Introduction
Liver cancer is a major public health issue, and the
incidence as well as the mortality of liver cancer are on the
increase. Globally, new cases of liver cancer have been annually
reported to be ~700,000, and East Asian countries, including Korea,
Japan and China, are considered to be high-risk areas (1–3). The
risk factors for liver cancer include chronic hepatitis virus,
dietary contaminants, fatty liver diseases, cirrhosis and host
genetic factors (4,5). Many signaling pathways are also
involved in hepatic tumorigenesis, including various growth factors
such as the Wnt, Hedgehog and TGF-β pathway (6–8).
Transforming growth factor-β (TGF-β) is a secreted
cytokine that regulates many biological processes in most cells. It
performs important functions in early and late embryogenesis, and
also plays a key role in adult tissues, including cellular
proliferation, apoptosis, differentiation, angiogenesis,
extracellular matrix synthesis and immune responses (9,10).
According to the cell or tissue type and the microenvironment, the
cellular responses to TGF-β are different. Various target genes,
including collagen type I, α-smooth muscle actin, SNAI1 and p15,
may be induced in the liver after activation of the TGF-β pathway.
These genes may induce diverse responses, including fibrosis,
cellular migration and suppression of cellular proliferation
(11,12). Several studies have shown that the
TGF-β pathway may suppress cellular proliferation, including early
tumorigenesis and liver regeneration in most cases. However, the
role of TGF-β in various pathological conditions is still unclear
(13–15).
The TGF-β/Smad pathway initiates with activation of
secreted TGF-β and the ligand assembles TGF-β receptor type I and
II (TGF-β RI and II) in the cellular membrane. Then, the TGF-β RII
phosphorylates and activates the TGF-β RI. Smad2 (ser465/476) and
Smad3 (ser433/435) are then phosphorylated by activating TGF-β RI
and are also called receptor-Smads (R-Smads). Activated R-Smads
bind Smad4, also referred to as common Smads (Co-Smads), and pass
the nuclear membrane. In the nucleus, the Smad complex binds with
co-factors and positively and negatively regulates the expression
of target genes (10,13,16,17).
The expression levels of p15, p21, KLF10 and Smad2 genes are also
increased and Smad7, cMyc, cyclin D1 genes or proteins are
downregulated in the proliferation control by the TGF-β/Smad
pathway (8,10). In the non-Smad TGF-β pathway, TGF-β
RI may also activate other signaling systems, including PI3K, RAS
and FAS (9,17–19).
KLF10 is an early target gene of the TGF-β/Smad
(Sma- and Mad-related) signaling pathway and it is reported that it
may inhibit cellular proliferation in various cell types (20,21).
At first, KLF10 was observed in normal human osteoblasts after
TGF-β treatment. Thus, KLF10 was initially named as TGF-β inducible
early gene-1 (TIEG1) (22). Later,
considering three zinc finger domains of the TIEG1 protein, it was
classified and renamed as Krüppel-like family of transcription
factors 10 (20,21,23).
Recent studies have shown that KLF10 expression may activate the
TGF-β/Smad pathway and mimic the roles of TGF-β in several cell
types. KLF10 may enhance the transcription of TGF-β-regulated
genes, including Smad2, p21, TGF-β1 and plasminogen activator
inhibitor-1 and suppress the transcription of the Smad7 gene,
inhibitory Smads (13,24,25).
However, there has been limited information concerning the role of
KLF10.
KLF10, one of the target genes of TGF-β, plays an
important role in numerous biological processes, including
tumorigenesis. However, there has been limited information
concerning KLF10, and the actual functions of KLF10 in liver
tumorigenesis are still unknown. In the present study, to elucidate
the role of KLF10 in chemically induced hepatic carcinogenesis,
histopathological and molecular analyses were performed using KLF10
KO mice injected with diethylnitrosamine (DEN) via intraperitoneal
route.
Materials and methods
Mice
Male KLF10-deficient C57BL/6J mice were kindly
provided by Dr Woon-Kyu Lee (Inha University, Incheon, Korea)
(26), and the C57BL/6J mice were
obtained from the Korea Research Institute of Bioscience and
Biotechnology (KRIBB, Daejeon, Korea). All mice were bred in a
laboratory animal breeding room under specific pathogen-free
conditions. For genotyping, the DNA samples were isolated from all
mouse tails using a genomic DNA extraction kit (Bioneer, Daejeon,
Korea) and subjected to polymerase chain reaction. The DNA primers
for genotyping were KLF10 F, CCTTCCTGCCAACAACTCTC; R, TCTGAGGAGGAC
CCTTGCT and KLF10 KO F, TCGCCTTCTTGACGAG TTCT. The size of the
KLF10 KO gene is 658 base pairs (bp) and that of the wild-type (WT)
gene is 248 bp. All the procedures were approved by the IACUC
(Institutional Animal Care and Use Committee, KU 09023, Seoul,
Korea) of Konkuk University.
Experimental design
For the liver carcinogenesis study, 25 mg/kg of DEN
was single injected in the mice via intraperitoneal route at 12
days of age. KLF10 KO and WT mice (13–14 mice/group) were
sacrificed at 26 and 36 weeks, respectively, after the carcinogen
administration (at the age of 28 and 38 weeks, respectively). Three
mice injected with PBS were used as control animals in each group.
At the time of the sacrifice, all the mice were grossly examined,
and blood was collected from the caudal vena cava. The liver was
excised and weighed. Some portion of the liver tissue was fixed in
10% neutral-buffered formalin and the rest of the hepatic tissue
was frozen for further analysis.
Hematoxylin and eosin (H&E) staining
and histopathological analysis
Fixed liver tissues were processed by standard
methods, embedded in paraffin and then cut in 4-μm sections. The
sections were deparaffinized, rehydrated and stained with H&E.
Furthermore, the sections were dehydrated, cleared, mounted and
viewed by light microscopy. The hepatic lesions were classified as
hepatic foci, hepatocellular adenoma (HCA) or hepatocellular
carcinoma (HCC). Two veterinary pathologists independently reviewed
all the lesions.
