Introduction
Colorectal cancer (CRC) is the fourth most fatal
cancer in Korea (1). Although the
5-year survival rate of CRC overall has been reported to be as high
as 71.3% (1), the rate for patients
with recurrence is <40% (2).
Epithelial-mesenchymal transition (EMT) is critical for
carcinogenesis, tumor invasion, metastasis, chemo-resistance and
acquisition of stem cell properties. The EMT phenotype is
indicative of a poor prognosis in CRC (3,4).
Combined treatment to attenuate or reverse EMT enhances the
sensitivity to conventional anti-neoplastic agents and improves
oncologic outcomes (5).
Both genetic and epigenetic changes are required for
the transformation, promotion and progression of CRC (6). Downregulation of tumor-suppressor
genes due to unbalanced histone deacetylase (HDAC) hyperactivity
has been reported in CRC. The histone acetylation status may play a
role in colon tumorigenesis and re-expression of these genes can be
induced by treatment with an HDAC inhibitor in CRC. HDAC inhibitors
have been reported to be effective as anticancer agents and, in
combination with conventional chemotherapeutics, have been reported
to synergistically enhance growth arrest, apoptosis and
differentiation in cancer cells (6). These effects in colon cancer cells
have currently been demonstrated for many HDAC inhibitors including
valproic acid (VPA) (7) and
trichostatin A (TSA) (8–11). TSA, a hydroxamate, may have the
highest potency against HDAC1, 3 and 8. VPA, a short chain fatty
acid, appears to have the highest activity against HDAC1 and 2, but
appears also to affect HDAC3, 4, 5 and 7 at higher doses, showing
non-specific sensitivity to HDACs (12). TSA and VPA reduce the growth and
survival, induce differentiation, enhance radiosensitivity and
reduce chemoresistance (13).
Histone modification has been shown to play a key
role in controlling EMT. Many studies have reported that various
HDAC inhibitors reverse or attenuate EMT through the upregulation
of E-cadherin in different solid tumors such as HCC (14), breast (15,16),
esophageal cancer (17) and ovarian
tumors (18). In addition, they
attenuate TGF-β1-induced EMT in different cells: hepatocytes
(19), retinal pigment epithelial
cells (20), lens epithelial cells
(21) and renal epithelial cells
(22). These results suggest that
HDAC inhibitors may have therapeutic roles to reduce EMT. However,
recent studies have also reported that HDAC inhibitors induce EMT,
reverse stem cell properties and enhance metastasis and invasion in
prostate cancer (23), head and
neck squamous cell carcinoma (24)
and nasopharyngeal carcinoma (25).
Whether HDAC inhibitors inhibit or induce EMT in CRC has not yet
been reported, and needs to be clarified.
We investigated the effect on the EMT of colon
cancer cells induced by the HDAC inhibitors TSA or VPA, alone, and
in combination with TGF-β1. TGF-β1 induced the EMT of colon cancer
cells in DNA microsatellite stable (MSS) cells, whereas cells with
DNA microsatellite instability (MSI) showed variable responses to
TGF-β1 (26). Tumor cells with MSI
having mutant TGF-β receptor type II (TGFBR2) exhibited less EMT
and a more favorable prognosis compared to cells with MSS. We
selected four CRC cell lines including MSI cells (DLD1 and HCT116)
and MSS cells (HT29 and SW480). We evaluated the expression of
E-cadherin as an epithelial marker, and vimentin as a mesenchymal
marker. The EMT phenotype was also evaluated by cell morphology,
migration and invasion assays.
Materials and methods
Cell lines and culture conditions
We used four colon carcinoma cell lines. DLD1, HT29,
HCT116 and SW480 were purchased from the Korean Cell Line Bank
(KCLB, Seoul, Korea). These cells were maintained in Dulbecco’s
modified Eagle’s medium (DMEM), containing 10% heat inactivated
fetal bovine serum (FBS), potassium penicillin 100 U/ml,
streptomycin 100 g/ml, 2 mM glutamine and 20 mM sodium bicarbonate.
The cells were incubated in 5% CO2 and 95% humidity in a
37°C chamber. The growth medium was changed every 3 days.
Western blot analysis
Total protein was extracted, and the protein
concentration was measured by the Bradford DC protein assay
(Bio-Rad Laboratories, Hercules, CA, USA). Then, 20–40 μg
protein from each sample was separated on 12% Bis-Tris
polyacrylamide gel through electrophoresis and blotted on immobilon
transfer membranes (Millipore, Temecula, CA, USA). Blots were
immunostained with vimentin (1:1,000; Sigma-Aldrich, St. Louis, MO,
USA) and E-cadherin (1:1,000; Invitrogen, Eugene, OR, USA). The
primary antibody was added at 4°C overnight, and then the secondary
antibody at room temperature for 1 h. Proteins were visualized by
using ECL Plus Western Blotting detection reagents (Intron
Biotechnology, Seoul, Korea) and measured by image analysis of the
Fuji Image Quant software (FujiFilm Las-3000 mini, Tokyo, Japan),
according to the manufacturer’s instructions.
Quantitative real-time RT-PCR
The total RNA was isolated using the RNase Mini kit
(Qiagen, Valencia, CA, USA). The residual DNA was removed using an
RNase Free NDase kit (Qiagen). High capacity RNA-to-cDNA kit
(Applied Biosystems, Foster City, CA, USA) was used to reverse
transcribe 1 μg of RNA into cDNA according to the
manufacturer’s instructions. Real-time PCR was performed using
specific primers to quantify gene expression using SYBR-Green RT
PCR reagents (Applied Biosystems). The relative amount of mRNA was
normalized to the expression of GAPDH. The primer sequences used in
the present study were as follows; GAPDH primer sequences: forward,
5′-CATCAATGGAAA TCCCATCA-3′ and reverse, 5′-TTCTCCATGGTGGTGAA
GAC-3′; E-cadherin primer pair: forward, 5′-TTCTGCTGC
TCTTGCTGTTT-3′ and reverse, 5′-TGGCTCAAGTCAAA GTCCTG-3; Vimentin
primer pair: forward, 5′-GCCCTTA AAGGAACCAATGA-3′ and reverse,
5′-AGCTTCAACGG CAAAGTTCT-3′. The PCR reactions were repeated three
times in three independent experiments.
Immunofluorescence microscopy
Individual sterile coverslips were placed in the
wells of a 4-well plate and colon carcinoma cells were added and
incubated for 24 h. Subsequently, the cells were treated with 10
ng/ml TGF-β1, 100 or 400 nM TSA, 0.5 mM VPA diluted in DMEM media
with 1% FBS for 48 h. After washing with PBS, the cells were fixed
in 3.7% paraformaldehyde for 20 min at room temperature. The
paraformaldehyde was removed and cells were washed with PBS.
