo Molecular Technologies, Kumamoto, Japan).
Experiments were performed in octuplicate. For the colony formation
assay, the stable C33A cells were cultured in a 6-well tissue
culture plate at 500 cells/well. Cells were grown for 10–14 days.
After being fixed by 4% paraformaldehyde for 30 min, the colonies
were stained with 0.1% crystal violet for 15 min and washed. The
colonies were counted by Image J software.
Transwell migration assay
Stable C33A cells (200 μl) in RPMI-1640 medium
without FBS were loaded into the upper chambers of BD Falcon™ Cell
Culture Inserts (8-μm pore size; 1×105 cells/chamber).
The lower chambers were filled with 800 μl medium (RPMI-1640 plus
10% FBS). After a 24-h incubation at 37°C, the cells were fixed
with paraformaldehyde and stained with
4′,6-diamidino-2-phenylindole (DAPI). The stained cells on the
lower side were observed under a fluorescence microscope. Three
fields per chamber were photographed and the number of migrated
cells was counted by Image J software.
Pathway screening assay
Signaling pathway arrays were conducted by a
luciferase assay. Reporters HSF1-luc,
phos-phatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/AKT-luc and
STAT3-luc were purchased from Qiagen (Valencia, CA, USA). C33A
cells were cotransfected with reporter plasmids and pSicoR-PTPRJ or
shPTPRJ in 24-well plates. After 48 h, the cells were lysed in 1X
reporter lysis buffer (Promega, Madison, WI, USA) and the
luciferase activity was measured by a Dual-Luciferase®
Reporter assay system (Promega). Experiments were conducted in
triplicates and repeated at least three times.
Real-time PCR
Total RNA was isolated using a TRIzol RNA extraction
kit, and 1 μg of RNA was reverse transcribed into cDNA according to
the manufacturer’s instructions (RevertAid First strand cDNA
Synthesis kit; Thermo). Real-time PCR was performed to measure the
expression of cyclin D1, Bcl-2, Bax, VEGF and MMP9. The following
primers were used: GAPDH-F, 5′-TGCACCACCAACTGCTTAGC-3′ and GAPDH-R,
5′-GCATGGACTGTGGTCATGAG-3′; BAX-F, 5′-GAGGATGATTGCCGCCGTGGACA-3′and
