Introduction
Cigarette smoking is closely associated with the
development and progression of various respiratory diseases
including lung cancer, chronic obstructive pulmonary disease
(COPD), interstitial lung diseases and bronchial asthma (1,2). In
particular, lung cancer and COPD are the main causes of death
related to cigarette smoking in the world. Particles in cigarette
smoke are reported to induce various lung injuries including
inflammation and fibrosis (3,4).
Studies have shown that Wnt/β-catenin/T-cell factor
(TCF) signaling is activated during lung injury and promotes the
survival and migration of alveolar epithelial cells (5). β-catenin is a key player of canonical
Wnt signaling. The activation of this signaling pathway mainly
depends on the cytoplasmic accumulation and nuclear localization of
β-catenin (6,7). First, a Wnt ligand binds to a
seven-pass transmembrane Frizzled (Fz) receptor as well as its
coreceptor LRP6 or LRP5 (8). The
binding leads to depolymerization of glycogen synthase kinase-3β
(GSK-3β)/APC/Axin complexes in the cytoplasm, β-catenin release and
phosphorylation. It results in the accumulation and stabilization
of cytosolic β-catenin, which then travels to nuclei to form
complexes with members of the DNA-binding family TCF-1/lymphoid
enhancer factors (LEF-1, 3 and 4). Activation of β-catenin/TCF
signaling promotes the downstream target gene transcription
involved in cell proliferation, migration and differentiation.
These targets include cyclin D1, c-Myc and matrix
metalloproteinases (MMP-2, -3, -7, -9 and -13) (9).
RNA-binding motif protein 5 (RBM5, previously
referred to as g15, LUCA-15 or H37) is one of ~35 genes located in
the 370-kb tumor suppressor locus on chromosome 3p21.3 (10). RBM5 is reported to induce cell cycle
arrest and apoptosis by pre-mRNA alternative gene splicing
(11,12). It is also suggested to act as a
tumor-suppressor gene by inhibiting tumor growth and reducing
metastatic potential (11,12). Both the mRNA and protein levels of
RBM5 are significantly lower in non-small cell lung carcinomas
(NSCLCs) than those in normal tissues (13). The RBM5 level is negatively
correlated with the smoking status in patients with lung cancer
(12). In addition to lung cancer,
whether the RBM5 level is also reduced in other cigarette
smoke-induced lung injury has not been determined. Meanwhile, the
activation of the Wnt/β-catenin signaling pathway plays an
important role in diseases associated with cigarette smoking
(5,14,15).
Alveolar epithelial cells are the major components of the airway
epithelium that are directly affected by cigarette smoking. The
molecular mechanisms by which the Wnt/β-catenin signaling pathway
is activated during cigarette smoke-induced lung injury remains
unclear. Whether RBM5 is involved in the activation of
Wnt/β-catenin signaling during lung injury also remains
unknown.
The aims of the present study were to determine the
level of RBM5 in cigarette smoke-injured lung epithelium and the
involvement of RBM5 in the activation of Wnt/β-catenin signaling
during lung injury. A549 cells were treated with cigarette smoke
extract (CSE) at a series of concentrations and for various times.
The present study demonstrated that the level of RBM5 in cigarette
smoke-injured lung epithelium was significantly reduced and that
RBM5 acts as an upstream molecular regulator of Wnt/β-catenin
signaling activity during lung injury.
Materials and methods
Preparation of CSE
CSE was prepared using a popular type of cigarette
in China (Hong Shuang Xi, 12 mg of tar, 1.1 mg of nicotine), as
previously described (16).
Briefly, a syringe-driven apparatus device was designed and
operated to allow a stream of smoke to flow into a tube-shaped
trap, which was maintained at room temperature. The smoke then
entered a 1.5-liter flask submerged in liquid nitrogen. The amount
of smoke obtained was determined by the increase in the weight
inside the flask. The collected smoke particles were dissolved in
dimethyl sulfoxide (DMSO) at 40 mg/ml, and the solution was
sterile-filtered through a 0.22-μm syringe filter
(Millipore, Watford, UK). The CSE solution was prepared by
dissolving the condensate in DMSO, which was then stored in small
vials at −80°C.
Cell culture
The human alveolar epithelial cell line A549 was
obtained from Jilin University. The cells were cultured in
RPMI-1640 supplemented with 10% fetal bovine serum (FBS; Gibco
Waltham, MA, USA) and antibiotics (100 IU/ml penicillin and 100
μg/ml streptomycin) at 37°C in 5% CO2.
MTT assay
The cells (5×103/well) were plated in
96-well microtiter plates. After 24 h of culture in regular medium,
the cells were then cultured in serum-free medium either
supplemented with CSE (5, 10, 15, 20, 40, 60, 80 and 160
μg/ml, respectively) or DMSO for 6, 12, 24, 48 and 72 h,
respectively. Next, 10 μl of
3-(4,5-dimethylthiazol-2yl-)-2,5-diphenyltetrazolium bromide (MTT)
(Sigma-Aldrich, St. Louis, MO, USA) (stock concentration: 5 mg/ml
in phosphate-buffered saline; PBS) was added to each well. After
incubation for 4 h with MTT, 150 μl of DMSO was added. The
plate was agitated for 10 min, and the optical density (OD) value
was measured at 570 nm using a Vmax Microplate Reader
(Molecular Devices, Sunnyvale, CA, USA). Background control wells
containing the same volume of complete culture medium were included
in each assay, and the background was subtracted before data
analysis.