Blocking solution was then added for 30 min to prevent nonspecific
binding, and then incubation was carried out using anti-E-cadherin
(1:100; Cell Signaling Technology, Danvers, MA, USA) and
anti-vimentin (1:100; Leica Microsystems, Wetzlar, Germany) as the
primary antibodies, overnight, at 4°C, on a rocking platform. The
washed slides were incubated for 1 h at room temperature with 1:100
dilutions of Alexa-488 anti-rabbit IgG (H+L) (Molecular Probes
Inc., Eugene, OR) secondary antibody, and Alexa-568 goat anti-mouse
IgG(H+L) (Molecular Probes) secondary antibody. They were washed
again, mounted with Vectashield mounting medium (Dako, Carpinteria,
CA, USA), and examined using Leica Zeiss optics in the Core
Facility of Chungbuk National University. We also captured
immunofluorescence images at time-points of 6, 12, 24 and 48 h to
evaluate the protein translocalization.
Cell invasion and migration assays
We evaluated cell invasion and migration ability in
the MSI or MSS cell lines. DLD1 and SW480 were selected. DLD1 and
SW480 cells were seeded in 96-well plates over a homogeneous thin
layer of fibronectin (BD Biosciences, Bedford, MA, USA) in a
Millicell cell culture insert (Millipore) that contained
polycarbonate filter membranes with 8-mm diameter pores. Tumor
cells from the control group were maintained in DMEM supplemented
with 1% FBS and 1% antibiotics, and the HDAC inhibitor group
received 100 nM of TSA or 0.5 mM VPA diluted in media. The lower
chamber contained DMEM supplemented with 10% FBS and 1%
antibiotics. After plating, the cells were incubated for 24 h at
37°C in a 5% CO2-humidified incubator. Invasive cells in
the lower chamber were stained with hematoxylin and eosin
(H&E). Images were captured using a QImaging Exi Aqua
monochrome digital camera attached to a Nikon Eclipse 80i
microscope (Nikon Inc., Melville, NY, USA) and visual-ized using
QCapture Pro software.
The cells were allowed to grow in 10% FBS containing
DMEM to confluency in a 6-well plate. A central linear wound was
made with a 200-ml sterile pipette tip and then the cells were
washed with PBS twice. Afterwards, the HDAC inhibitor group
received 100 nM of TSA or 0.5 mM VPA and 10 ng/ml TGF-β1 diluted in
media with 1% FBS. Plates were photographed after 0, 24 and 48
h.
Cytotoxicity assay
Cell viability was assessed using Promega cell
proliferation MTS assay (Promega, Madison, WI, USA). Briefly, CRC
cells were seeded in 96-well plates at a density of
1×103/well and incubated with increasing concentrations
of TSA, VPA and TGF-β1 for 48 h to determine a dose-response curve.
Subsequently, dead cells were washed away, the attached cells were
incubated with MTS, and cell viability was detected using
microplate reader Model-680 (Bio-Rad Laboratories). All the
experiments were conducted in triplicate.
Statistical analysis
The SPSS 16.0 software (IMB, Armonk, NY, USA) was
used to analyze the data. The differences between groups were
evaluated using the Mann-Whitney U test and Wilcoxon signed-rank
test, with P<0.05 being considered to indicate a statistically
significant result.
Results
Cell morphology
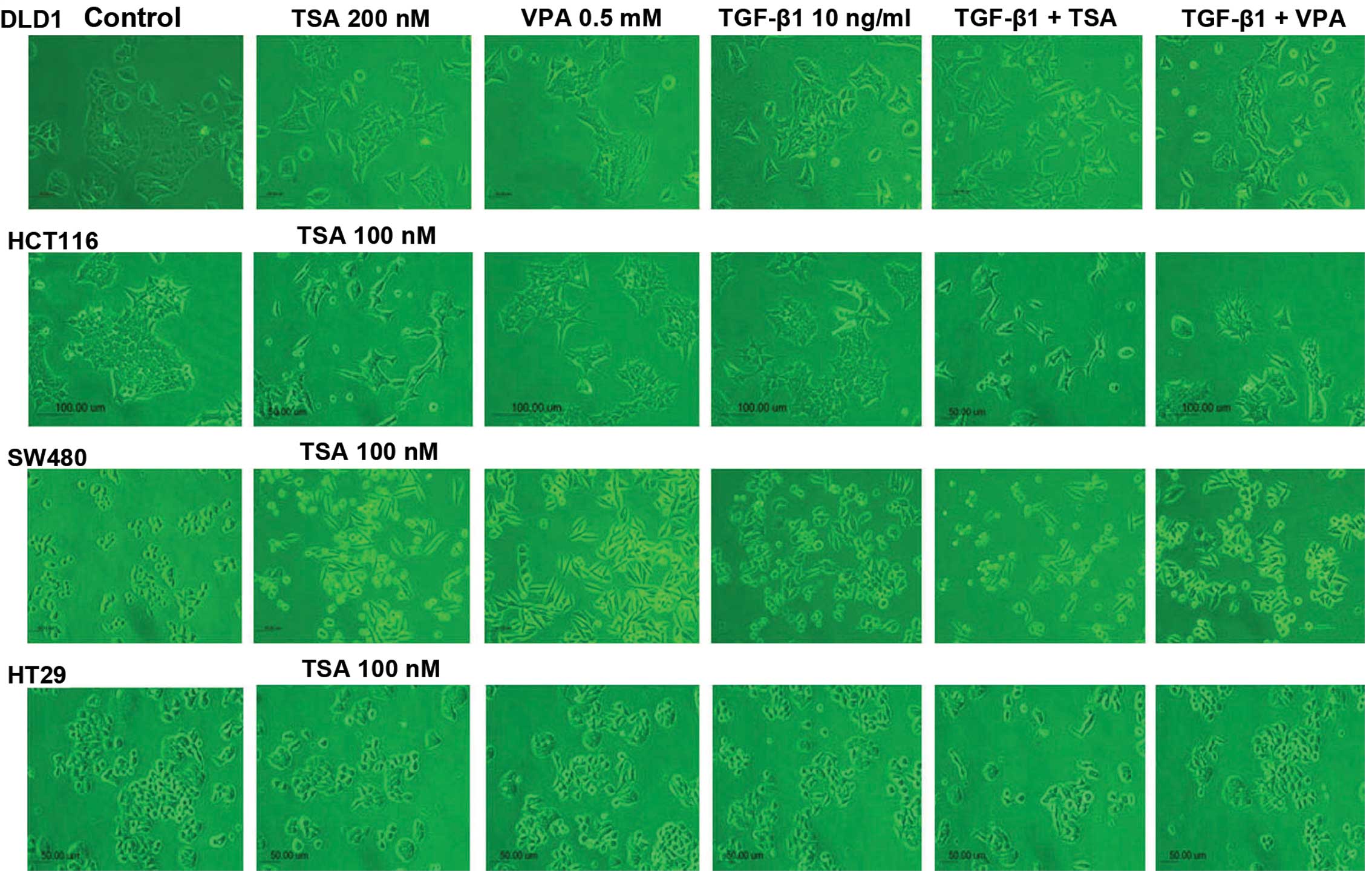
The four CRC cell lines were altered from round or
rectangular-shaped cells to spindle-shaped cells with loose
cell-cell contact following TSA or VPA treatment or TGF-β1
treatment. The response to TGF-β1 treatment in the DLD1 and HCT116
cells was not prominent compared to the SW480 and HT29 cells.