BAX-R, 5′-GGTGGGGGAGGAGGCTTGAGG-3′; Bcl-2-F, 5′-ATG
TGTGTGGAGAGCGTCAACC-3′ and Bcl-2-R, 5′-TGAGCAG
AGTCTTCAGAGACAGCC-3′; MMP9-F, 5′-CCTGGAGAC CTGAGAACCAATC-3′ and
MMP9-R, 5′-CCACCCGAGT GTAACCATAGC-3′; and VEGF-F, 5′-TGCCCACTGAGGA
GTCCAAC-3′ and VEGF-R, 5′-TGGTTCCCGAAACGC TGA G-3′. All samples
were read in triplicate, and values were normalized to GAPDH
expression.
Cell cycle analysis
The stable C33A cells were harvested and washed
twice with PBS and fixed in 70% cold ethanol at 4°C overnight.
Before the analysis, the cells were washed twice with PBS, and then
resuspended with 400 μl PBS and 100 μg/ml RNaseA (Sigma) and 50
μg/ml propidium iodide (PI) (Sigma). After incubation for 30 min at
37°C, the cells were subjected to DNA content analysis using a FACS
Calibur (Becton Dickinson, San Jose, CA, USA). The results were
analyzed with Flowjo software.
Statistical analysis
SPSS 19 software was used for statistical analyses.
Data are presented as means ± SD. Statistical significance was
determined by the Student’s t-test in some experiments. Differences
with P-values of <0.05 were considered significant.
Results
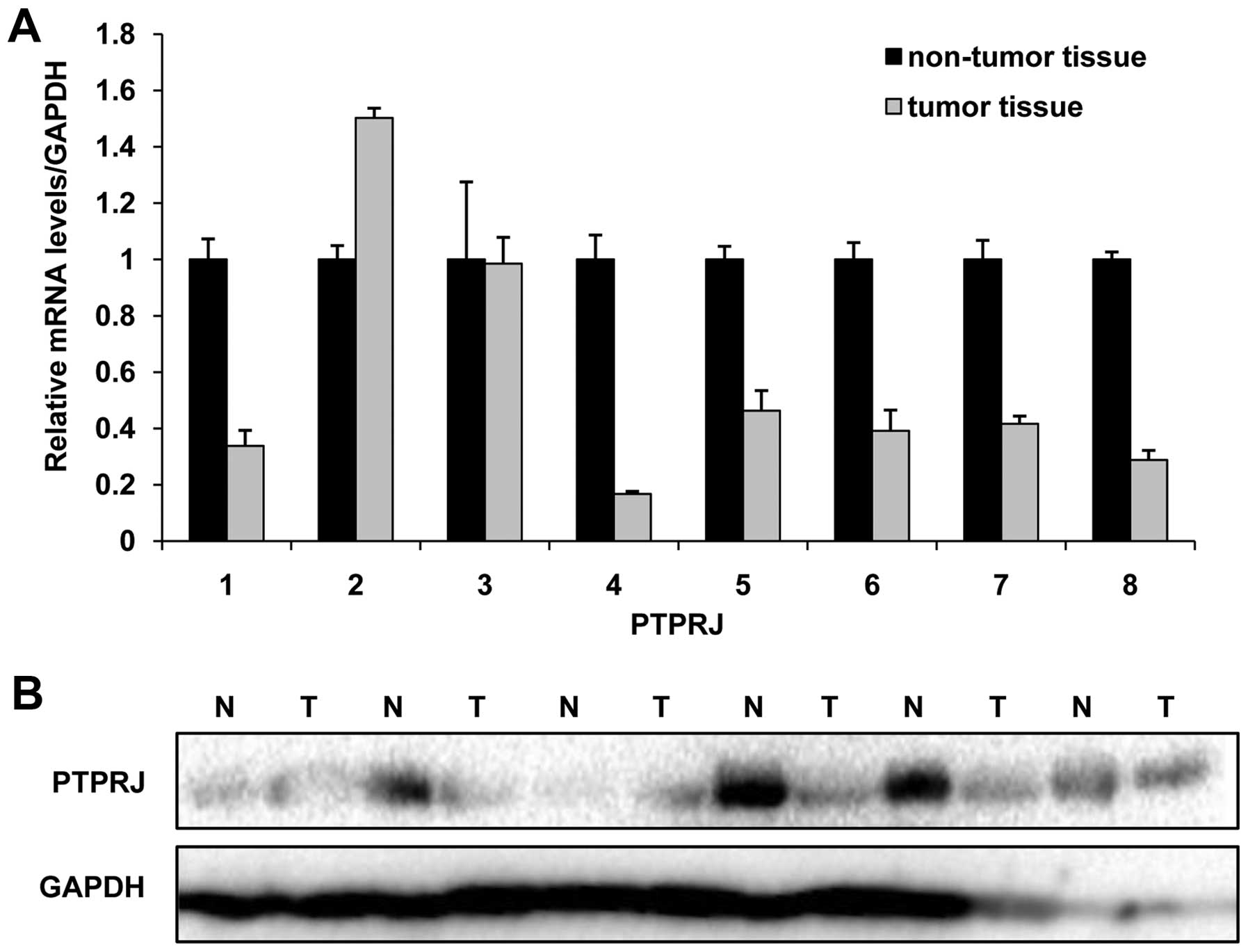
PTPRJ is downregulated in human cervical
tumor tissues
PTPRJ is widely regarded as a tumor suppressor and
its expression is decreased in various tumor types. In the present
study, we analyzed the mRNA and protein levels of PTPRJ in 8-paired
patient samples. We observed that both the mRNA and protein levels
of PTPRJ were decreased in the majority of the examined tumor
tissues (75%). There was a >50% decrease in 6-paired tumor and
non-tumor tissues (Fig. 1A and
B).
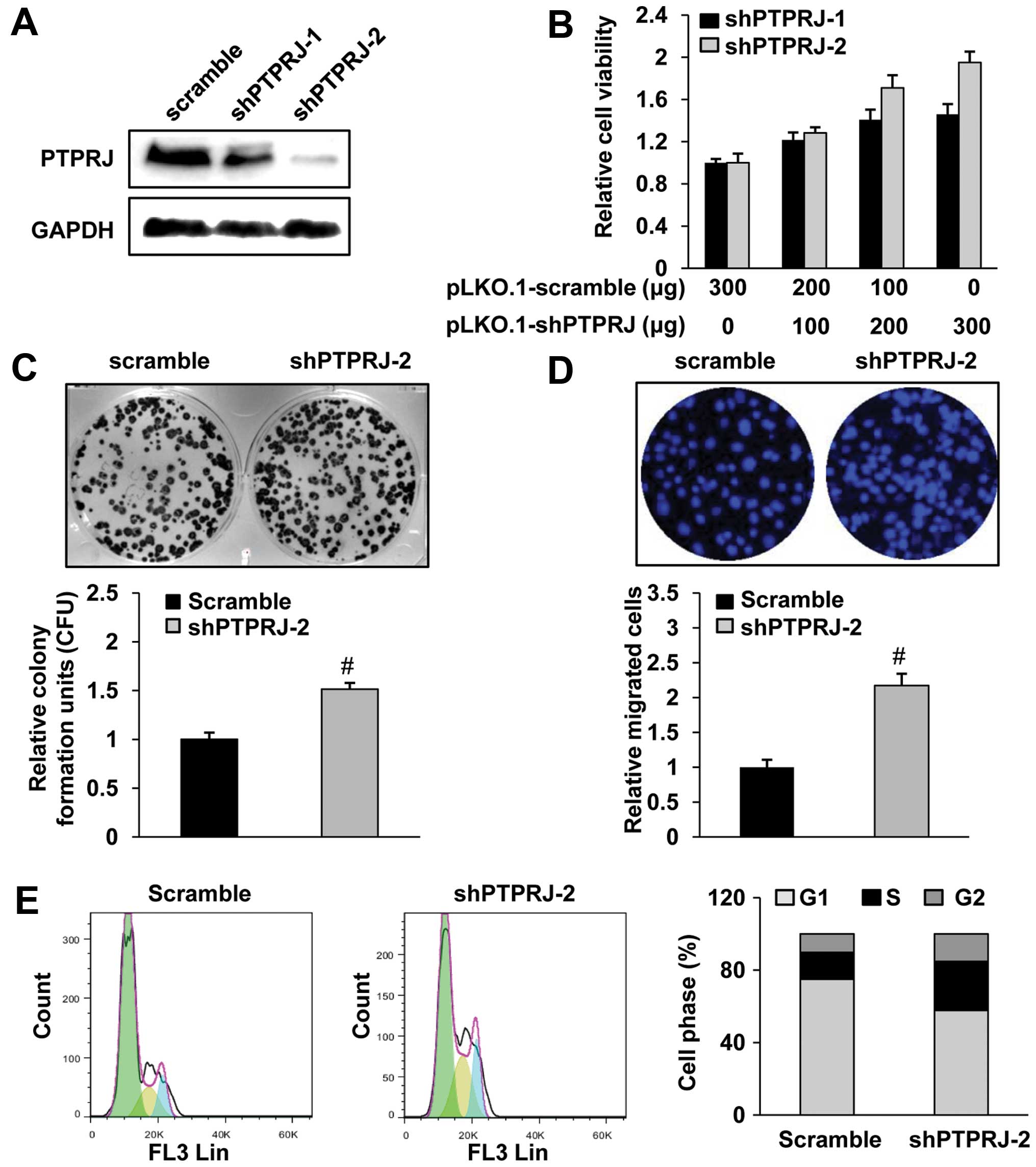
Suppression of PTPRJ promotes cell growth
and migration in the cervical cancer C33A cells
To investigate the role of PTPRJ in the C33A cell
line, we used construct codings for two different shRNAs against
PTPRJ or a control shRNA. After puromycin selection, we obtained
the stable cell lines expressing shPTPRJ. The PTPRJ shRNAs
effectively decreased the mRNA and protein levels of PTPRJ, as
determined by western blotting (Fig.