Semi-quantitative reverse
transcription-polymerase chain reaction (RT-PCR)
Cellular total RNA was isolated using TRIzol reagent
(Takara Biotechnology Co., Dalian, China), according to the
manufacturer’s instructions. cDNA was reverse transcribed using a
reverse transcription kit (Takara Biotechnology), according to the
manufacturer’s instructions. Semi-quantitative real-time PCR was
then performed using the following primers: RBM5, forward,
5′-ACACGATGGAT GGAAGCCA-3′ and reverse, 5′-TCTGCTCTGCCTCTGACTT-3′;
glyceraldehyde-3-phosphate dehydrogenase (GAPDH), forward,
5′-GGGTGATGCTGGTGCTGAGTATGT-3′ and reverse,
5′-AAGAATGGGAGTTGCTGTTGAAGTC-3′. The primer oligonucleotides were
synthesized by Shanghai Promega Biological Products, Ltd.
(Shanghai, China). For each set of PCRs, 5 μl of
reverse-transcribed RNA (cDNA) was added directly into a PCR
mixture (final volume of 50 μl) containing 1X Taq DNA
polymerase reaction buffer (Shanghai Promega Biological Products),
2.5 mM MgCl2, 0.2 mM dNTP mixture, 1 unit of Taq
DNA polymerase (Takara Biotechnology), and 1 μM of the
appropriate primer pair. The resulting amplified DNA fragments were
separated by electrophoresis through a 1.5% agarose gel, and the
resulting bands were visualized and scanned using a white
ultraviolet transilluminator (Ultra-Violet Products Ltd.,
Cambridge, UK) and quantified by densitometry.
Immunofluorescence staining
The cells were fixed with cold 100% methanol at
−20°C for 5 min and permeabilized with PBS-0.5% Triton X-100 for 5
min. After blocking with 5% bovine serum albumin for 1 h at 37°C,
the cultures were incubated with rabbit polyclonal anti-β-catenin
antibody (1:150 dilution) overnight at 4°C. After washing, the
cells were incubated with Cy3-conjugated goat anti-rabbit secondary
antibody (1:200 dilution) for 1 h at room temperature. Finally,
DAPI (Sigma-Aldrich) was used to stain the nuclei. Fluorescence
images were captured using a fluorescence microscope (Nikon Eclipse
E600; Nikon, Tokyo, Japan).
Western blot analysis
The cells were plated in culture dishes for 24 h in
RPMI-1640 and 10% FBS before exposure to CSE. The supernatant of
the cultured cells was discarded, and the attached cells were
washed twice with PBS. The cells were incubated with CSE (0, 10,
20, 40 and 80 μg/ml, respectively) or DMSO for 48 h. After
the treatments, for the preparation of crude cell lysates, the
cells were harvested in accordance with the treatment routines
described above, washed with cold PBS, and then incubated in
ice-cold radioimmunoprecipitation assay buffer. The cells were
sonicated on ice for 30 sec and lysed at 4°C for 60 min. The cell
lysates were then centrifuged for 30 min at 12,000 × g and 4°C. For
cell fractionation into the cytoplasmic and nuclear extracts, the
Cytoplasmic Extraction kit (CWBIO, Beijing, China) was used
according to the manufacturer’s instructions. The protein
concentrations were determined using a protein assay kit (Bio-Rad
Laboratories, Hercules, CA, USA). Equal amounts of lysates (40
μg) were separated using 12% SDS-PAGE and transferred onto
nitro cellulose membranes (Millipore, Bedford, MA, USA). The
membranes were blocked with 5% non-fat milk diluted in buffer (10
mM Tris-HCl, 100 mM NaCl and 0.1% Tween-20) for 1 h at room
temperature. The membranes were then probed with antibodies
including anti-RBM5 (ab85504), anti-β-catenin (E247, ab32572),
anti-His-H3 (acetyl K27, ab4729), anti-p-β-catenin and
(phospho-Y654, ab24925) and anti-β-actin [ACTN05 (C4), ab3280] from
Abcam (Cambridge, MA, USA), anti-Wnt-2 (11160-1-AP), anti-Wnt-7a
(10605-1-AP), anti-FRZB (12884-1-AP), anti-p-GSK-3β (phospho-Y216,
ab75745) and anti-GSK-3β from Proteintech Group, Inc. (Chicago, IL,
USA; 22104-1-AP), overnight at 4°C. Horseradish
peroxidase-conjugated secondary antibodies (Santa Cruz
Biotechnology) at a dilution of 1:500 were then added to the
solution and allowed to incubate for 1 h at room temperature. The
protein bands were then detected using an Enhanced
Chemiluminescence kit (Pierce Biotechnology Ltd., Rockford, IL,
USA). The protein levels were quantified by densitometry using
Quantity One software (Bio-Rad Laboratories).
Luciferase assay
The cells were plated onto a 12-well plate 1 day
prior to transfection. Following confirmation of 70–80% confluency,
the cells were transfected with 0.4 μg of TCF luciferase
reporter plasmids (wild-type, pGL3-OT; mutant, pGL3-OF,
respectively) for 24 h. Meanwhile, the cells in each group were
cotransfected with 0.1 μg of β-galactosidase expression
vector (pCH110) to normalize the transfection efficiency. The cells
were then treated with 40 μg/ml CSE and incubated for 48 h.