However, the morphological changes induced by TSA and VPA treatment
were similar in both the MSS and MSI cell lines. Morphological
changes were most prominent in the SW480 cells, which were
distributed as single round cells. However, the HT29 cells showed
minor changes compared to the SW480 cells. DLD1 and HCT116 cells
showed a wider gap between cells and were spindle-shaped. The
morphological changes were similar following TSA or VPA single
treatment or in combination with TGF-β1 treatment (Fig. 1).
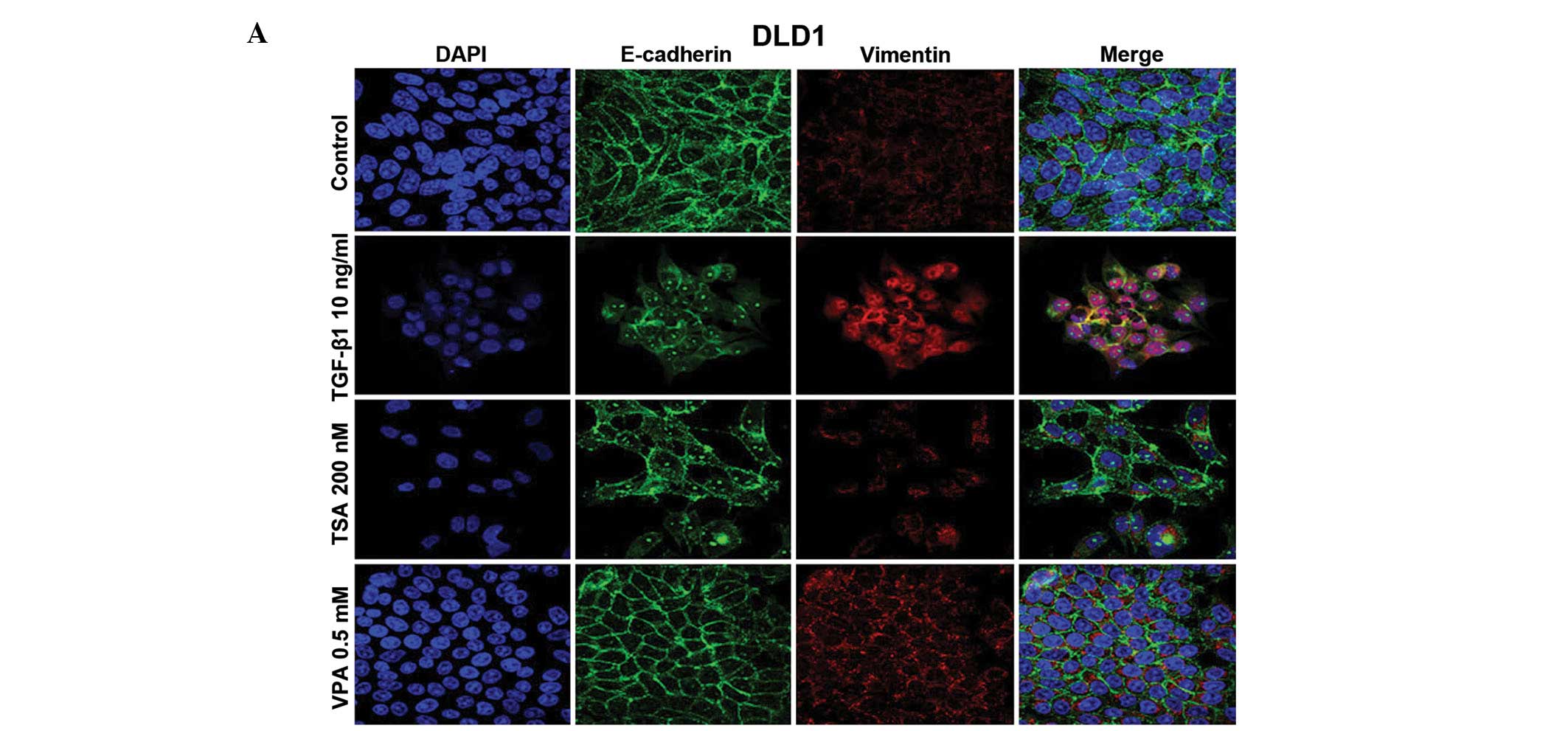
Nuclear localization of E-cadherin and
increased expression of vimentin in the colon cancer cells
The four CRC cell lines showed attenuation of
membrane expression and nuclear translocation of E-cadherin and/or
increased vimentin expression following TSA and VPA treatment in
both the MSI and MSS cell lines (Fig.
2A–D). The effects of TSA and VPA treatment on the DLD1 cells
differed; TSA induced changes in E-cadherin expression, whereas VPA
had effects on vimentin expression. TGF-β1 induced a mesenchymal
phenotype in the MSS cells (HT29 and SW480), whereas TGF-β1
treatment in MSI cells (DLD1 and HCT116) had variable results; a
mesenchymal phenotype in DLD1 cells was noted and increased
membrane expression of E-cadherin in the HCT116 cells was observed.
Nuclear localization of E-cadherin and increased expression of
vimentin occurred after 6 h and increased until 24 h in the HT29
cells (Fig. 2E).