Immunohistochemical staining
For immunohistochemistry, some serial sections
(4-μm) were deparaffinized, rehydrated, placed in 0.01 M citrate
buffer (pH 6.0) and heated in a microwave for 10 min. Then, the
slides were incubated for 10 min in 1.0%
H2O2. The slides were preincubated with
blocking serum (Vectastain ABC kit; Vector Laboratories,
Burlingame, CA, USA), incubated with proliferative cell nuclear
antigen (PCNA; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA)
antibody and then incubated with biotinylated secondary antibodies
(Vectastain ABC kit) followed by incubation with avidin-coupled
peroxidase (Vectastain ABC kit) (both from Vector Laboratories).
After development with 3,3′-diamino-benzidine (DAB, DAB Substrate
kit; Vector Laboratories) the sections were counterstained with
hematoxylin.
Quantitative reverse
transcription-polymerase chain reaction (RT-PCR) analyses
RT-PCR was performed on RNA samples that were
isolated from the liver tissues. Total RNA was prepared from frozen
liver tissues using TRIzol (Invitrogen) and reverse transcribed
into cDNA using M-MLV reverse transcriptase (both from Invitrogen,
Carlsbad, CA, USA). The cDNA was used as a template for
amplification in the PCR. Band intensities were quantified using
the Multi Gauge software V3.0 (Fujifilm, Tokyo, Japan) and
normalized to transcription levels of β-actin. The sequences of the
primer pairs were as follows: Smad2 F, CGGTTAGATGAGCTTGA GAA and R,
TCACTGATATATCCAGGTGG; Smad3 F, TCACTGGATGGTCGGCTGCA and R,
CTTCACTCAGGT AGCCAGGA; Smad4 F, CGATGCCTGTCTGAGCATTG and R,
CTCCGTTGATGCGCGATTAC; Smad7 F, ATCGGT CACACTGGTGCGTG and R,
TCCAGTGTGGCGGACTT GAT; p15 F, CTGGATGTGTGTGACGCCTG and R, CTT
CGTGCTTGCAGTCTTCC; p21 F, AGCTAGGATGACAG TGAAGC and R,
CAAGTGCCTAGATATGCCTC; TGF-β RI F, TATGATATGACAACATCAGG and R,
AACCACAGC TGCGTCCATGT; TGF-β RII F, GTGTGCCTGTAACATGG AAG and R,
AGGTGTTCTGCTTCAGCTTG; c-myc F, TAG TGCTGCATGAGGAGACA and R,
GTTGCTGATCTGCT TCAGGA; cyclin D1 F, GGATGCTGGAGGTCTGTGAG and R,
GAGTTGTCAGTGTAGATGCAC, β-actin F, TGGAAT CCTGTGGCATCA TGAAA and R,
TAAAACGCAGCTCAG TAACAGTCC.
Western blot analysis
Protein was extracted from the liver with extraction
solution (Pro-PrepTM; Intron Biotechnology, Seoul,
Korea). The protein concentrations were determined using the BCA
kit (Pierce Biotechnology Inc., Rockford, IL, USA). After being
transferred onto nitrocellulose membranes, the proteins were
blocked and incubated overnight with specific antibodies against
β-actin, Smad4, Smad7, p15, p21, TGF-β RI, cMyc (Santa Cruz
Biotechnology, Inc.), Smad2, Smad3, p-Smad3, p27, TGF-β RII and
cyclin D1 (Cell Signaling Technology Inc., Beverly, MA, USA) at
4°C. Then, the membranes were washed with TBST and incubated with
either anti-rabbit or anti-mouse secondary antibodies (Santa-Cruz
Biotechnology, Inc.), which were horseradish-peroxidase linked.
Specific antibodies were detected with an ECL test kit (KPL,
Gaithersburg, MD, USA). The band intensities were quantified using
Imagequant Software (Image Lab V4.0; Bio-Rad Inc., San Diego, CA,
USA) and normalized to β-actin expression.
Statistical analysis
For statistical analysis, all data obtained were
analyzed using SPSS V14.0 software (SPSS Inc., Chicago, IL, USA).
Statistically significant differences between the studied groups
were evaluated using the unpaired Student’s t-test or the Fisher’s
exact test. Results were determined to be statistically significant
for values of P<0.05 (P<0.05 and P<0.01 are indicated in
the figure legends).
Results
Gross findings and histopathological
analysis
KLF10 KO mice (n=14 and 14, respectively) and WT
mice (n=14 and 13, respectively) were sacrificed at 26 and 36 weeks
after DEN injection (at the age of 28 and 38 weeks, respectively),
and the body and liver weights were determined. The number and
diameter of the masses in the liver were determined. All the masses
were divided into foci (<10 mm) and nodules (≥10 mm) depending
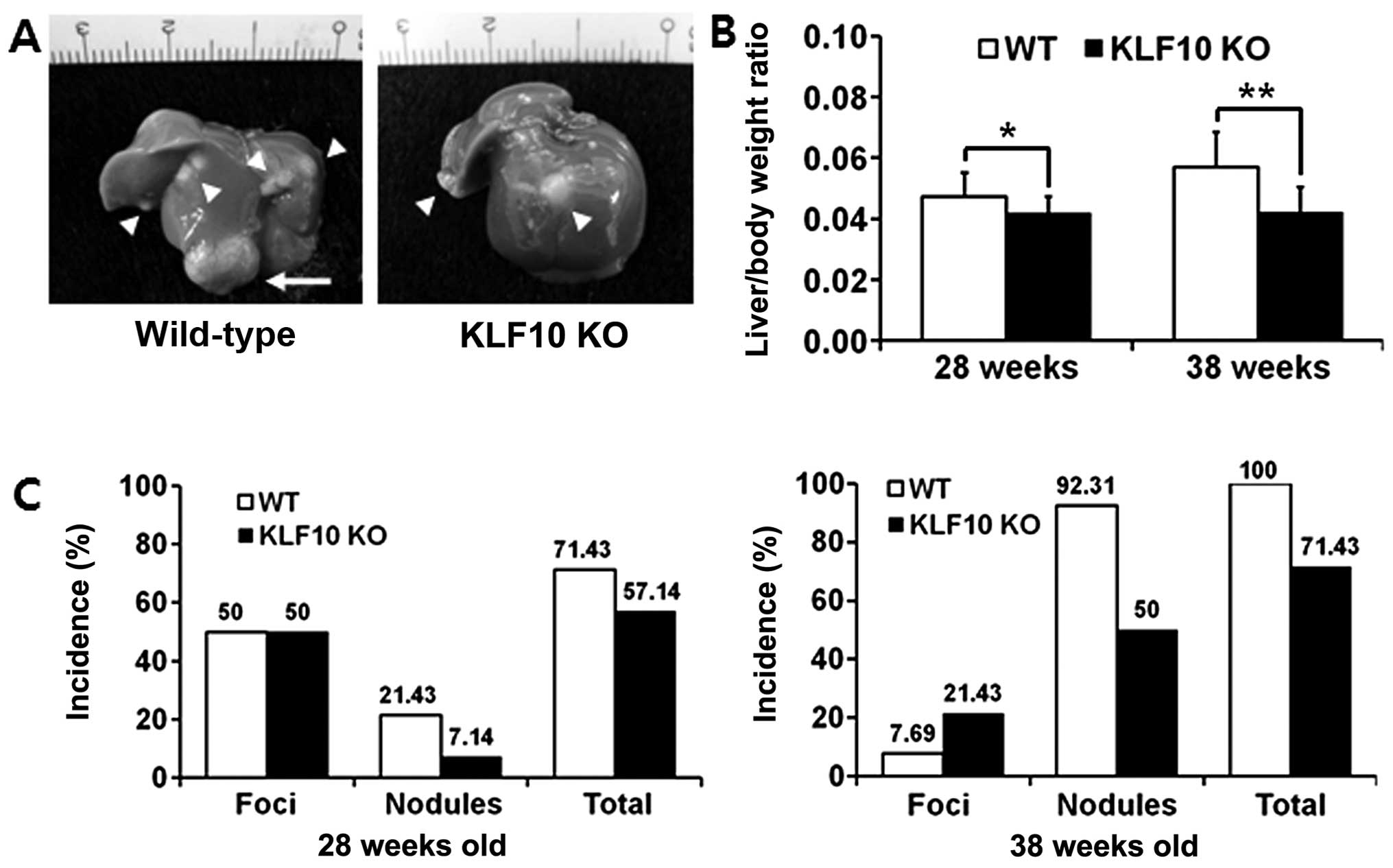
on their size (Fig. 1A).