Finally, the luciferase reporter assay and β-galactosidase assay
were performed using commercial kits as directed by the
manufacturer. The luciferase activity was read using a Lumat LB
9507 luminometer (EG&G Berthold, Bad Wildbad, Germany) and
normalized for β-galactosidase activity (OD420).
Overexpression of RBM5 in the cells
The cells (4×105/well) were plated onto a
6-well plate and were transfected with 4 μg of DNA from the
expression vector pcDNA3 or pcDNA3-RBM5 using Lipofectamine 2000
(Life Technologies, Carlsbad, CA, USA). After transfection for 6 h,
the cell culture medium was replaced with fresh medium. The cells
were harvested at 48 h following transfection.
The pcDNA3.1 and pcDNA3.1-RBM5 plasmids were
provided by Dr Leslie Sutherland (Research Program, Northeast
Cancer Centre, Health Sciences North, Toronto, ON, Canada).
CONO77-shRBM5 was provided by Shanghai Genechem Co., Ltd.
(Shanghai, China).
Silencing of RBM5 in the cells
A short hairpin RNA (shRNA) sequence targeting human
RBM5 and a non-target sequence were obtained from Shanghai
Genechem. The RBM5 siRNA sequences used were forward,
5′-CCACCAAAGAUGGCAUUGATT-3′ and reverse,
5′-UCAAUGCCAUCUUUGGUGGTT-3′; and those of the non-target shRNA
(Scramble) were forward, 5′-UUCUCCGAACGUGUCACGUTT-3′ and reverse,
5′-ACGUGACACGUUCGGAGAATT-3′. The transfections with siRNA were
performed using Lipofectamine 2000 according to the manufacturer’s
protocol. Briefly, the A549 cells were seeded into 6-well plates
and transfected the next day with 100 pmol of shRBM5 or shScramble
using 10 μl of Lipofectamine 2000. The cells were harvested
48 h after transfection.
Treatment of the cells with the
β-catenin/TCF inhibitor
The novel small-molecule ICG-001 (S2662;
Selleckchem, Houston, TX, USA) which selectively inhibits
β-catenin/TCF-mediated gene transcription, was previously described
(23,25). A549 cells were seeded onto 6-well
plates and treated with 12.5 μl of ICG-001 or DMSO on the
second day. After treatment for 24 h, the cell culture medium was
replaced with medium containing the same dose of ICG-001 or DMSO.
The cells were continuously incubated for another 24 h. Then, the
protein was extracted from the cells and subjected to western blot
analysis.
Results
Cytotoxicity of CSE on cell viability and
growth
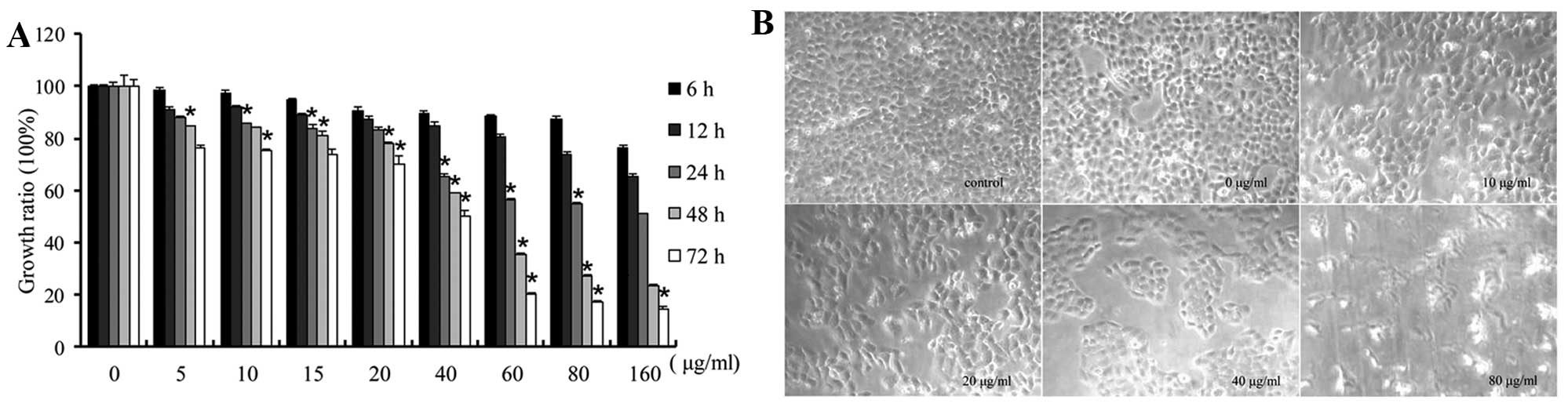
The effects of CSE on cell growth in vitro
were evaluated using an MTT assay. A549 cells were treated with
different concentrations of CSE (5, 10 15, 20, 40, 80 and 160
μg/ml, respectively) for 6, 12, 24, 48 or 72 h,
respectively. The results showed that CSE exposure inhibited cell
growth and viability (Fig. 1A).
Compared with the DMSO-treated control cells, which were closely
adherent, the CSE-exposed cells showed delayed confluence and
larger interspaces between the cells under light micro-scopy. The
shrinkage and death of cells were more apparent after exposure to
80 μg/ml CSE for 48 h (Fig.
1B).