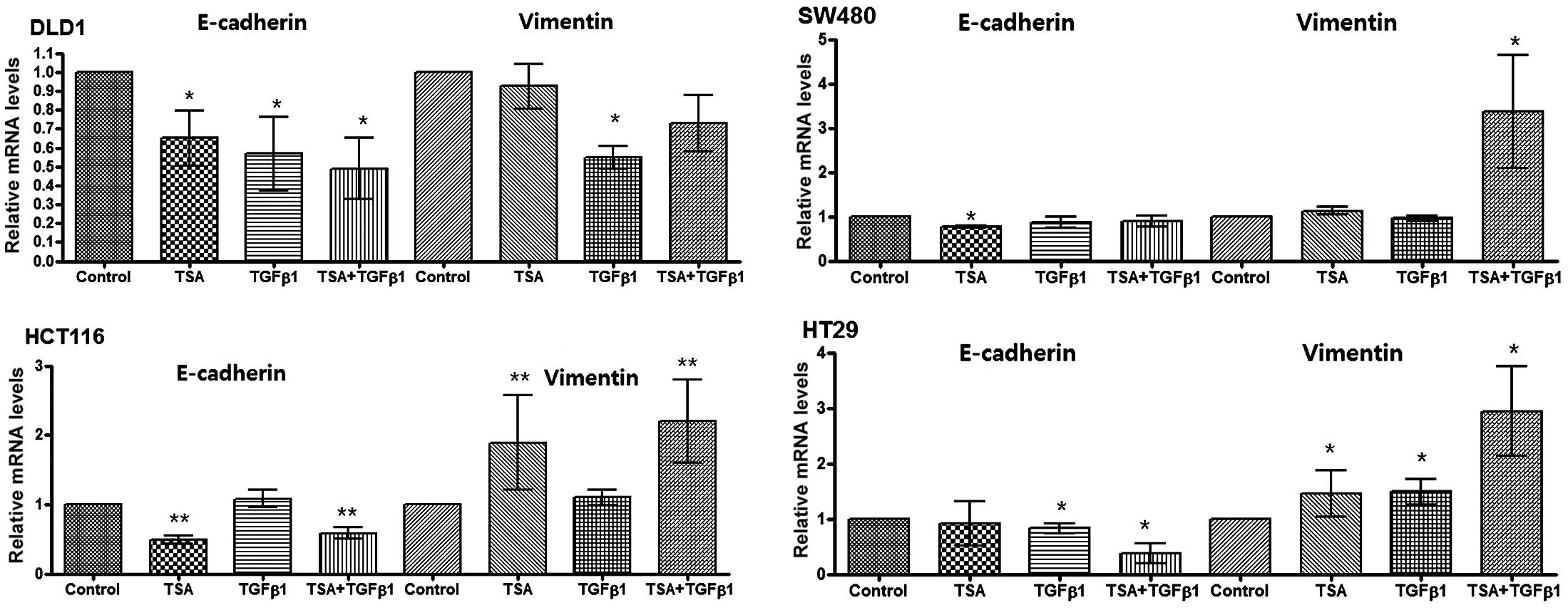
mRNA expression of E-cadherin and
vimentin in the colon cancer cell lines
The four colon cancer cell lines showed decreased
E-cadherin or increased vimentin expression following TSA or TGF-β1
treatment. These effects were greater in the presence of TGF-β1.
However, the mRNA expression among the four cell lines showed
slight differences (Fig. 3).
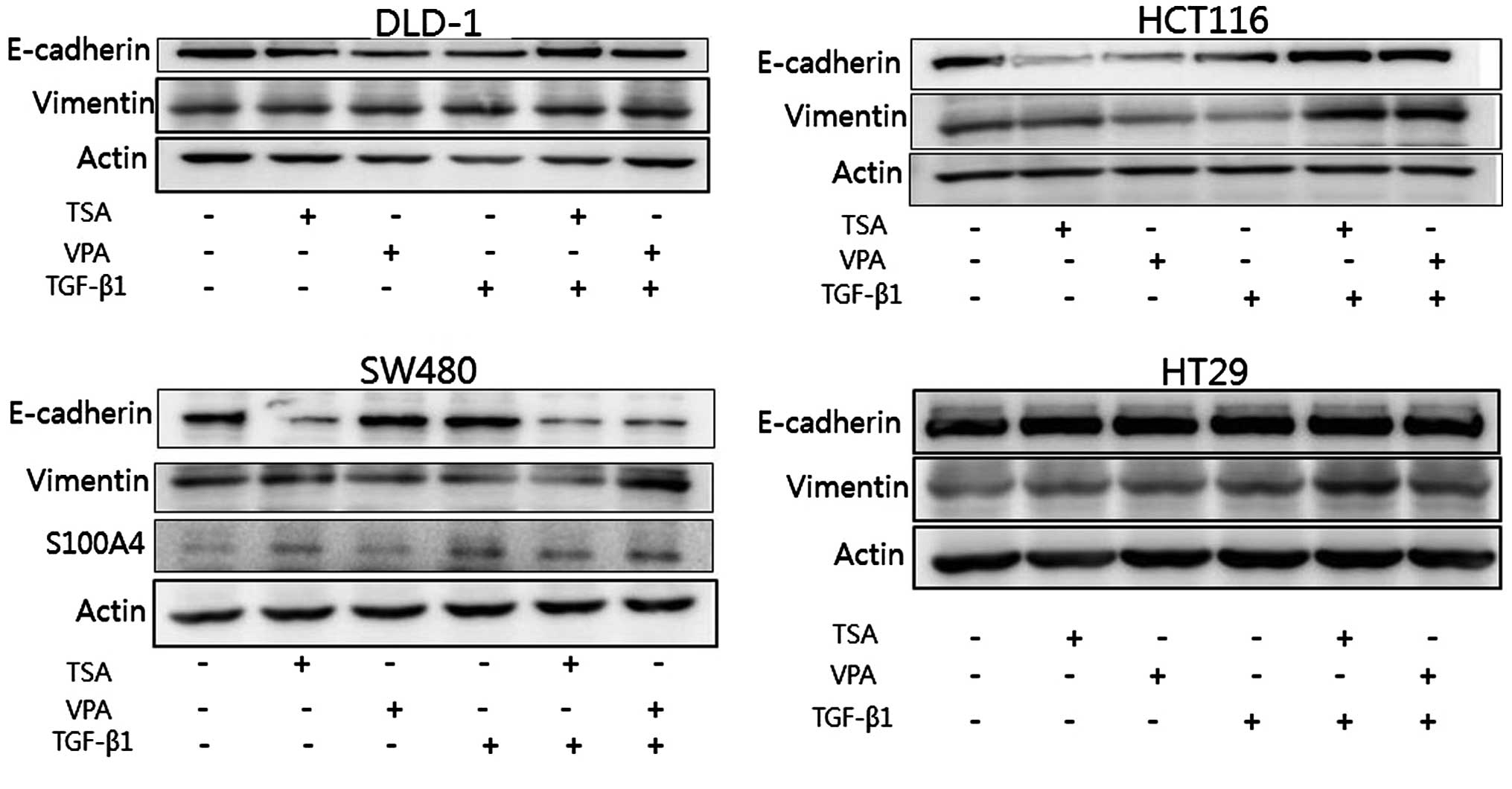
Protein expression of E-cadherin and
vimentin in the colon carcinoma cells
After treatment with HDAC inhibitors, a decrease in
E-cadherin and an increase in S100A4 and vimentin expression were
noted in the SW480 cells. These changes were enhanced by
co-treatment with TGF-β1. A decrease in E-cadherin expression was
demonstrated following TSA or VPA treatment in the HCT116 and DLD1
cells. Vimentin was increased by co-treatment with the HDAC
inhibitors and with TGF-β1 in the four cell lines (Fig. 4).