2A).
To assess whether the modulation of PTPRJ level
influences the tumorigenetic properties of the C33A cells, we
measured the cell viability, proliferation and migration
capabilities by using these stable cell lines or transfected C33A
cells with various doses of the indicated plasmids. By the CCK8
assay, we observed that the shPTPRJ-transfected cells showed an
increased cell viability in a dose-dependent manner (Fig. 2B). By a colony formation assay, we
observed that the shPTPRJ-infected C33A cells exhibited increased
proliferation (Fig. 2C). To further
investigate the role of PTPRJ in cell migration, we performed an
invasion assay by using a Transwell chamber under serum-starved
conditions. We found that the shPTPRJ infection significantly
enhanced the C33A cell invasive ability by ~2.2-fold relative to
the scramble infection (Fig. 2D).
Finally, the cell cycle distribution of the
shPTPRJ/scramble-infected C33A cells was measured by use of
propidium iodide staining. As shown in Fig. 2E, the percentage of shPTPRJ-infected
cells in the G1 phase was considerably decreased with significantly
increased percentages of cells in the S and G2/M phases, in
comparison to the scramble-infected cells.
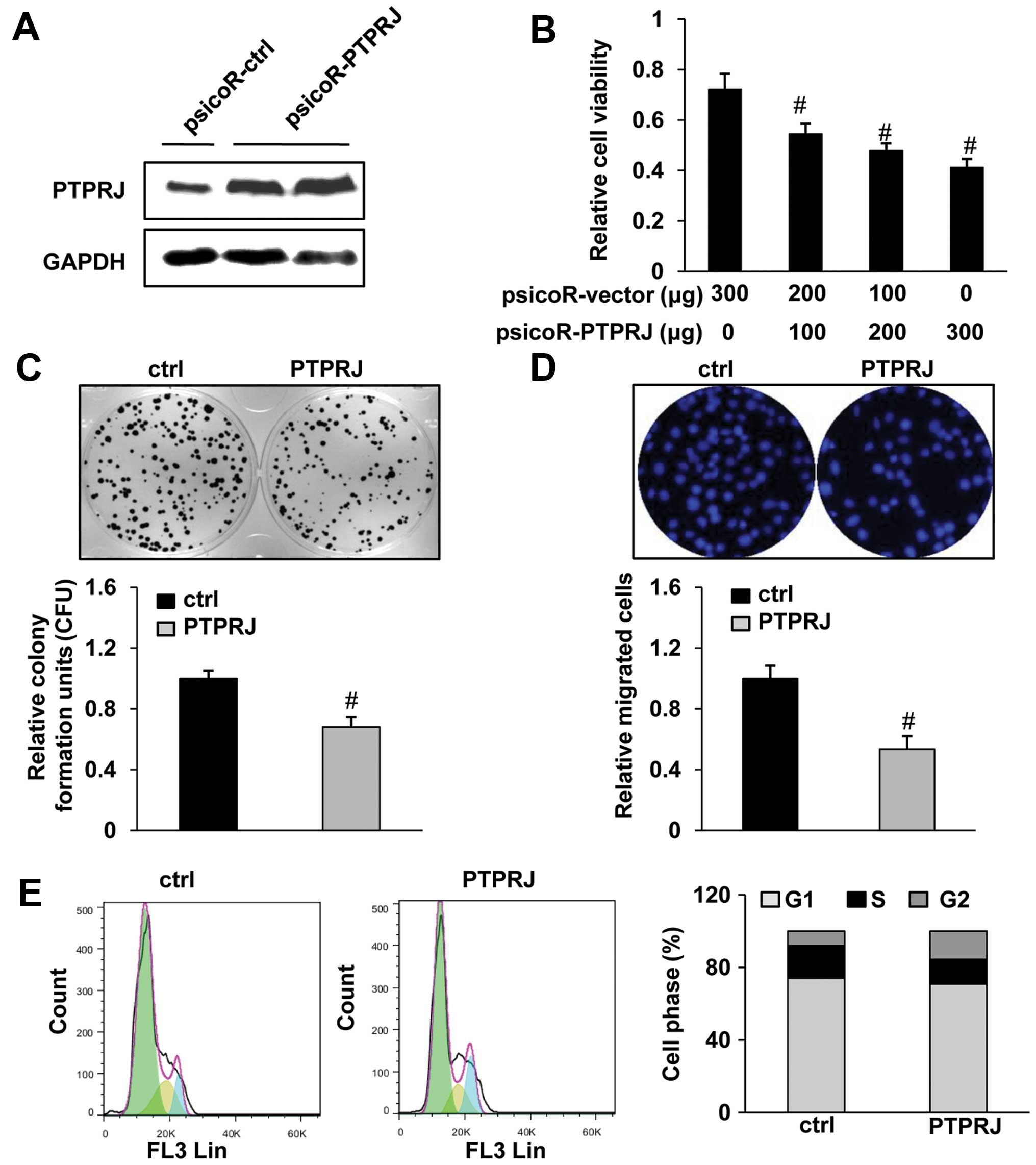
Re-expression of PTPRJ suppresses cell
viability, proliferation and migration of the C33A cells
To identify the effects of PTPRJ re-expression on
the cell proliferation of C33A cells, we generated a stable cell
line with high endogenous PTPRJ levels (Fig. 3A). C33A cells with an increased
PTPRJ expression had a growth disadvantage, as assessed by the
CCK-8 assay (Fig. 3B). Consistent
with the previous studies in other cervical cancer cells such as
HeLa (16), cell proliferation in