Increased cytoplasmic accumulation and
nuclear translocation of β-catenin in the A549 cells following CSE
treatments
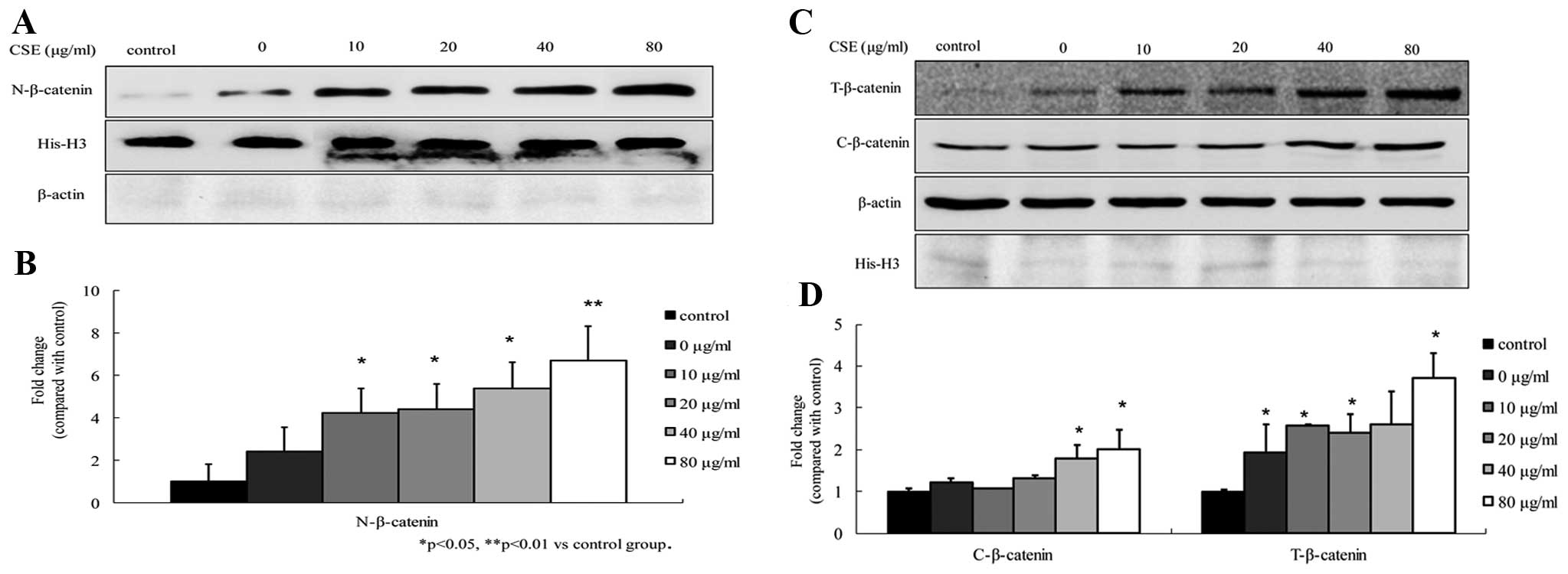
To determine the effects of CSE on the β-catenin
levels in A549 cells, the β-catenin protein levels were analyzed by
western blot analysis. The results showed that the total, cytosolic
and nuclear β-catenin levels were increased upon exposure to CSE,
compared to those in the control cells. The increases in nuclear
β-catenin were more significant at concentrations of 10–80
μg/ml, and total β-catenin and cytosolic β-catenin at
concentrations of 40–80 μg/ml (all P<0.05). In addition,
the increases were dose-dependent (Fig.
2). The levels of β-catenin in the A549 cells exposed to CSE
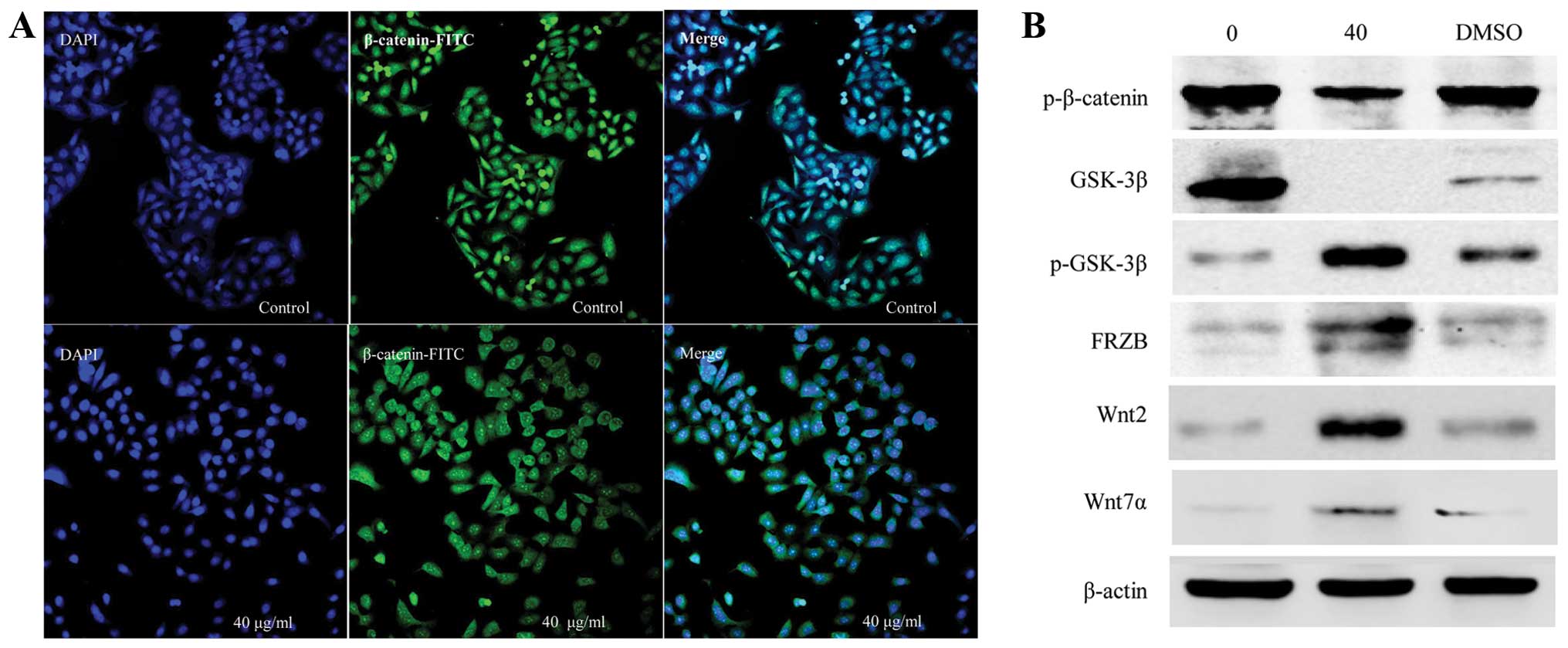
were also examined by immunofluorescence staining. The results
showed that strongly positive β-catenin cells were detected in both
the cytoplasm and nuclei of the CSE-treated cells and that the