Migration and invasion assays
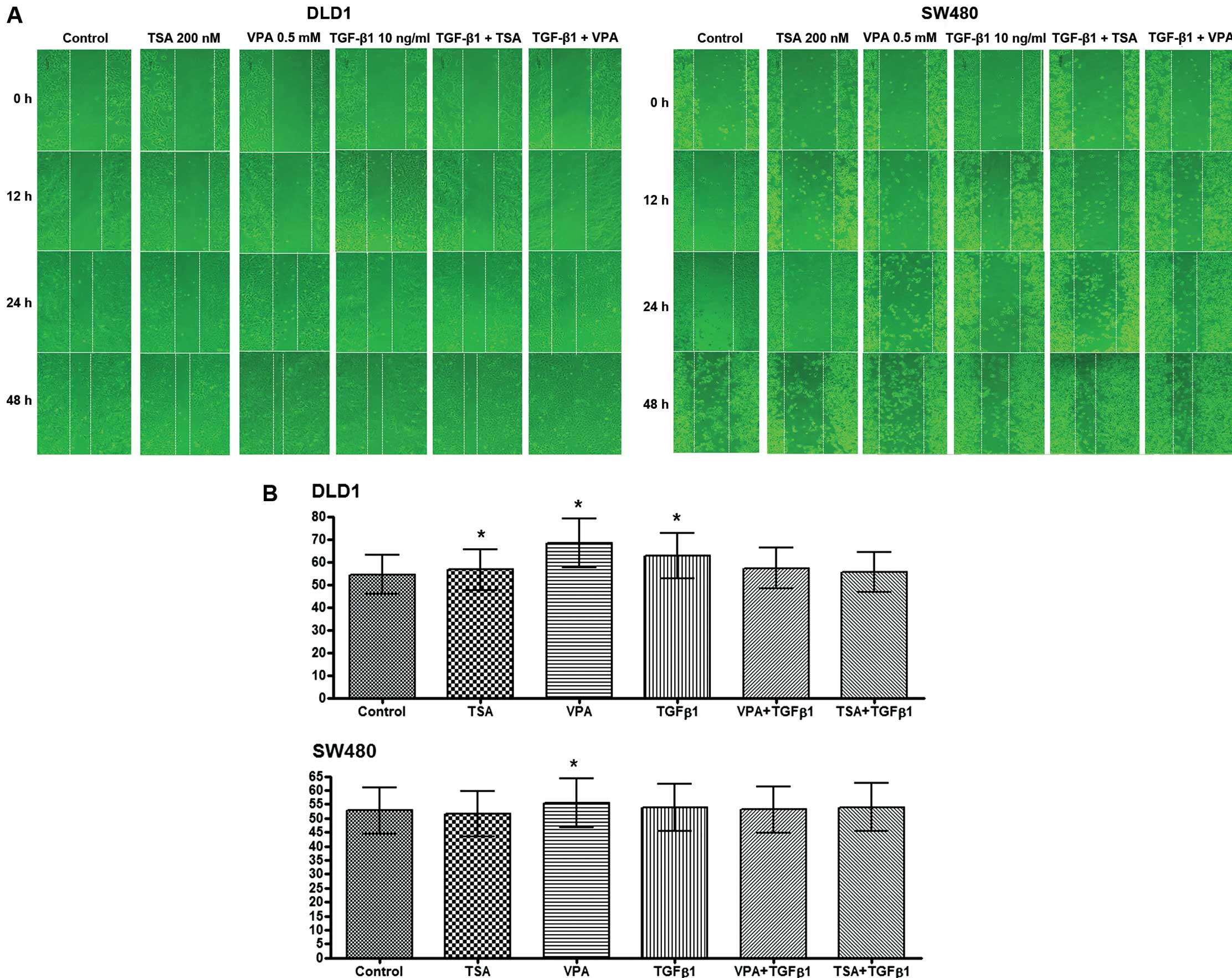
Single treatment with TSA or VPA did not show a
difference in effects compared to the controls. However, the
migratory ability increased following VPA and TGF-β1 treatment in
the DLD1 cells and it increased following the TSA or VPA treatment
combined with TGF-β1 in the SW480 cells (Fig. 5A). These results demonstrated that
EMT was enhanced following TSA or VPA treatment in combination with
TGF-β1.
Invasion was increased by TSA, VPA or TGF-β1
treatment compared to no treatment in the DLD1 cells (P=0.002,
P=0.002 and P=0.001, respectively). However, invasive ability did
not increase following co-treatment with HDAC inhibitors and TGF-β1
in the DLD1 cells. Invasion was increased following VPA treatment
only when compared to no treatment in the SW480 cells (P=0.039)
(Fig. 5B).
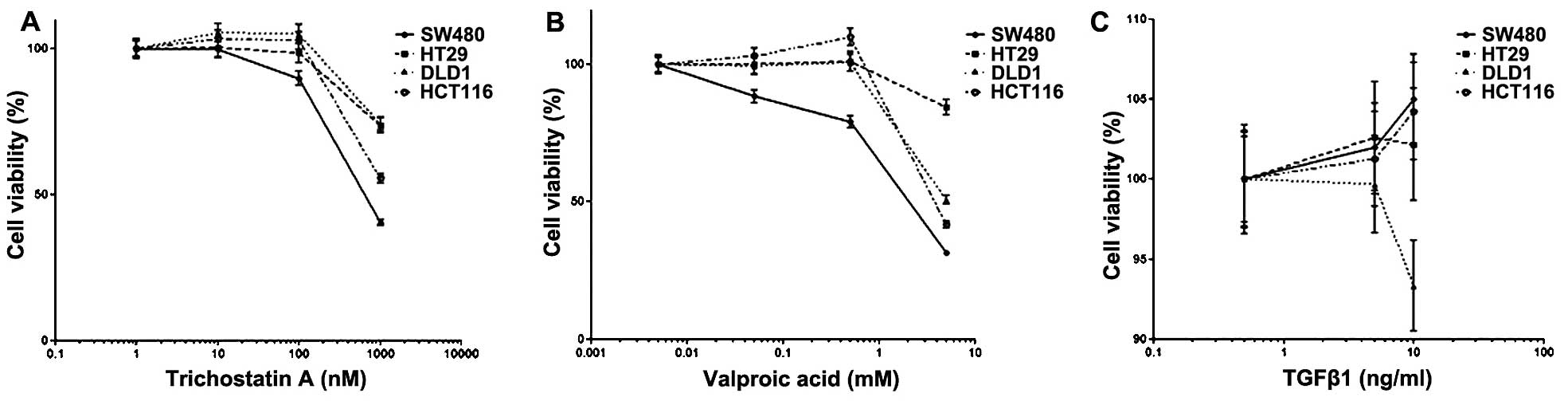
Cell proliferation
Cell proliferation of colon carcinoma cells was
assessed following treatment with TSA, VPA or TGF-β1 (Fig. 6). TSA (100 nM) or VPA (0.5 mM) did
not influence cell proliferation except in the SW480 cells, which
exhibited slightly suppressed cell proliferation. TGF-β1 (5 ng/ml)
did not have an influence on cell proliferation. However, TGF-β1
(10 ng/ml) enhanced cell proliferation compared to the control
except for DLD1 cells, which showed suppressed cell
proliferation.