The liver/body weight ratios were significantly
lower in the KLF10 KO than these ratios in the WT mice. At the age
of 28 weeks, the mean ± standard deviation (SD) values of the
ratios of the WT and KLF10 KO mice were 0.047±0.0079 and
0.042±0.0054, respectively (P=0.046). At the age of 38 weeks, the
mean ± SD ratios of the WT and KLF10 KO mice were 0.056±0.00076 and
0.042±0.0083, respectively (P=0.00077) (Fig. 1B). The incidence of masses was also
lower in the KLF10 KO than that in the WT mice. The total incidence
of masses was 71.43 vs. 57.14% at 26 weeks after DEN treatment, and
100 vs. 71.43% at 36 weeks after DEN treatment in the WT and KLF10
KO mice, respectively. In addition, the WT mice had more nodules
than those in the KLF10 KO mice. The total incidence of nodules was
21.43 vs. 7.14% at the age of 26 weeks and 92.31 vs. 50%, at the
age of 36 weeks in the WT and KLF10 KO mice, respectively, after
DEN treatment (Fig. 1C).
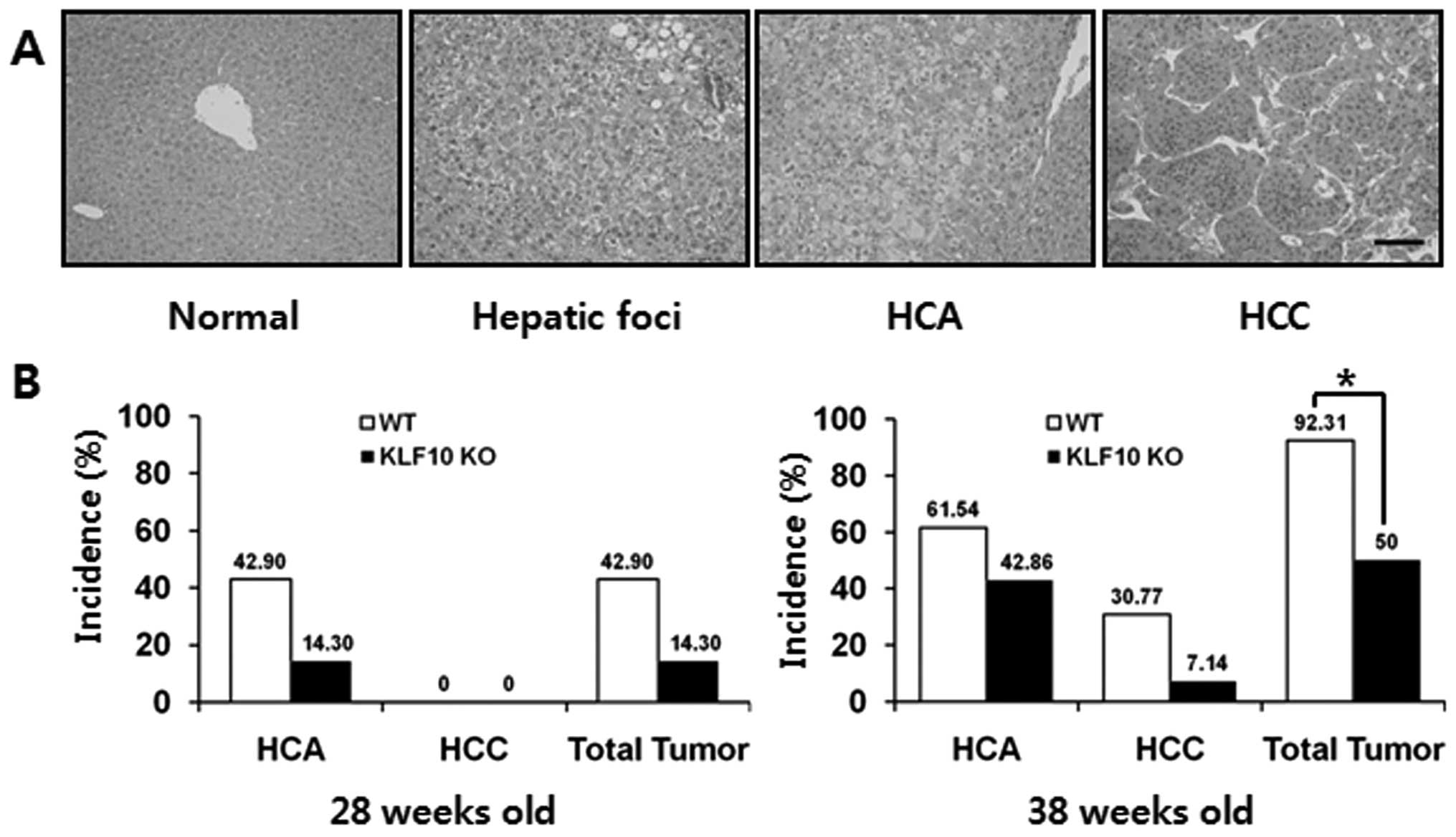
In the DEN-treated mice, the hepatic lesions
progressed from hepatic foci, HCA to HCC. In the microscopic
observation, the hepatic foci showed irregular cellular size. Most
hepatic foci consisted primarily of clear or vacuolated cells, and
partially of basophilic cells. HCAs displayed well-circumscribed
lesions, composed of hepatocytes with cellular pleomorphism,
anisokaryosis and basophilic or vacuolated cytoplasm containing
hepatocytes. Most of the adenomas did not have normal lobular
architecture and their vasculatures were compressed or altered. In
HCCs, cellular atypia with malignancy, an increase in the
nuclear-cytoplasmic ratio and mitotic figures were common and a
trabecular growth pattern was the most prominent feature (Fig. 2A). At the age of 28 weeks, the
incidence of HCA in the KLF10 KO vs. the WT mice was 14.30 (2/14)
and 42.90% (6/14), respectively, but there were no malignant
lesions observed in any mice. At the age of 38 weeks, the incidence
of HCA, HCC and total tumors was 42.86 (6/14), 7.14 (1/14) and
50.00% (7/14) in the KLF10 KO mice and 61.54 (8/13), 30.77 (4/13)
and 92.31% (12/13) in the WT mice, respectively. The tumor
incidence was significantly higher in the WT than that in the KLF10
KO mice (Fig. 2B; P=0.021 by
Fisher’s exact test).