expression was more significant in the nuclei of cells exposed to
40 μg/ml CSE for 48 h, while the β-catenin expression was
only slightly positive in the control cells (Fig. 3A). To further study the mechanisms,
other molecules involved in the Wnt/β-catenin/TCF pathway were
examined. Compared to the untreated control groups, western blot
analysis showed that the levels of phosphorylated β-catenin and
total GSK-3β were decreased, while phosphorylated GSK-3β, FRB, Wnt2
and Wnt7a were increased following treatment with 40 μg/ml
CSE (Fig. 3B).
Activation of β-catenin/TCF signaling in
the A549 cells following CSE treatments
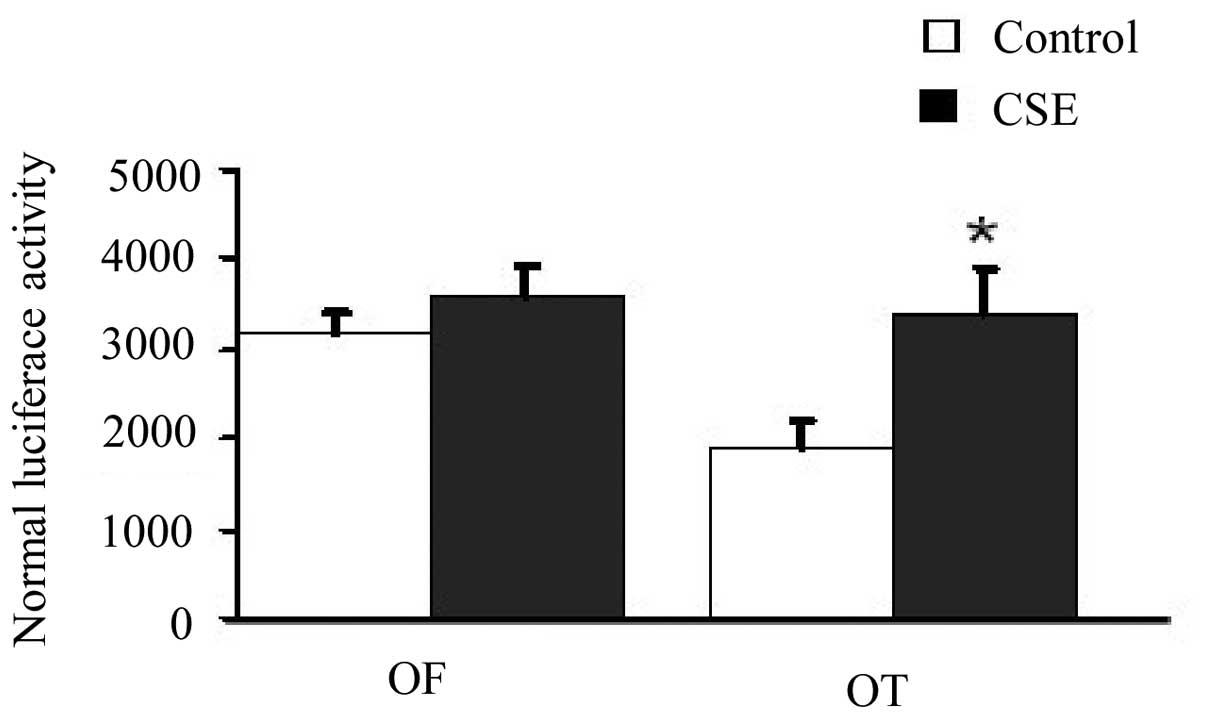
The luciferase activity assay showed that the
luciferase activity in the the CSE-exposed cells transfected with
the TCF luciferase reporter wild-type plasmid (pGL3-OT) was
significantly greater than that in the cells without CSE exposure
(33,167±3,085 vs. 19,978±1,916, respectively; P<0.05).