Discussion
The present study demonstrated that HDAC inhibitors
TSA and VPA induced EMT, which was enhanced following TGF-β1
co-treatment in the colon cancer cells. The mesenchymal phenotype
was determined by reduced expression and nuclear translocation of
E-cadherin, and decreased expression of vimentin in the MSS (SW480
and HT29) and MSI (DLD1 and HCT116) cells with variable
susceptibility. TSA or VPA also increased cell migration and
invasion ability.
EMT is a biological process that is encountered in
primary mesenchyme, wound healing and carcinoma (27). A hallmark of EMT is the
disintegration and disassembly of cell-cell junctions by loss of
proteins associated with the polarized epithelial phenotype, which
normally interacts with the basement membrane via its basal
surface, initiating invasive and metastatic behavior. EMT is
characterized by loss of proteins, including E-cadherin, ZO-1 and
tight junctional molecules, whereas it is characterized by
upregulation of vimentin, which is a biomarker of the mesenchymal
phenotype.
Many studies have reported that HDAC inhibitors
reverse or attenuate EMT through upregulation of E-cadherin in
different solid tumors and attenuate TGF-β-induced EMT in normal
epithelial cells. They suggest that HDAC inhibitors may have
therapeutic roles in solid cancers and fibrotic disorders by
inhibition of EMT.
However, several recent studies reported opposite
results that HDAC inhibitors can induce EMT. HDAC inhibitors TSA
and vorinostat (suberoylanilidehydroxamic acid, SAHA) were found to
induce the EMT phenotype, increasing mesenchymal markers such as
vimentin and N-cadherin, concomitant with an increase in the
expression of Sox2 and Nanog, which is associated with cancer
stem-cell characteristics in prostate cancer cells (23). They suggest that the mechanisms of
EMT induction by HDAC inhibitors are through the activation of
promoters of Snail, Slug and Twist1. Inhibition of HDAC by TSA also
induced EMT in head and neck squamous cell carcinomas, accumulation
of BMI-1, an oncogene associated with tumor aggressiveness, and
expression of vimentin (24).
Another study showed that HDAC inhibitors induced fibroblast-like
morphology, upregulated Snail and vimentin and downregulated
E-cadherin in nasopharyngeal carcinoma cells following sodium
butyrate (NaB) or SAHA treatment (25). HDAC inhibitor TSA enhanced vimentin
gene expression requiring the proximal promoter region including
GC-box1, a known Sp1/Sp3 binding site (28). A recent study revealed that SAHA
induced EMT during human embryo implantation (29).
The different results in regards to the EMT response
to HDAC inhibitor treatment may be related to the balance of gene
expression, which includes EMT inducers and EMT suppressors.
Studies with cDNA arrays have shown that treatment with HDAC
inhibitors leads to a 2-fold or greater change in the expression of
~7–10% of the genes examined (30).
HDAC inhibitor was found to induce about as many genes as were
repressed. Whether or not these changes in gene expression result
in cell death probably depends upon the cellular context. In
addition, the gene expression pattern caused by HDACs on tumors is
relatively non-specific (12),
which suggests that individual HDAC inhibitors show differing
degrees of effectiveness in their action on EMT, even in the same
cell type.
The present study found that TSA and VPA induced
EMT-like phenotypes in all the CRC cell lines irrespective of MSI,
mutation of TGFBR2, K-RAS or APC genes, and tumor aggressiveness.
HCT116 cells have wild-type APC and higher malignant potential.
DLD1, HT29 and SW480 cells have mutated APC and mutated P53. All
cell lines, except HT29, also carry activating mutations in the
KRAS gene. Malignant potential was also different in the four CRC
cell lines; DLD-1, HT29, HCT116 and SW480, in ascending order
(31). The changes in EMT phenotype
induced by TGF-β1 and HDAC inhibitors were variable according to
the different cell type. Mutant or wild-type TGFBR2, APC, and K-RAS
proteins were different in the four CRC cell lines. EMT induction
by TGF-β1 was greater in the MSS (SW480 and HT29) cells than that
in the MSI (DLD1 and HCT116) cells.
The expression of E-cadherin, which suppresses EMT,
may prevent invasiveness and metastasis. However, recent studies
showed that the shift of E-cadherin localization from the membrane
to the cytoplasm or nucleus in CRC was associated with metastasis
and poor outcomes in human tissues (32) and animal models (33). A colonic cancer metastasis model
showed that E-cadherin proteolysis and nuclear localization were
associated with aggressive growth foci in the peritoneal
microenvironment. The localization of E-cadherin was also
demonstrated in various tumors including Merkel cell carcinoma
(34), stomach (35), renal (36), esophageal (37) and solid pseudo-papillary tumors of
the pancreas (38). However, most
reports of nuclear localization of E-cadherin in cancer concerned
studies that utilized immunohistochemistry only, which must be
interpreted with caution (39). The
mechanism by which the E-cadherin molecule is translocated to the
nucleus is currently not clear. The present study showed that TSA
and VPA induced nuclear translocation of E-cadherin in the four CRC
cell lines with preserved or reduced membrane expression. We
suggest that the most important E-cadherin change during EMT
process was not the mRNA or protein amounts but the location. The
amount of E-cadherin reduction following treatment of HDAC
inhibitors was not enough to reach statistical analysis in several
cell lines in the present study. A similar result was noted in the
previous study, which showed that SAHA treatment in breast cancer
cells induced redistribution of E-cadherin from the cell surface to
the cytoplasm while preserving total E-cadherin levels (40).
TSA was previously found to prevent TGF-β (5
ng/ml)-induced EMT in a concentration-dependent manner in human
renal epithelial cells (22) and in
mouse hepatocytes (41). TSA
treatment induced a fundamental increase in the mRNA levels of ZO-1
and E-cadherin in a concentration-dependent manner, which started
to be expressed at doses of 20 ng/ml of TSA and was most effective
at the dose of 400 nM in mouse hepatocytes (41). Cell cytotoxicity and apoptosis were
induced at higher doses (800 nM), and had no effect at lower doses
(200 nM) (19). These results
suggest that the effect of TSA on EMT begins at a low dosage, prior
to apoptosis and inhibition of proliferation, although the dosage
differs according to cell type and cancer progression. We used TSA
(100 or 200 nM) at a level that induces mild suppression of cell
proliferation while preserving cell viability. The concentration of
VPA was fixed at 0.5 mM, which is in the range of low toxicity in
the human (42).