the PTPRJ-overexpressing C33A cells was obviously reduced as
compared to the corresponding control cells, confirming the
antiproliferative activity of PTPRJ in the cervical cancer cell
line C33A (Fig. 3C). We further
used the PTPRJ-expressing C33A cells to perform a cell migration
assay. Our results revealed that PTPRJ upregulation effectively
reduced cell migration toward the serum starvation (Fig. 3D). By cell cycle analysis, we
observed that the PTPRJ-overexpressing cells had a clearly
decreased S-phase proportion and increased G2/M arrest (Fig. 3E).
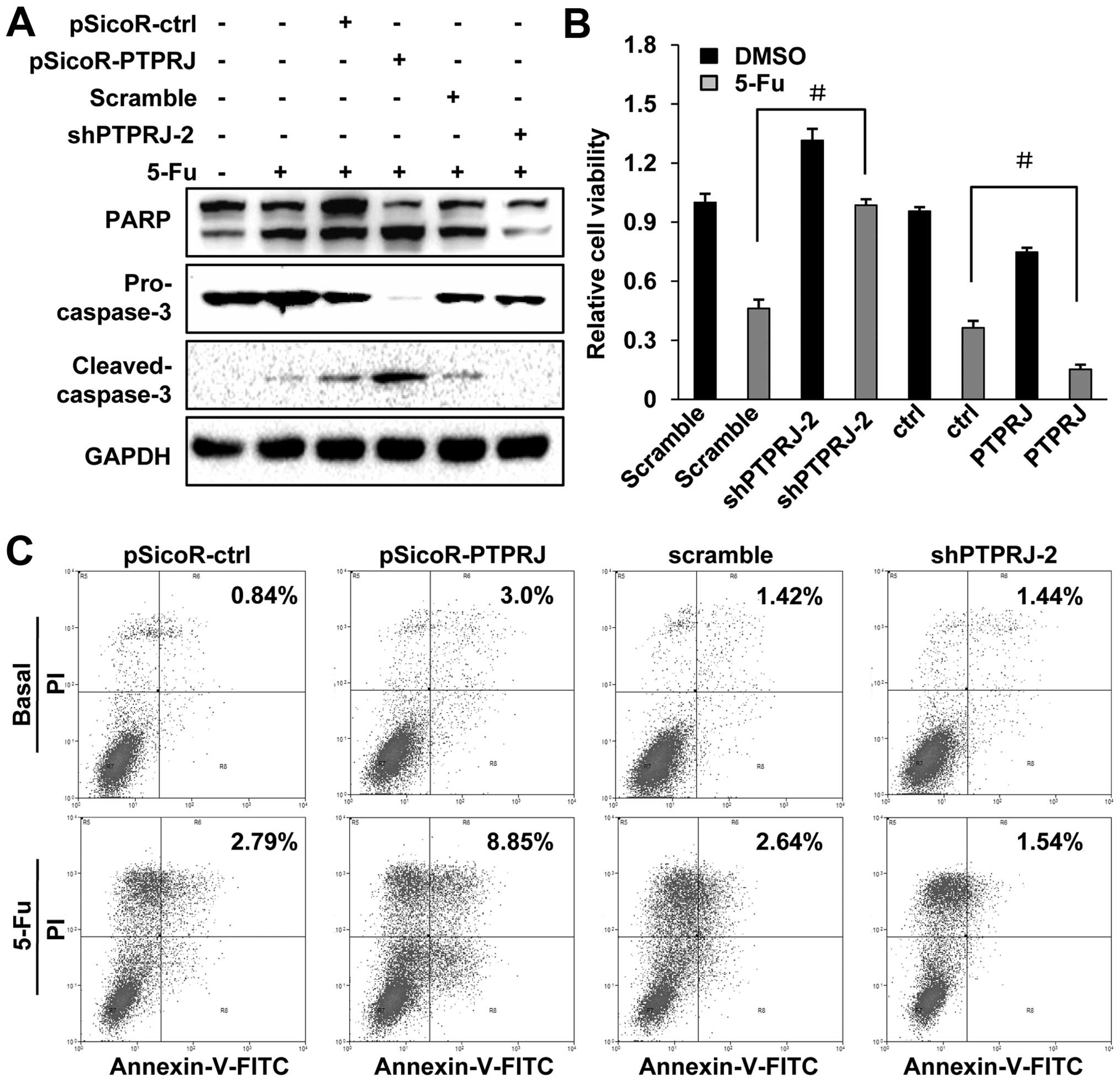
Downregulation of PTPRJ enhances
5-FU-induced cell apoptosis in the C33A cells
5-FU is an important and widely used anticancer
drug, including cervical cancer. However, tumor cell resistance to
5-FU-induced cell death often occurs during clinical treatment
(17). The PTPRJ agonist has been
shown to induce cell apoptosis (18). To investigate the role of PTPRJ in
5-FU-induced cell apoptosis, we conducted western blotting to
detect the expression of caspase-3 and PARP, the effective markers
for cell apoptosis. Our results revealed that cleaved-caspase-3 and
cleaved-PARP were upregulated after treating the wild-type C33A
cells with 50 μg/ml 5-FU, while the levels of pro-caspase-3 and
full-length PARP were decreased. In the stable C33A cell line that
overexpressed PTPRJ, the levels of pro-caspase-3 and full-length
PARP were significantly lower, and the levels of cleaved-caspase-3
and cleaved-PARP were higher than the levels in the control group,
after treatment with an equal dose of 5-FU (50 μg/ml). In contrast,
pro-caspase-3 and full-length PARP were increased, while
cleaved-caspase-3 and cleaved-PARP were decreased in the stable
cell line expressing shPTPRJ (Fig.