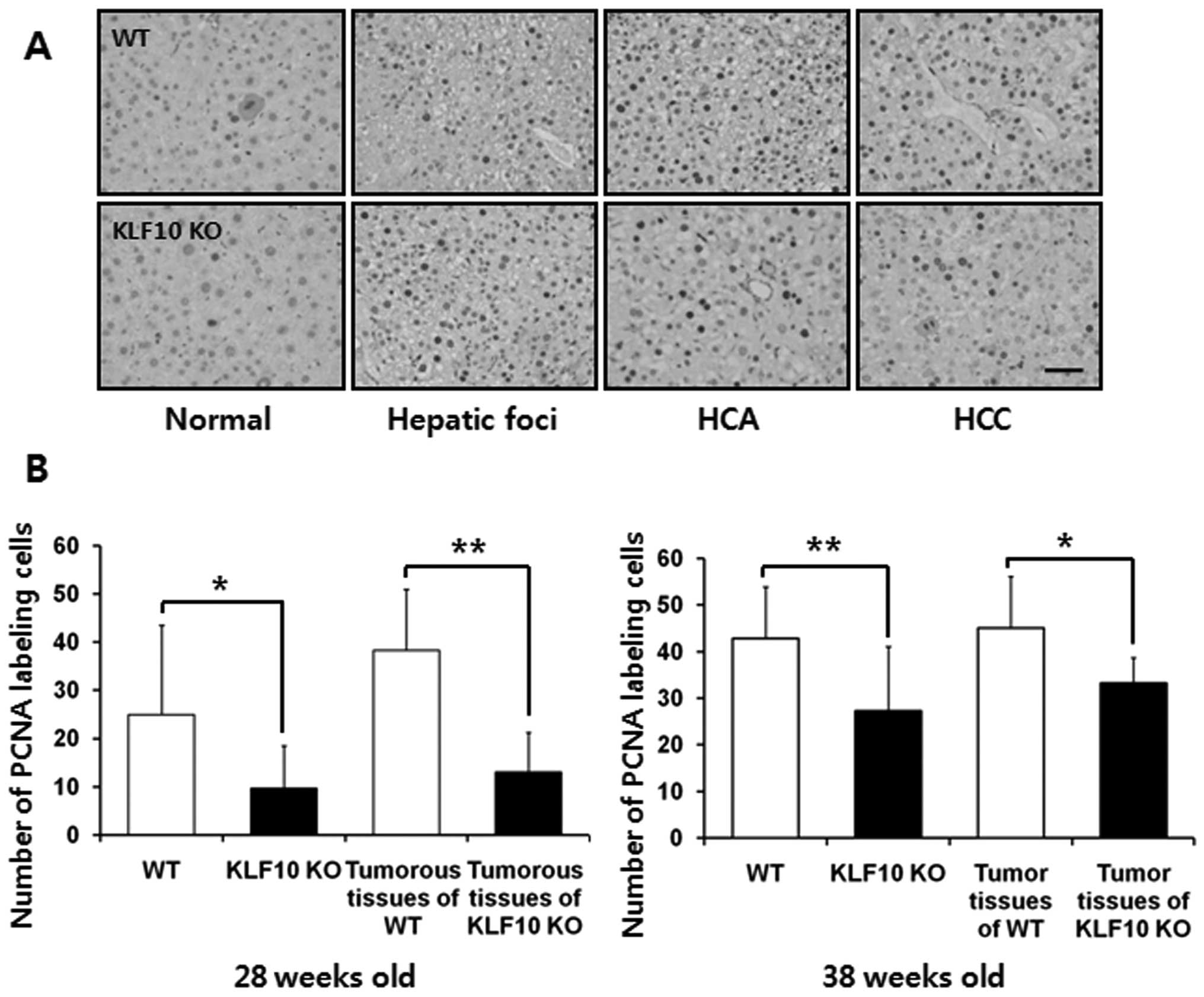
Comparison of the cellular proliferative
potential of liver tissues in the WT and KLF10 KO mice
The immunohisto-chemical staining for PCNA was
assessed to analyze the proliferative potential of hepatocytes. The
number of PCNA-positive nuclei was counted in an area of 0.1
mm2 under a light microscope. The PCNA labeling index
was significantly higher in the WT than that in the KLF10 KO mice.
At 26 weeks after DEN treatment, the mean ± SD values of the WT and
KLF10 KO mice were 24.9±18.6 and 9.64±8.83, respectively (P=0.010).