Meanwhile, the luciferase activity in the CSE-exposed cells
transfected with the TCF luciferase reporter mutant plasmid
(pGL3-OF) was similar to that in the cells without CSE exposure
(35,657±2,301 vs. 32,908±2,350, respectively, P>0.05). These
results suggest that the β-catenin/TCF signaling pathway was
activated in cells following CSE treatment (Fig. 4).
Downregulation of RBM5 in the A549 cells
following CSE treatments
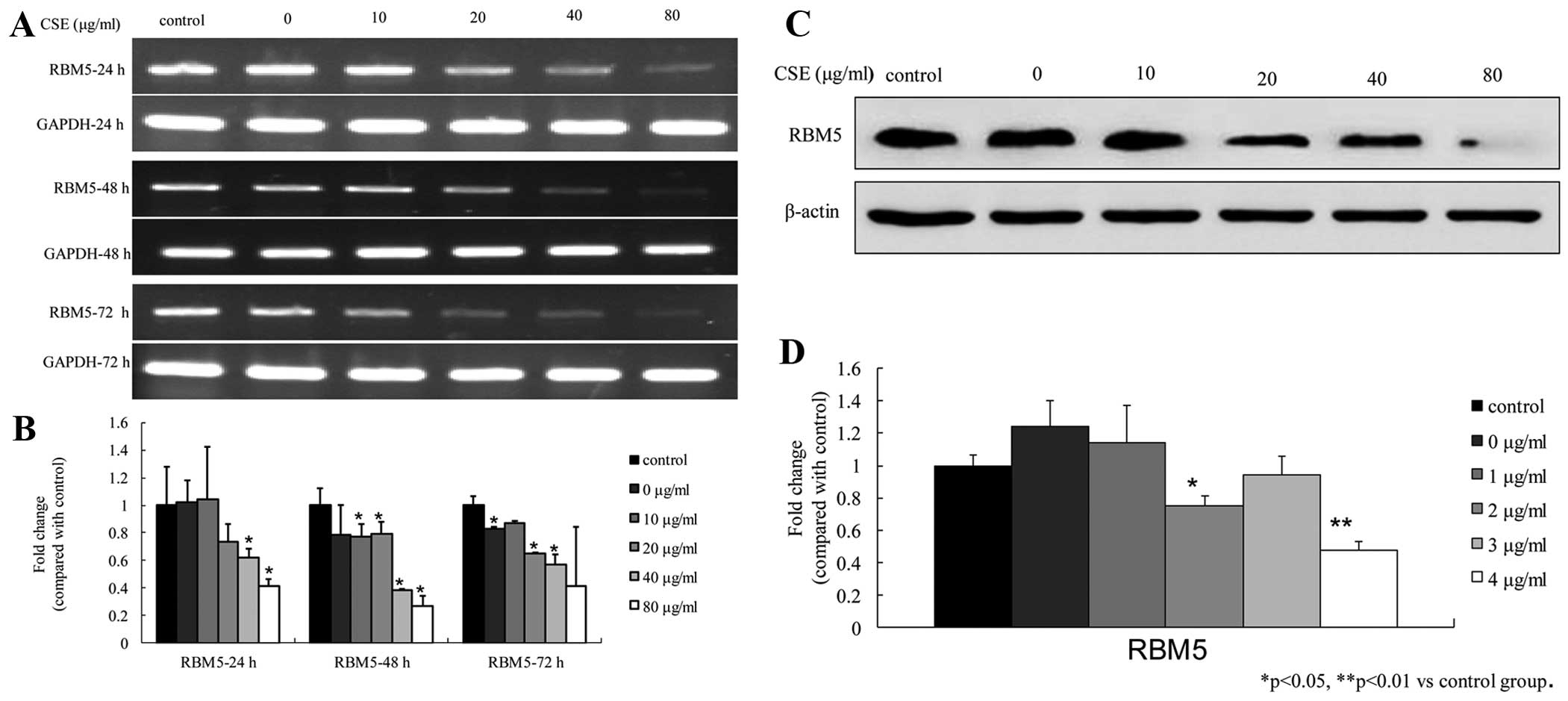
To determine the effect of CSE on the RBM5 level in
cells, we examined both the mRNA and protein levels of RBM5 in the
A549 cells following treatment with various concentrations of CSE
at different time-points. The real-time PCR results showed that CSE
inhibited the RBM5 mRNA levels in both a dose- and time-dependent
manner. The fold-change in gene expression was significantly
reduced at 40 and 80 μg/ml at 24 h; at 10, 20, 40 and 80
μg/ml at 48 h; and at 0, 20, 40 and 80 μg/ml at 72 h
(all P<0.05) (Fig. 5A and B). In
addition, the western blot analysis showed that the RBM5 protein
levels were reduced in the cells treated with CSE for 48 h and that
the levels were significantly reduced at 20 and 80 μg/ml.
The RBM5 expression level after treatment with 40 μg/ml CSE
was slightly greater than that after treatment with 20 μg/ml
CSE. These results suggest that CSE inhibited RBM5 protein
expression (Fig. 5C and D).