Pharmacological inhibitors of class I and II HDAC
activity (HDAC inhibitors) are potent inducers of growth arrest,
differentiation and apoptosis of colon cancer cells in vitro
and in vivo. Studies of HDAC inhibitors on colon cancer
cells have attempted to identify the improvements in response, and
potential efficacy with conventional chemotherapeutics and their
tolerable cytotoxicity. Many studies have reported that HDAC
inhibitors have an effective action against colon cancer cells with
a synergistic effect together with radiation or targeted therapies
(6). VPA and TSA inhibit
proliferation in colon cancer cells and have been shown to have far
greater selective toxicity to tumor cells (43). However, the present study
demonstrated that HDAC inhibitor TSA or VPA can induce the EMT of
CRC cells, alone or in combination with TGF-β1. These results
should be considered when using combination treatment of HDAC
inhibitors and other chemotherapies.
Acknowledgments
The present study was supported by a grant from the
National R&D Program for Cancer Control, Ministry of Health and
Welfare, Republic of Korea (1120330) and a National Research
Foundation of Korea grant funded by the Korea government (NRF-2013R
1A 1A 2063994).
References
|
1
|
Jung KW, Park S, Kong HJ, Won YJ, Lee JY,
Seo HG and Lee JS: Cancer statistics in Korea: incidence,
mortality, survival, and prevalence in 2009. Cancer Res Treat.
44:11–24. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tsai HL, Chu KS, Huang YH, Su YC, Wu JY,
Kuo CH, Chen CW and Wang JY: Predictive factors of early relapse in
UICC stage I-III colorectal cancer patients after curative
resection. J Surg Oncol. 100:736–743. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee SJ, Choi SY, Kim WJ, Ji M, Lee TG, Son
BR, Yoon SM, Sung R, Lee EJ, Youn SJ and Park SM: Combined aberrant
expression of E-cadherin and S100A4, but not β-catenin is
associated with disease-free survival and overall survival in
colorectal cancer patients. Diagn Pathol. 8:992013. View Article : Google Scholar
|
|
4
|
He X, Chen Z, Jia M and Zhao X:
Downregulated E-cadherin expression indicates worse prognosis in
Asian patients with colorectal cancer: evidence from meta-analysis.
PLoS One. 8:e708582013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Findlay VJ, Wang C, Watson DK and Camp ER:
Epithelial-to-mesenchymal transition and the cancer stem cell
phenotype: insights from cancer biology with therapeutic
implications for colorectal cancer. Cancer Gene Ther. 21:181–187.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mariadason JM: HDACs and HDAC inhibitors
in colon cancer. Epigenetics. 3:28–37. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen X, Wong P, Radany E and Wong JY: HDAC
inhibitor, valproic acid, induces p53-dependent radiosensitization
of colon cancer cells. Cancer Biother Radiopharm. 24:689–699. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Meng J, Zhang HH, Zhou CX, Li C, Zhang F
and Mei QB: The histone deacetylase inhibitor trichostatin A
induces cell cycle arrest and apoptosis in colorectal cancer cells
via p53-dependent and -independent pathways. Oncol Rep. 28:384–388.
2012.PubMed/NCBI
|
|
9
|
Hsu YF, Sheu JR, Lin CH, Yang DS, Hsiao G,
Ou G, Chiu PT, Huang YH, Kuo WH and Hsu MJ: Trichostatin A and
sirtinol suppressed survivin expression through AMPK and p38MAPK in
HT29 colon cancer cells. Biochim Biophys Acta. 1820:104–115. 2012.
View Article : Google Scholar
|
|
10
|
Xiong H, Du W, Zhang YJ, Hong J, Su WY,
Tang JT, Wang YC, Lu R and Fang JY: Trichostatin A, a histone
deacetylase inhibitor, suppresses JAK2/STAT3 signaling via inducing
the promoter-associated histone acetylation of SOCS1 and SOCS3 in
human colorectal cancer cells. Mol Carcinog. 51:174–184. 2012.
View Article : Google Scholar
|
|
11
|
Habold C, Poehlmann A, Bajbouj K, Hartig
R, Korkmaz KS, Roessner A and Schneider-Stock R: Trichostatin A
causes p53 to switch oxidative-damaged colorectal cancer cells from
cell cycle arrest into apoptosis. J Cell Mol Med. 12:607–621. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Marchion D and Munster P: Development of
histone deacetylase inhibitors for cancer treatment. Expert Rev
Anticancer Ther. 7:583–598. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mologni L, Cleris L, Magistroni V, Piazza
R, Boschelli F, Formelli F and Gambacorti-Passerini C: Valproic
acid enhances bosutinib cytotoxicity in colon cancer cells. Int J
Cancer. 124:1990–1996. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
DI Fazio P, Montalbano R, Quint K, Alinger
B, Kemmerling R, Kiesslich T, Ocker M and Neureiter D: The
pan-deacetylase inhibitor panobinostat modulates the expression of
epithelial-mesenchymal transition markers in hepatocellular
carcinoma models. Oncol Lett. 5:127–134. 2013.
|
|
15
|
Shah P, Gau Y and Sabnis G: Histone
deacetylase inhibitor entinostat reverses epithelial to mesenchymal
transition of breast cancer cells by reversing the repression of
E-cadherin. Breast Cancer Res Treat. 143:99–111. 2014. View Article : Google Scholar
|
|
16
|
Srivastava RK, Kurzrock R and Shankar S:
MS-275 sensitizes TRAIL-resistant breast cancer cells, inhibits
angiogenesis and metastasis, and reverses epithelial-mesenchymal
transition in vivo. Mol Cancer Ther. 9:3254–3266. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Taylor MD, Liu Y, Nagji AS, Theodosakis N
and Jones DR: Combined proteasome and histone deacetylase
inhibition attenuates epithelial-mesenchymal transition through
E-cadherin in esophageal cancer cells. J Thorac Cardiovasc Surg.
139:1224–1232. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Meng F, Sun G, Zhong M, Yu Y and Brewer
MA: Anticancer efficacy of cisplatin and trichostatin A or
5-aza-2′-deoxycytidine on ovarian cancer. Br J Cancer. 108:579–586.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kaimori A, Potter JJ, Choti M, Ding Z,
Mezey E and Koteish AA: Histone deacetylase inhibition suppresses
the transforming growth factor beta1-induced
epithelial-to-mesenchymal transition in hepatocytes. Hepatology.