When comparing only tumorous tissues including pre-neoplastic
hepatic foci, the values were 38.3±12.7 and 13.1±8.19, respectively
(P=0.0023). At 36 weeks after DEN administration, the mean ± SD
values of the WT and KLF10 KO mice were 42.8±11.2 and 27.2±13.8,
respectively (P=0.0060). When comparing only tumorous tissues
excluding hepatic foci tissues, the values were 45.1±11.1 and
33.2±5.40, respectively (P=0.041) (Fig.
3).
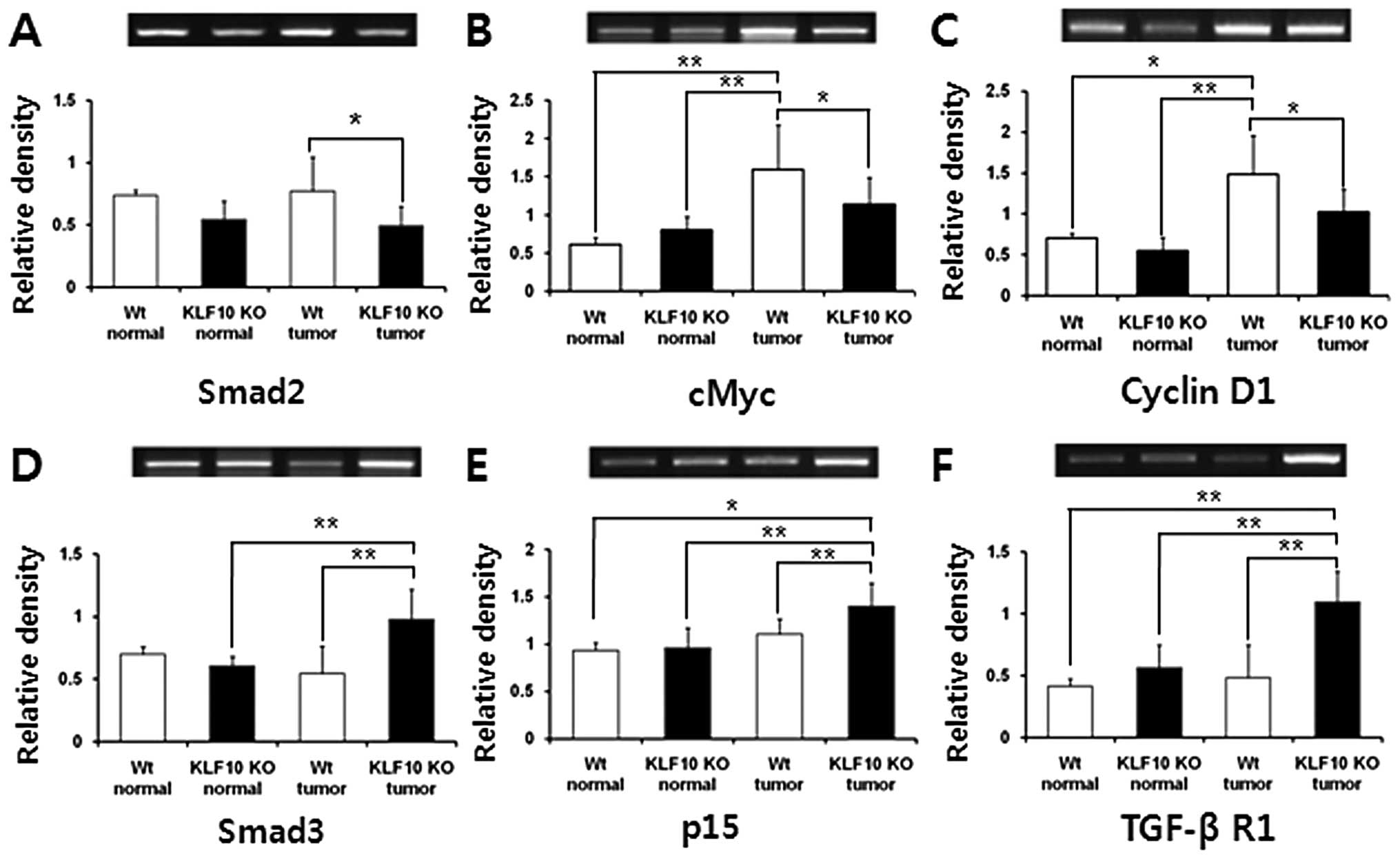
Analyses of expression of genes involved
in the TGF-β/Smad signaling pathway
To investigate the mRNA transcription levels of the
genes (Smad2, Smad3, Smad4, Smad7, p15, p21, TGF-β RI, TGF-β RII,
cMyc and cyclin D1) involved in the TGF-β/Smad pathway, RT-PCR
analyses were performed. The mRNA levels of Smad2, one of the KLF10
target genes, were higher in the WT than levels in the KLF10 KO
mice and a significant difference was obtained in the tumor
tissues. The levels of Smad3 and p15, antiproliferative genes, were
significantly higher in the tumor tissues of the KLF10 KO than
levels in the tumor tissues of the WT mice. TGF-β RI, which is
involved in the early stage of the TGF-β pathway and may
potentially inhibit cellular proliferation (10), was expressed prominently in the
KLF10 KO tumor tissues when compared with the other tissues. In
contrast, cMyc and cyclin D1 genes associated with cellular
multiplication showed significantly higher expression in the WT
tumors than levels in the other tissues (Fig. 4). Other genes, Smad2, Smad4, Smad7,
p21 and TGF-β RII, did not have any significant difference in mRNA
levels (data not shown).
Western blot analysis of proteins
involved in the TGF-β/Smad signaling pathway
To examine the expression levels of the TGF-β
pathway target or associated proteins [Smad2, Smad3, Smad4, Smad7,
phospho-Smad2 (p-Smad2), phospho-Smad3 (p-Smad3; ser433/435), p15,
p21, p27, TGF-β1, TGF-β RI, TGF-β RII and cyclin D1], western blot
analysis of each protein was assessed and normalized to the β-actin
level. Similar to the quantitative RT-PCR results, the protein
levels of Smad2 were significantly higher in the WT than levels in
the KLF10 KO tissues. Likewise, Smad3 and p15 levels were
significantly higher in the KLF10 KO than levels in the WT tumor
and the normal KLF10 KO tissues. The TGF-β RI protein level was
also significantly higher in the KLF10 KO than levels in the WT
tumor and normal tissues. TGF-β1, the initiator of the TGF-β
pathway and one of the KLF10 target genes, was higher in the KLF10
KO tumors than that in the other tissues. In contrast, the cyclin
D1 level was significantly higher in the WT tumors than this level
in the other tissues (Fig. 5).