RBM5 regulates Wnt/β-catenin signaling in
the A549 cells
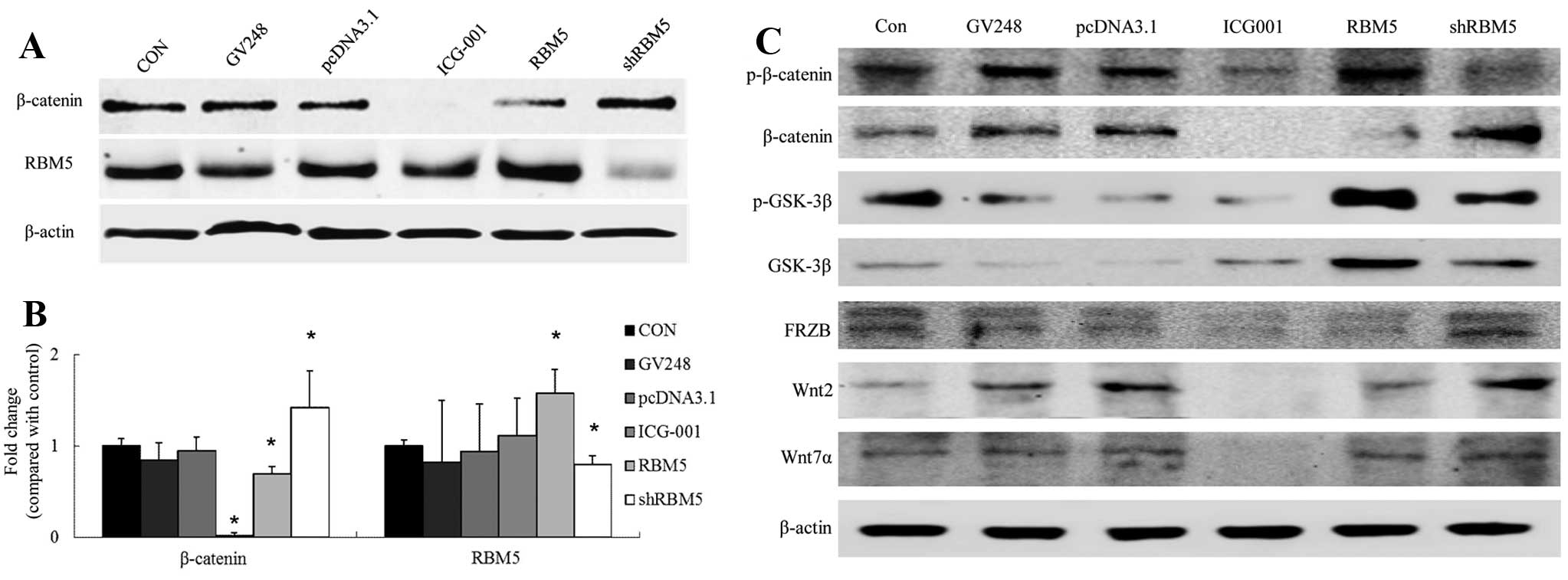
To determine the effects of RBM5 on Wnt/β-catenin
signaling in the A549 cells, we either overexpressed wild-type RBM5
or knocked down RBM5 using RBM5 shRNA in the cells. As shown in
Fig. 6, compared with the control
group, overexpression of RBM5 significantly inhibited β-catenin
expression in the A549 cells (P<0.05), increased phosphorylation
of β-catenin and GSK-3β, and increased total GSK-3β expression. In
contrast, silencing of RBM5 significantly enhanced the β-catenin
level in the A549 cells (P<0.05), reduced phosphorylation of
β-catenin and GSK-3β and reduced total GSK-3β expression.
Furthermore, the cells were treated with the β-catenin/TCF
signaling inhibitor ICG-001. The data showed that ICG-001 had no
apparent effect on the RBM5 expression level in the cells. These
studies suggest that RBM5 is an upstream molecule that regulates
Wnt/β-catenin signaling and negatively regulates this pathway in
A549 cells (Fig. 6).
Discussion
The primary purpose of the present study was to
determine whether the RBM5 level is reduced and whether it
regulates Wnt/β-catenin signaling in cigarette smoke-induced lung
injury. The A549 cell line has been considered to be a preferred
cellular model for studies related to type II alveolar epithelial
cells, which secrete specific type II cell markers including
surfactant proteins and some other markers (17,18).
In the present study, A549 cells were exposed to various
concentrations of CSE for different periods of time. We observed
that CSE is cytotoxic to alveolar epithelial cells as it inhibited
the viability of cells in a dose- and time-dependent manner. Our
study is consistent with previous reports (19,20).
Cigarette smoke is a complex mixture of harmful chemicals, some of
which are known carcinogens. Recent research has shown that A549
cells are more vulnerable to harmful chemicals in CSE than other
types of cells (21).
In the present study, we observed that both
cytosolic and nuclear β-catenin levels in the CSE-treated cells
were significantly elevated following CSE exposure for 48 h,
compared with those in the control cells. According to the western
blot results, there were increased nuclear β-catenin levels in the
cytoplasm as well as increased cytosolic β-catenin and total
β-catenin levels in the nuclei. These results were also confirmed
by an increased number of β-catenin-positive cells in both the
cytoplasm and nuclei as examined by immunofluorescence staining.