4A).
Next, using the CCK-8 assay, we measured the cell
viability of the wild-type C33A cells and PTPRJ/shPTPRJ stable cell
lines after treatment with 5-FU. As expected, when given the 5-FU
treatment, the viability of the stable cell line expressing shPTPRJ
was obviously higher than the wild-type cells, while cells
overexpressing PTPRJ showed a decreased cell viability, when
compared with the wild-type cells (Fig.
4B).
In addition, the stable transfectants were treated
with DMSO or 5-FU and subjected to FACS assays. At the basal state,
the apoptotic rates of the cells transfected with the indicated
genes remained unchanged. When the cells were exposed to 5-FU,
ectopic overexpression of PTPRJ obviously increased the percentage
of apoptotic cells, while inhibition of PTPRJ by shPTPRJ-2
attenuated the 5-FU-induced cell apoptosis, compared with the cells
transfected with pSicoR-control or scramble, respectively (Fig. 4C). All things considered, the
inhibition of PTPRJ decreased the sensitivity of C33A to
5-FU-induced cell apoptosis.
The activities of pJAK1 and pSTAT3 are
modulated by PTPRJ in the C33A cells
PTPRJ is a receptor-like protein-tyrosine
phosphatase that directly regulates the dephosphorylation of
several receptor tyrosine kinases, including FLT3, ERK, PDGFR.
Omerovic et al (19)
reported that PTPRJ negatively regulated serine/threonine protein
kinase B (AKT) phosphorylation in Ras-mutated cancer cells. In
accordance with previous studies, during the pathway screening
assay, we observed that the luciferase activity of the PI3K/AKT
pathway was decreased in the C33A cells transfected with PTPRJ,
while inhibition of PTPRJ by shRNA led to an apparent increase in
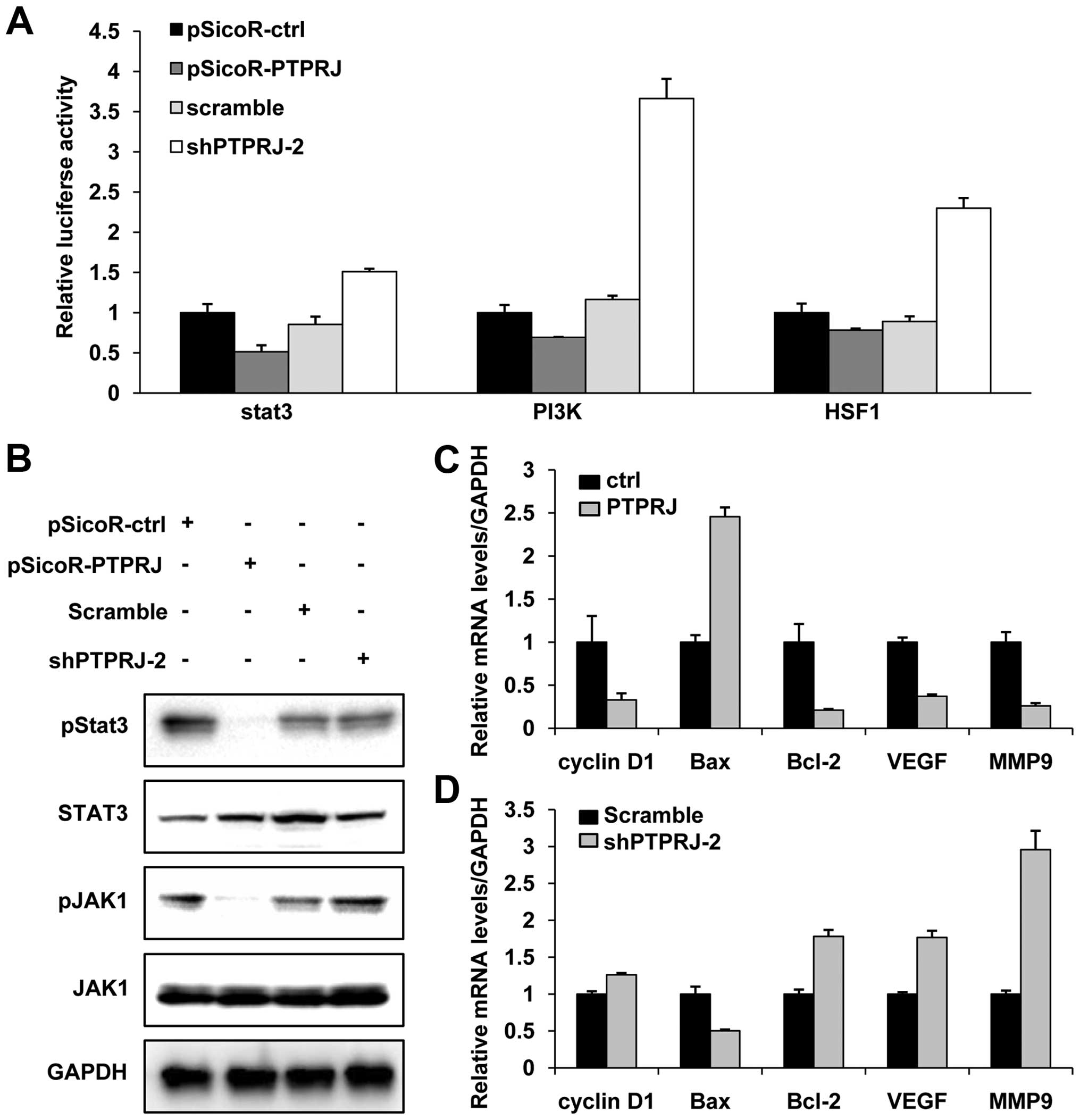
the activity of the PI3K/AKT pathway (Fig. 5A). Interestingly, we observed a
similar change of heat shock transcription factor 1 (HSF1) and
STAT3 pathway (Fig. 5A). STAT3 is
crucial for cell proliferation, differentiation and apoptosis
(20–22). STAT3 is regulated by JAK1 and other
stimuli or proteins. In cervical cancer, STAT3 was reported to be
correlated with clinical stage and positive-pSTAT3 patients have a
favorable prognosis (23,24). Given the dephosphorylation activity
of PTPRJ, we analyzed the phosphorylated levels of JAK1 and STAT3.
The western blotting showed that the levels of pJAK1 and pSTAT3
were decreased while the total protein levels of JAK1 and STAT3
were constant when the cells were transfected with PTPRJ. On the
contrary, shPTPRJ transfection caused the upregulation of the
levels of pJAK1 and pSTAT3 (Fig.