Other proteins, Smad4, Smad7, p-Smad2, p21, p27 and TGF-β RII, did
not have any meaningful difference in protein levels (data not
shown). Additional analysis of p-Smad3, the activated form of
Smad3, was performed, and the expression level in the KLF10 KO
tumors was more dominant than that in the WT tumors (P=0.042)
(Fig. 5C). The p-Smad3/Smad3 ratio
used to analyze the degree of Smad3 activation was also
significantly higher in the KLF10 KO, than this ratio in the WT
tumor tissues (Fig. 5H).
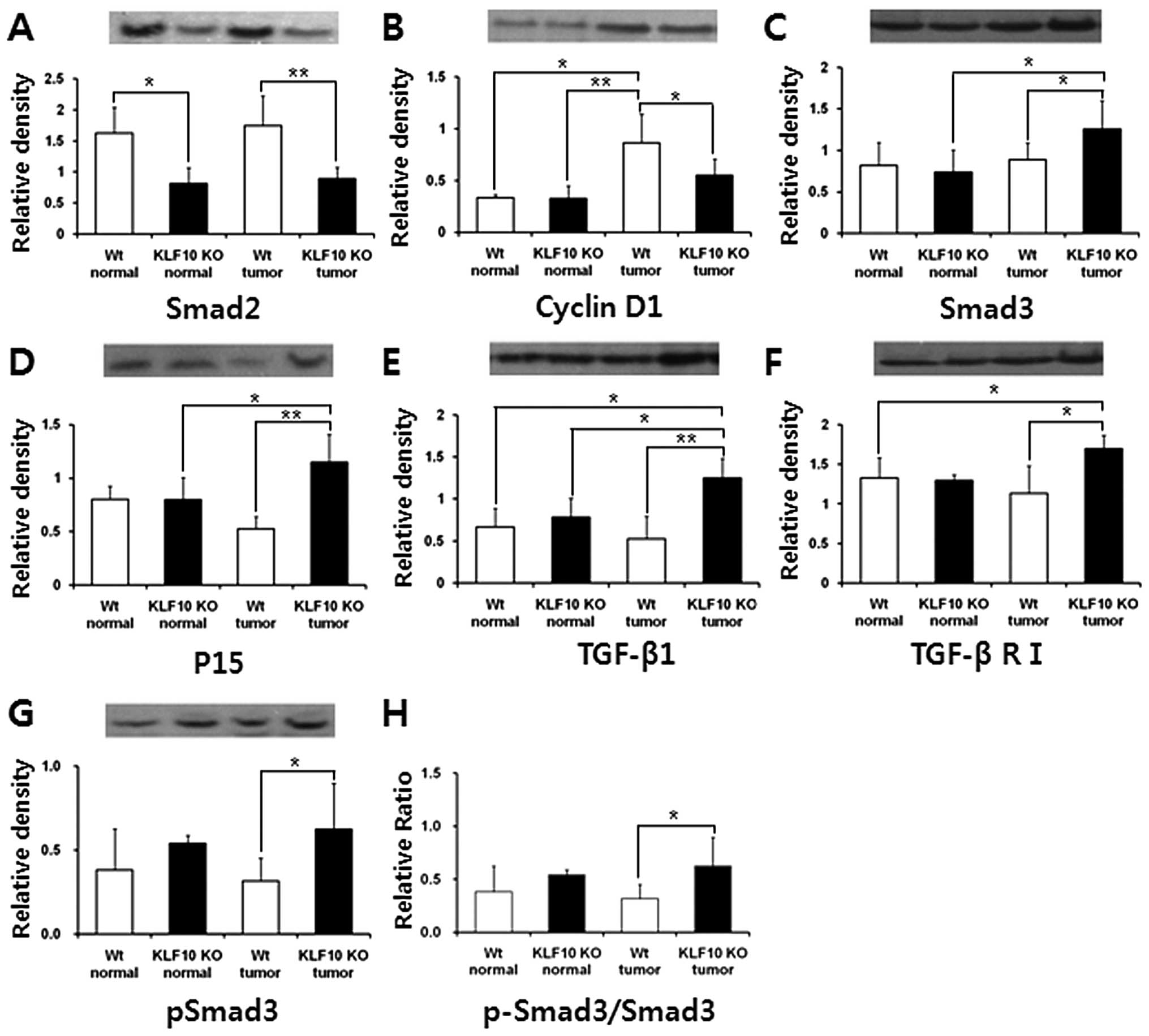 | Figure 5Quantification of the protein
expression levels of Smad2, cyclin D1, Smad3, p15, TGF-β1, TGF-β R1
and pSmad3, which are involved in the TGF-β/Smad pathway. (A and B)
Smad2 and cyclin D1 were expressed significantly higher in the WT
groups. (C–F) Smad3, P15, TGF-β1 and TGF-β RI were expressed
significantly higher in the KLF10 KO mice. (G) pSmad3 expression
and (H) Smad3 activation were significantly higher only in the
KLF10 KO tumor group. The band intensities were quantified and
normalized to the β-actin level. *P<0.05 and
**P<0.01 by the unpaired Student’s t-test. TGF-β,
transforming growth factor-β; WT, wild-type. |
Discussion
KLF10, first observed in 1995, is known to induce
upregulation of TGF-β target genes by enhancement of R-Smad and
co-Smad activation. It may also enhance TGF-β signaling
transduction by induction of the expression of several genes
involved in TGF-β signaling, Smad2 and TGF-β1 (21,27,28).
TGF-β may regulate many biological processes in most cells, and
cellular responses to TGF-β are different under various conditions
(11,12). The actual functions of TGF-β with
KLF10 in various pathophysiological conditions still remain unclear
(13,20,24).
Particularly, there have not been any in vivo studies to
elucidate the roles of KLF10 in the liver. In the present study,
functions of the KLF10 gene in chemically induced hepatic
tumorigenesis were investigated using KLF10 KO mice. The mouse
model of DEN-induced HCC, which was also employed in the present
study, has been extensively used for liver carcinogenesis studies
since it is genetically and histologically similar to the human one
(29,30).