52:1033–1045. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xiao W, Chen X, Liu X, Luo L, Ye S and Liu
Y: Trichostatin A, a histone deacetylase inhibitor, suppresses
proliferation and epithelial-mesenchymal transition in retinal
pigment epithelium cells. J Cell Mol Med. 18:646–655. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen X, Xiao W, Chen W, Luo L, Ye S and
Liu Y: The epigenetic modifier trichostatin A, a histone
deacetylase inhibitor, suppresses proliferation and
epithelial-mesenchymal transition of lens epithelial cells. Cell
Death Dis. 4:e8842013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yoshikawa M, Hishikawa K, Marumo T and
Fujita T: Inhibition of histone deacetylase activity suppresses
epithelial-to-mesenchymal transition induced by TGF-beta1 in human
renal epithelial cells. J Am Soc Nephrol. 18:58–65. 2007.
View Article : Google Scholar
|
|
23
|
Kong D, Ahmad A, Bao B, Li Y, Banerjee S
and Sarkar FH: Histone deacetylase inhibitors induce
epithelial-to-mesenchymal tranasition in prostate cancer cells.
PLoS One. 7:e450452012. View Article : Google Scholar
|
|
24
|
Giudice FS, Pinto DS Jr, Nör JE, Squarize
CH and Castilho RM: Inhibition of histone deacetylase impacts
cancer stem cells and induces epithelial-mesenchyme transition of
head and neck cancer. PLoS One. 8:e586722013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jiang GM, Wang HS, Zhang F, Zhang KS, Liu
ZC, Fang R, Wang H, Cai SH and Du J: Histone deacetylase inhibitor
induction of epithelial-mesenchymal transitions via up-regulation
of Snail facilitates cancer progression. Biochim Biophys Acta.
1833:663–671. 2013. View Article : Google Scholar
|
|
26
|
Pino MS, Kikuchi H, Zeng M, Herraiz MT,
Sperduti I, Berger D, Park DY, Iafrate AJ, Zukerberg LR and Chung
DC: Epithelial to mesenchymal transition is impaired in colon
cancer cells with microsatellite instability. Gastroenterology.
138:1406–1417. 2010. View Article : Google Scholar :
|
|
27
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu Y, Zhang X, Salmon M and Zehner ZE: The
zinc finger repressor, ZBP-89, recruits histone deacetylase 1 to
repress vimentin gene expression. Genes Cells. 12:905–918. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Uchida H, Maruyama T, Nishikawa-Uchida S,
Oda H, Miyazaki K, Yamasaki A and Yoshimura Y: Studies using an in
vitro model show evidence of involvement of epithelial-mesenchymal
transition of human endometrial epithelial cells in human embryo
implantation. J Biol Chem. 287:4441–4450. 2012. View Article : Google Scholar :
|
|
30
|
Xu WS, Parmigiani RB and Marks PA: Histone
deacetylase inhibitors: molecular mechanisms of action. Oncogene.
26:5541–5552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gujral TS and MacBeath G: A system-wide
investigation of the dynamics of Wnt signaling reveals novel phases
of transcriptional regulation. PLoS One. 5:e100242010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Elzagheid A, Algars A, Bendardaf R, Lamlum
H, Ristamaki R, Collan Y, Syrjanen K and Pyrhonen S: E-cadherin
expression pattern in primary colorectal carcinomas and their
metastases reflects disease outcome. World J Gastroenterol.
12:4304–4309. 2006.PubMed/NCBI
|
|
33
|
Céspedes MV, Larriba MJ, Pavón MA, Alamo
P, Casanova I, Parreño M, Feliu A, Sancho FJ, Muñoz A and Mangues
R: Site-dependent E-cadherin cleavage and nuclear translocation in
metastatic colorectal cancer model. Am J Pathol. 177:2067–2079.
2010. View Article : Google Scholar
|
|
34
|
Han AC, Soler AP, Tang CK, Knudsen KA and
Salazar H: Nuclear localization of E-cadherin expression in Merkel
cell carcinoma. Arch Pathol Lab Med. 124:1147–1151. 2000.PubMed/NCBI
|
|
35
|
Moon KC, Cho SY, Lee HS, Jeon YK, Chung
JH, Jung KC and Chung DH: Distinct expression patterns of
E-cadherin and beta-catenin in signet ring cell carcinoma
components of primary pulmonary adenocarcinoma. Arch Pathol Lab
Med. 130:1320–1325. 2006.PubMed/NCBI
|
|
36
|
Gervais ML, Henry PC, Saravanan A, Burry
TN, Gallie BL, Jewett MA, Hill RP, Evans AJ and Ohh M: Nuclear
E-cadherin and VHL immunoreactivity are prognostic indicators of
clear-cell renal cell carcinoma. Lab Invest. 87:1252–1264. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Salahshor S, Naidoo R, Serra S, Shih W,
Tsao MS, Chetty R and Woodgett JR: Frequent accumulation of nuclear
E-cadherin and alterations in the Wnt signaling pathway in
esophageal squamous cell carcinomas. Mod Pathol. 21:271–281. 2008.
View Article : Google Scholar
|
|
38
|
Serra S, Salahshor S, Fagih M, Niakosari
F, Radhi JM and Chetty R: Nuclear expression of E-cadherin in solid
pseudopapillary tumors of the pancreas. JOP. 8:296–303.
2007.PubMed/NCBI
|
|
39
|
Ordonez NG: Value of E-cadherin and
N-cadherin immunostaining in the diagnosis of mesothelioma. Hum
Pathol. 34:749–755. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Robertson FM, Woodward WA, Pickei R, Ye Z,
Bornmann W, Pal A, Peng Z, Hall CS and Cristofanilli M:
Suberoylanilide hydroxamic acid blocks self-renewal and homotypic
aggregation of inflammatory breast cancer spheroids. Cancer.
116:2760–2767. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lei W, Zhang K, Pan X, Hu Y, Wang D, Yuan
X, Shu G and Song J: Histone deacetylase 1 is required for
transforming growth factor-beta1-induced epithelial-mesenchymal
transition. Int J Biochem Cell Biol. 42:1489–1497. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Manoguerra AS, Erdman AR, Woolf AD, Chyka
PA, Caravati EM, Scharman EJ, Booze LL, Christianson G, Nelson LS,
Cobaugh DJ and Troutman WG: Valproic acid poisoning: an
evidence-based consensus guideline for out-of-hospital management.
Clin Toxicol (Phila). 46:661–676. 2008. View Article : Google Scholar
|
|
43
|
Insinga A, Monestiroli S, Ronzoni S,
Gelmetti V, Marchesi F, Viale A, Altucci L, Nervi C, Minucci S and
Pelicci PG: Inhibitors of histone deacetylases induce
tumor-selective apoptosis through activation of the death receptor
pathway. Nat Med. 11:71–76. 2005. View
Article : Google Scholar
|