5B).
To confirm the effect of PTPRJ on the JAK1/STAT3
pathway, the mRNA levels of cyclin D1, Bax, Bcl-2, VEGF and MMP9,
downstream effectors of STAT3, were measured by real-time PCR. The
levels of cyclin D1, Bcl-2, VEGF and MMP9 were decreased, and the
expression of Bax was increased in the cells transfected with PTPRJ
(Fig. 5C). We obtained the opposite
results in the cells transfected with shPTPRJ (Fig. 5D). Collectively, these results
revealed a negative regulation of PTPRJ on the JAK/STAT3 signaling
pathway.
Discussion
Cervical cancer is a leading cause of cancer-related
deaths. PTPRJ is known to negatively regulate cell growth in many
malignant tumors by phosphorylation of different substrates, but
the understanding of its signaling transduction in cervical cancer
is limited. The present study reports for the first time that PTPRJ
plays an important role in proliferation and migration of the
cervical cancer cell line C33A. PTPRJ regulates cell cycle
progression and cell apoptosis induced by 5-FU and these processes
are mediated via suppression of the phosphorylated levels of AKT
and ERK. Our results suggest that PTPRJ may be a promising
therapeutic target for cervical cancer treatment.
The tumor-suppressor effect of PTPRJ is well
documented in breast tumors (25),
liver cancer (14), acute myeloid
leukemia (11) and colorectal
cancer (26). Recent studies showed
that the PTPRJ agonist effectively inhibits cell proliferation and
triggers apoptosis of breast cancer cells (16). Accordingly, the present study showed
that downregulation of PTPRJ is correlated with promotion of cell
proliferation, migration and the G1-S phase transition. In
contrast, the ectopic overexpression of PTPRJ effectively inhibited
cancer cell growth and induced cell cycle arrest. The present study
established the tumor-suppressor role of PTPRJ in C33A cells.
PTPRJ and other members of the PTP family have been
reported to be involved in the cell susceptibility to a variety of
human-related cancers (27). 5-FU
is the most extensively studied agent in cancer treatment. However,
acquired drug resistance and severe side effects limit the
effectiveness of chemotherapy (17). Previous research showed that the
anti-cancer efficacy of 5-FU can be synergistically promoted when
used in combination with gene therapy, which may be tumor
suppressor-based or RNAi-based (28–31).
In the present study, knockdown of PTPRJ increased the C33A cell
resistance to 5-FU-induced apoptosis, whereas ectopic expression of
PTPRJ increased the cell apoptosis in the C33A cells.