In the necropsy and histopathological results, KLF10
KO mice exhibited a lower incidence of tumors (Figs. 1C and 2B). Additionally, PCNA labeling indices
were significantly higher in the WT than indices in the KLF10 KO
mice (Fig. 3B). Considering these
results, KLF10 KO mice may benefit from antiproliferative activity
following chemically induced hepatic tumorigenesis. To elucidate
the causes behind the increased antiproliferative effects in KLF10
KO mice, the expression levels of genes involved in the TGF-β/Smad
pathway were examined by quantitative RT-PCR. The expression level
of the Smad2 gene, one of the KLF10 target gene, was higher in the
WT than that in the KLF10 KO mice, and the significant difference
was obtained between the tumor tissues of the WT mice and those of
the KLF10 KO (Fig. 4A). This may be
caused by malfunction of the KLF10 gene in KO mice, and the
difference was intensified in the tumor tissues. However, Smad7 and
p21, previously also reported as target genes of KLF10, did not
show any significant differences. Smad4 and TGF-β RII, mediators of
TGF-β signaling, did not have any significant differences either
(data not shown). This may indicate that KLF10 ablation did not
affect the expression of the genes mentioned above in hepatic
carcinogenesis, except for Smad2. In contrast, mRNA levels of
Smad3, TGF-β RI and p15 were increased in the tumor tissues of the
KLF10 KO mice, and cMyc, cyclin D1 were expressed prominently in
the tumor tissues of the WT mice (Fig.
4B–F). In the western blot analysis, similar results were
obtained, and TGF-β1 was expressed significantly higher in the
tumor tissues of the KLF10 KO mice than in other tissues. In
addition, the p-Smad3/Smad3 ratio was significantly higher in the
tumor tissues of the KLF10 KO mice (Fig. 5). Considering these results, the
KLF10 null condition induced abundant expression of TGF-β R1,
TGF-β1, Smad3 and increased activation of Smad3. It reinforced the
TGF-β/Smad pathway in chemically induced carcinogenesis. As a
result, p15, one of the positively regulated target genes of the
TGF-β pathway, was increased in the KLF10-deficient mice and it
suppressed transcription and function of proliferation-related
genes, cMyc and cyclin D1. The activated Smad complex may also
directly suppress transcription of the cMyc gene. Thus, lack of
KLF10 suppressed cellular proliferation of hepatocytes during liver
tumorigenesis by reinforcement of the TGF-β/Smad pathway.
TGF-β induces a great variety of cellular responses
dependent on the type of cells and the microenvironments (15,18,31).
KLF10, one of the early target genes of TGF-β, is also involved in
various biological processes and diseases. Most previous studies
have shown that the effects of KLF10 were similar to those of
TGF-β, including antiproliferation; however, this information
remains insufficient and fractionary (21,32–34).
There is one study that examined the roles of KLF10 in liver cancer
and it revealed that upregulation of KLF10 in an HCC cell line may
induce inhibition of cellular proliferation (35). However, dissimilar to the cell
culture environment, the KLF10 null condition in the liver of a
living organism may induce various other responses, as in the
present study, due to differences in the composition of the cells,
the architecture of the tissue and the microenvironment.
Likewise, various biological responses may be
induced in the same knockout mouse. For example, tumorigenesis of
the lymphoid system was accelerated, while hepatic carcinogenesis
was suppressed in ataxia telangiectasia-mutated gene (ATM) KO mice.
This may be caused by a dissimilar process of malignancy derived
from differences in the cell type that participate in tumorigenesis
and the microenvironment (36–38).
One study concerning tumorigenesis using KLF10 KO mice showed that
the incidence of skin cancer was increased in KLF10 KO mice
(26). As mentioned above, diverse
responses may be induced in tumorigenesis according to the type of
tissues, although the same genetically engineered mice were used.
Therefore, results showing tumor suppressor activity can be
obtained in liver carcinogenesis study using KLF10 KO mice,
although previous studies were mainly focused on the
tumor-suppressor effects of KLF10.
TGF-β may activate Ras, Rho, TAK1 and PP2A except
for Smad proteins. In addition, KLF10 may be induced by bone
morphogenetic proteins, estrogen and epidermal growth factor except
for TGF-β (17,19,39).
Furthermore, KLF11 was recently found to exhibit similar effects as
KLF10 (21,40). Considering these studies, some
compensatory mechanisms for the KLF10 null condition may exist in
the non-Smad TGF-β pathways or KLF10 signaling. Thus, to elucidate
the exact mechanisms in KLF10 KO mice, additional analyses of
intracellular signaling pathways (except for Smads) are needed, and
also the effects of other inducers of KLF10 (except for TGF-β) are
warranted. In addition, to confirm the antiproliferative effect of
the KLF10 null condition, in vivo analysis concerning the
KLF10 blockade using WT mice is also needed.
In conclusion, the KLF10 KO mice exhibited a lower
tumor incidence and cellular proliferation than the WT mice
following DEN-induced liver tumorigenesis. Although the expression
of the Smad2 gene was decreased in the hepatic tissues of the KLF10
KO mice, the TGF-β/Smad signaling pathway was reinforced by
upregulation of Smad3, TGF-β1, TGF-β R1 and increased activation of
Smad3. This induced increased expression of p15 and the suppression
of proliferation-related genes, cMyc and cyclin D1. Based on the
results, KLF10 KO mice may benefit from antiproliferative
activities following liver tumorigenesis by enhancement of the
TGF-β pathway. This study provides the first observation concerning
the mito-inhibitory effects in liver cells using KLF10 KO mice.
However, the exact mechanisms and effects of the tumor-suppressor
activities in KLF10 KO mice require further elucidation. To confirm
these mechanisms, additional analysis of the non-Smad TGF-β pathway
and in vivo examinations using WT mice with KLF10 blocking
agents are needed.
Acknowledgements
This study was supported by Konkuk University in
2014.
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shin HR, Carlos MC and Varghese C: Cancer
control in the Asia Pacific region: current status and concerns.
Jpn J Clin Oncol. 42:867–881. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maluccio M and Covey A: Recent progress in
understanding, diagnosing, and treating hepatocellular carcinoma.
CA Cancer J Clin. 62:394–399. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Villanueva A, Newell P, Chiang DY,
Friedman SL and Llovet JM: Genomics and signaling pathways in
hepatocellular carcinoma. Semin Liver Dis. 27:55–76. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Farazi PA and DePinho RA: Hepatocellular
carcinoma pathogenesis: from genes to environment. Nat Rev Cancer.