Sufficient evidence has emerged that AKT is a key
regulator of cell survival and it contributes to cell
susceptibility to chemotherapy in different types of tumors
(32,33). The activation of AKT could be
regulated by PTPRJ. Consistent with the previous study, we observed
that PTPRJ significantly reduced the activity of the PI3K/AKT
pathway. A novel finding of the present study was the regulatory
effect of PTPRJ on the JAK1/STAT3 axis. STAT3 is a key
transcription factor involved in numerous cell signal transduction
networks and numerous studies have implicated the potential of
STAT3 as a therapeutic target for the treatment of different types
of cancer (34,35). We found that PTPRJ modulated the
phosphorylation levels of JAK1 and STAT3. The mRNA levels of the
downstream effectors of the STAT3 pathway were altered when the
cells were transfected with PTPRJ or shPTPRJ. Notably, the
expression of Bcl-2, an important anti-apoptotic factor, was
downregulated and the pro-apoptotic factor, Bax, was upregulated
when PTPRJ was overexpressed in C33A cells. This may explain why
the ectopic overexpression of PTPRJ accelerated 5-FU-induced cell
apoptosis. Collectively, these results suggest that STAT3 is a
substrate of PTPRJ and further study is needed to reveal whether
this effect is due to the direct interaction between PTPRJ and
STAT3.
All things considered, the present study
demonstrated that blockage of PTPRJ significantly promoted tumor
biological features and upregulation of PTPRJ led to inhibition of
the growth of the C33A cells. Furthermore, our findings provide
novel evidence that PTPRJ is a regulator of cell apoptosis induced
by 5-FU. Sustained PTPRJ overexpression modulated the
phosphorylated levels of JAK1/STAT3, and the downstream factors
such as Bcl-2, Bax, cyclin D1, which were critical mediators of
cell processes like cell apoptosis and proliferation. These data
suggest that PTPRJ may serve as a potential target for cervical
cancer therapy.
Acknowledgements
We thank Professor Jun Zhu and Dr Zhong Tian Bai for
their assistance with the construction of the lentiviral vectors
and the establishment of the stable cell lines and Yong Xiu Yang
for his staff support.
Abbreviations:
|
PTPRJ
|
protein tyrosine phosphatase receptor
J
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
JAK1
|
Janus kinase 1
|
|
Bax
|
B cell lymphoma 2-associated X
protein
|
|
Bcl-2
|
B cell lymphoma 2
|
|
PI3K
|
phosphatidylinositol-4,5-bisphosphate
3-kinase
|
|
AKT
|
serine/threonine protein kinase B
|
|
HSF1
|
heat shock transcription factor 1
|
|
5-FU
|
5-fluorouracil
|
|
VEGF
|
vascular endothelial growth factor
|
|
MMP9
|
matrix metalloproteinase 9
|
References
|
1
|
Parkin DM and Bray F: Chapter 2: The
burden of HPV-related cancers. Vaccine. 24(Suppl 3): S3/11–25.
2006. View Article : Google Scholar
|
|
2
|
Ronco G, Dillner J, Elfström KM, et al:
Efficacy of HPV-based screening for prevention of invasive cervical
cancer: follow-up of four European randomised controlled trials.
Lancet. 383:524–532. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pinion SB, Kennedy JH, Miller RW and
MacLean AB: Oncogene expression in cervical intraepithelial
neoplasia and invasive cancer of cervix. Lancet. 337:819–820. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jeon YH, Lee HW, Lee YL, et al: Combined
E7-dendritic cell-based immunotherapy and human sodium/iodide
symporter radioiodine gene therapy with monitoring of antitumor
effects by bioluminescent imaging in a mouse model of uterine
cervical cancer. Cancer Biother Radiopharm. 26:671–679. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Koch M and Wiese M: Gene expression
signatures of angiocidin and darapladib treatment connect to
therapy options in cervical cancer. J Cancer Res Clin Oncol.
139:259–267. 2013. View Article : Google Scholar
|
|
6
|
Zheng Y, Chen H, Zeng X, et al: Surface
modification of TPGS-b-(PCL-ran-PGA) nanoparticles with
polyethyleneimine as a co-delivery system of TRAIL and endostatin
for cervical cancer gene therapy. Nanoscale Res Lett. 8:1612013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Keane MM, Lowrey GA, Ettenberg SA, Dayton
MA and Lipkowitz S: The protein tyrosine phosphatase DEP-1 is
induced during differentiation and inhibits growth of breast cancer
cells. Cancer Res. 56:4236–4243. 1996.PubMed/NCBI
|
|
8
|
Iuliano R, Trapasso F, Le Pera I, et al:
An adenovirus carrying the rat protein tyrosine phosphatase eta
suppresses the growth of human thyroid carcinoma cell lines in
vitro and in vivo. Cancer Res. 63:882–886. 2003.PubMed/NCBI
|
|
9
|
Massa A, Barbieri F, Aiello C, et al: The
expression of the phosphotyrosine phosphatase DEP-1/PTPeta dictates
the responsivity of glioma cells to somatostatin inhibition of cell
proliferation. J Biol Chem. 279:29004–29012. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Trapasso F, Iuliano R, Boccia A, et al:
Rat protein tyrosine phosphatase eta suppresses the neoplastic
phenotype of retrovirally transformed thyroid cells through the
stabilization of p27(Kip1). Mol Cell Biol. 20:9236–9246. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Godfrey R, Arora D, Bauer R, et al: Cell
transformation by FLT3 ITD in acute myeloid leukemia involves
oxidative inactivation of the tumor suppressor protein-tyrosine
phosphatase DEP-1/ PTPRJ. Blood. 119:4499–4511. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kovalenko M, Denner K, Sandstrom J, et al:
Site-selective dephosphorylation of the platelet-derived growth
factor beta-receptor by the receptor-like protein-tyrosine
phosphatase DEP-1. J Biol Chem. 275:16219–16226. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lampugnani MG, Orsenigo F, Gagliani MC,
Tacchetti C and Dejana E: Vascular endothelial cadherin controls
VEGFR-2 internalization and signaling from intracellular
compartments. J Cell Biol. 174:593–604. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Palka HL, Park M and Tonks NK: Hepatocyte
growth factor receptor tyrosine kinase met is a substrate of the
receptor protein-tyrosine phosphatase DEP-1. J Biol Chem.
278:5728–5735. 2003. View Article : Google Scholar
|
|
15
|
Sacco F, Tinti M, Palma A, et al: Tumor
suppressor density-enhanced phosphatase-1 (DEP-1) inhibits the RAS
pathway by direct dephosphorylation of ERK1/2 kinases. J Biol Chem.
284:22048–22058. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ortuso F, Paduano F, Carotenuto A, et al:
Discovery of PTPRJ agonist peptides that effectively inhibit in
vitro cancer cell proliferation and tube formation. ACS Chem Biol.
8:1497–1506. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Longley DB, Harkin DP and Johnston PG:
5-fluorouracil: mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Paduano F, Ortuso F, Campiglia P, et al:
Isolation and functional characterization of peptide agonists of
PTPRJ, a tyrosine phosphatase receptor endowed with tumor
suppressor activity. ACS Chem Biol. 7:1666–1676. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Omerovic J, Clague MJ and Prior IA:
Phosphatome profiling reveals PTPN2, PTPRJ and PTEN as potent
negative regulators of PKB/Akt activation in Ras-mutated cancer
cells. Biochem J. 426:65–72. 2010. View Article : Google Scholar
|
|
20
|
Aggarwal BB, Kunnumakkara AB, Harikumar
KB, et al: Signal transducer and activator of transcription-3,
inflammation, and cancer: how intimate is the relationship? Ann NY
Acad Sci. 1171:59–76. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aggarwal BB, Sethi G, Ahn KS, et al:
Targeting signal-transducer-and-activator-of-transcription-3 for
prevention and therapy of cancer: modern target but ancient
solution. Ann NY Acad Sci. 1091:151–169. 2006. View Article : Google Scholar
|
|
22
|
Ihle JN: Cytokine receptor signalling.
Nature. 377:591–594. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sobti RC, Singh N, Hussain S, Suri V,
Bharti AC and Das BC: Overexpression of STAT3 in HPV-mediated
cervical cancer in a north Indian population. Mol Cell Biochem.
330:193–199. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Choi CH, Song SY, Kang H, et al:
Prognostic significance of p-STAT3 in patients with bulky cervical
carcinoma undergoing neoadjuvant chemotherapy. J Obstet Gynaecol
Res. 36:304–310. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Smart CE, Askarian Amiri ME, Wronski A, et
al: Expression and function of the protein tyrosine phosphatase
receptor J (PTPRJ) in normal mammary epithelial cells and breast
tumors. PLoS One. 7:e407422012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Toland AE, Rozek LS, Presswala S, Rennert
G and Gruber SB: PTPRJ haplotypes and colorectal cancer risk.
Cancer Epidemiol Biomarkers Prev. 17:2782–2785. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mita Y, Yasuda Y, Sakai A, et al: Missense
polymorphisms of PTPRJ and PTPN13 genes affect susceptibility to a
variety of human cancers. J Cancer Res Clin Oncol. 136:249–259.
2010. View Article : Google Scholar
|
|
28
|
Lv XG, Ji MY, Dong WG, et al: EBP50 gene
transfection promotes 5-fluorouracil-induced apoptosis in gastric
cancer cells through Bax- and Bcl-2-triggered mitochondrial
pathways. Mol Med Rep. 5:1220–1226. 2012.PubMed/NCBI
|
|
29
|
Taiyoh H, Kubota T, Fujiwara H, et al: NK4
gene expression enhances 5-fluorouracil-induced apoptosis of murine
colon cancer cells. Anticancer Res. 31:2217–2224. 2011.PubMed/NCBI
|
|
30
|
Tian F, Fan T, Jiang Y, Zhang X and Wang
X: A small interfering RNA targeting NF-kappaB p65 alone or
combined with 5-FU inhibits growth of esophageal squamous cell
carcinoma in nude mice. Pathol Res Pract. 208:32–38. 2012.
View Article : Google Scholar
|
|
31
|
Xie Q, Liang BL, Wu YH, et al: Synergistic
anticancer effect of rAd/P53 combined with 5-fluorouracil or
iodized oil in the early therapeutic response of human colon cancer
in vivo. Gene. 499:303–308. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
McCubrey JA, Steelman LS, Abrams SL, et
al: Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in
malignant transformation and drug resistance. Adv Enzyme Regul.
46:249–279. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang LH, Yin AA, Cheng JX, et al: TRIM24
promotes glioma progression and enhances chemoresistance through
activation of the PI3K/Akt signaling pathway. Oncogene. Jan
27–2014.(Epub ahead of prin). View Article : Google Scholar
|
|
34
|
Kamran MZ, Patil P and Gude RP: Role of
STAT3 in cancer metastasis and translational advances. Biomed Res
Int. 2013:4218212013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gong J, Muñoz AR, Chan D, Ghosh R and
Kumar AP: STAT3 down regulates LC3 to inhibit autophagy and
pancreatic cancer cell growth. Oncotarget. 5:2529–2541.
2014.PubMed/NCBI
|