6:674–687. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sia D and Villanueva A: Signaling pathways
in hepatocellular carcinoma. Oncology. 81:18–23. 2011. View Article : Google Scholar
|
|
8
|
Villanueva A, Chiang DY, Newell P, et al:
Pivotal role of mTOR signaling in hepatocellular carcinoma.
Gastroenterology. 135:1972–1983. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kaminska B, Wesolowska A and Danilkiewicz
M: TGF beta signaling and its role in tumour pathogenesis. Acta
Biochim Pol. 52:329–337. 2005.
|
|
10
|
Inman GJ: Switching TGF-β from a tumor
suppressor to a tumor promoter. Curr Opin Genet Dev. 21:93–99.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Breitkopf K, Weng H and Dooley S:
TGF-β/Smad-signaling in liver cells: Target genes and inhibitors of
two parallel pathways. Signal Transduction. 6:329–337. 2006.
View Article : Google Scholar
|
|
12
|
Achyut BR and Yang L: Transforming growth
factor-β in the gastrointestinal and hepatic tumor
microenvironment. Gastroenterology. 141:1167–1178. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Massagué J: TGFbeta in cancer. Cell.
134:215–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fausto N, Campbell JS and Riehle KJ: Liver
regeneration. Hepatology. 43:S45–S53. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Itoh S and ten Dijke P: Negative
regulation of TGF-β receptor/Smad signal transduction. Curr Opin
Cell Biol. 19:176–184. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ellenrieder V: TGFbeta regulated gene
expression by Smads and Sp1/KLF-like transcription factors in
cancer. Anticancer Res. 28:1531–1539. 2008.PubMed/NCBI
|
|
17
|
Kang JS, Liu C and Derynck R: New
regulatory mechanisms of TGF-beta receptor function. Trends Cell
Biol. 19:385–394. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Derynck R, Akhurst RJ and Balmain A:
TGF-beta signaling in tumor suppression and cancer progression. Nat
Genet. 29:117–129. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Moustakas A and Heldin CH: Non-Smad
TGF-beta signals. J Cell Sci. 118:3573–3584. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Subramaniam M, Hawse JR, Rajamannan NM,
Ingle JN and Spelsberg TC: Functional role of KLF10 in multiple
disease processes. Biofactors. 36:8–18. 2010.PubMed/NCBI
|
|
21
|
Spittau B and Krieglstein K: Klf10 and
Klf11 as mediators of TGF-beta superfamily signaling. Cell Tissue
Res. 347:65–72. 2012. View Article : Google Scholar
|
|
22
|
Subramaniam M, Harris SA, Oursler MA,
Rasmussen K and Riggs BL: Identification of a novel
TGF-beta-regulated gene encoding a putative zinc finger protein in
human osteoblasts. Nucleic Acids Res. 23:4907–4912. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cook T, Gebelein B, Belal M, Mesa K and
Urrutia R: Three conserved transcriptional repressor domains are a
defining feature of the TIEG subfamily of Sp1-like zinc finger
proteins. J Biol Chem. 274:29500–29504. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Johnsen SA, Subramaniam M, Janknecht R and
Spelsberg TC: TGF-beta inducible early geneenhances
TGFbeta/Smad-dependent transcriptional responses. Oncogene.
21:5783–5790. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Subramaniam M, Hawse JR, Johnsen SA and
Spelsberg TC: Role of TIEG1 in biological processes and disease
states. J Cell Biochem. 102:539–548. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Song KD, Kim DJ, Lee JE, Yun CH and Lee
WK: KLF10, transforming growth factor-β-inducible early gene 1,
acts as a tumor suppressor. Biochem Biophys Res Commun.
419:388–394. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dooley S, Weng H and Mertens PR:
Hypotheses on the role of transforming growth factor-beta in the
onset and progression of hepatocellular carcinoma. Dig Dis.
27:93–101. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Buenemann CL, Willy C, Buchmann A,
Schmiechen A and Schwarz M: Transforming growth
factor-beta1-induced Smad signaling, cell-cycle arrest and
apoptosis in hepatoma cells. Carcinogenesis. 22:447–452. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Newell P, Villanueva A, Friedman SL, Koike
K and Llovet JM: Experimental models of hepatocellular carcinoma. J
Hepatol. 48:858–879. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bakiri L and Wagner EF: Mouse models for
liver cancer. Mol Oncol. 7:206–223. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Heldin CH and Miyazono K: Transforming
growth factor-beta. An interesting candidate for clinical use.
Lakartidningen. 92:1569–1572. 1995.(In Swedish). PubMed/NCBI
|
|
32
|
Bensamoun SF, Hawse JR, Subramaniam M, et
al: TGF-beta inducible early gene-1 knockout mice display defects
in bone strength and microarchitecture. Bone. 39:1244–1251. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tsubone T, Moran SL, Subramaniam M, Amadio
PC, Spelsberg TC and An KN: Effect of TGF-beta inducible early gene
deficiency on flexor tendon healing. J Orthop Res. 24:569–575.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bos JM, Subramaniam M, Hawse JR, et al:
TGFβ-inducible early gene-1 (TIEG1) mutations in hypertrophic
cardiomyopathy. J Cell Biochem. 113:1896–1903. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiang L, Lai YK, Zhang JF, et al:
Transactivation of the TIEG1 confers growth inhibition of
transforming growth factor-β-susceptible hepatocellular carcinoma
cells. World J Gastroenterol. 18:2035–2042. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Reliene R and Schiestl RH: Antioxidants
suppress lymphoma and increase longevity in Atm-deficient mice. J
Nutr. 137:S229–S232. 2007.
|
|
37
|
Teoh N, Pyakurel P, Dan YY, et al:
Induction of p53 renders ATM-deficient mice refractory to
hepatocarcinogenesis. Gastroenterology. 138:1155–1165. 2010.
View Article : Google Scholar
|
|
38
|
Stracker TH, Roig I, Knobel PA and
Marjanović M: The ATM signaling network in development and disease.
Front Genet. 4:372013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Derynck R and Zhang YE: Smad-dependent and
Smad independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gohla G, Krieglstein K and Spittau B:
Tieg3/Klf11 induces apoptosis in OLI-neu cells and enhances the
TGF-beta signaling pathway by transcriptional repression of Smad7.
J Cell Biochem. 104:850–861. 2008. View Article : Google Scholar : PubMed/NCBI
|