The changes were dose-dependent. Unfortunately, proteins were only
collected from cells treated for 48 h. Thus, we were unable to
determine whether the changes were also time-dependent. These
findings suggest that CSE upregulates β-catenin levels and promotes
β-catenin cytosolic accumulation and nuclear translocation. In
addition, the luciferase activity assay showed that the luciferase
activity in the CSE-exposed cells transfected with the TCF
luciferase reporter wild-type plasmid (pGL3-OT) was significantly
greater than that in cells without CSE exposure. These results
suggest that Wnt/β-catenin signaling was activated in the
CSE-exposed lung epithelial cells. Previous studies have shown that
Wnt/β-catenin signaling is activated in lung injury (5,14,15,22).
Moreover, aberrant activation of WNT signaling also has been
reported in the airway of cigarette smoke-associated COPD (23). Thus, the present study is in line
with these reports, confirming that aberrant activation of
Wnt/β-catenin signaling exists in cigarette smoke-associated airway
injury and is responsible for cell proliferation and tissue
remodeling at the late stage of injury in response to CSE stimuli.
However, one report has contradicted these studies, suggesting that
smoking downregulates the Wnt/β-catenin pathway in the human airway
epithelium and contributes to the dysregulation of airway
epithelium differentiation observed in smoking-related airway
disorders (24).
Previous studies suggest that frequently reduced
RBM5 levels exist in different types of tumors including breast
cancer (25), schwannoma (26), and ~75% of primary lung cancers
(27). We previously reported that
the protein levels of RBM5 are lower in NSCLC compared with
non-tumorous tissues (13). In
addition, we studied the possible involvement of RBM5 in drug
resistance during chemotherapy using a cisplatin-sensitive and a
cisplatin-resistant NSCLC cell line (10). These studies suggest that RBM5 plays
a role in suppressing tumor development and progression.
Additionally, the RBM5 level has been shown to be higher in the
adult thymus compared with that in the fetal thymus, which suggests
that RBM5 may also be involved in normal development (28). Furthermore, the inhibitory effects
of RBM5 on tumor growth both in vitro and in vivo
have been suggested due to its induction of G1 cell cycle arrest
and apoptosis (25). In the present
study, for the first time, we observed that both the mRNA and
protein levels of RBM5 in the CSE-injured lung epithelium also were
significantly reduced compared to the controls. The reductions in
human alveolar epithelial cells were both time-and dose-dependent.
Similarly, a study from another group showed that a decreased RBM5
level was more frequently observed in the alveolar epithelium of
smokers than of nonsmokers (13).
Together, these data suggest that loss of RBM5 contributes to cell
proliferation and tumor transformation. The possible molecular
mechanisms may involve cell cycle arrest and induction of
apoptosis.
Given that RBM5 was downregulated and Wnt/β-catenin
signaling was activated in the CSE-exposed alveolar epithelial
cells, we postulated that RBM5 may be involved in the activation of
Wnt/β-catenin signaling in CSE-induced lung injury. To further
study whether RBM5 regulates β-catenin signaling, we examined the
protein level of β-catenin in the A549 cells following the
overexpression of wild-type RBM5 or the silencing of RBM5 using
RBM5 shRNA in these cells. The present study revealed that
overexpression of RBM5 significantly inhibited Wnt/β-catenin
signaling in A549 cells, while silencing of RBM5 significantly
enhanced Wnt/β-catenin signaling in the A549 cells. However, the
β-catenin/TCF signaling inhibitor ICG-001 had no apparent effect on
the RBM5 level in cells. These results suggest that RBM5 acted as
an upstream molecule of Wnt/β-catenin/TCF signaling and that RBM5
negatively regulates this pathway in A549 cells.
In summary, we demonstrated that CSE leads to
epithelial injury by directly inhibiting the viability of alveolar
epithelial cells. Downregulation of RBM5 and activation of
Wnt/β-catenin signaling are involved in CSE-induced alveolar
epithelial injury. In addition, RBM5 acts as an upstream molecule
that negatively regulates the activity of Wnt/β-catenin
signaling.
Acknowledgments
We thank Medjaden Bioscience Limited for assisting
in the preparation of this manuscript.
References
|
1
|
Feldman C and Anderson R: Cigarette
smoking and mechanisms of susceptibility to infections of the
respiratory tract and other organ systems. J Infect. 67:169–184.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Niewoehner DE: Cigarette smoking, lung
inflammation, and the development of emphysema. J Lab Clin Med.
111:15–27. 1988.PubMed/NCBI
|
|
3
|
Sangani RG and Ghio AJ: Lung injury after
cigarette smoking is particle related. Int J Chron Obstruct Pulmon
Dis. 6:191–198. 2011.PubMed/NCBI
|
|
4
|
Ye Q, Huang K, Ding Y, et al: Cigarette
smoking contributes to idiopathic pulmonary fibrosis associated
with emphysema. Chin Med J (Engl). 127:469–474. 2014.
|
|
5
|
Flozak AS, Lam AP, Russell S, et al:
Beta-catenin/T-cell factor signaling is activated during lung
injury and promotes the survival and migration of alveolar
epithelial cells. J Biol Chem. 285:3157–3167. 2010. View Article : Google Scholar :
|
|
6
|
Clevers H: Wnt/beta-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu F and Millar SE: Wnt/beta-catenin
signaling in oral tissue development and disease. J Dent Res.
89:318–330. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
He X, Semenov M, Tamai K and Zeng X: LDL
receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling:
arrows point the way. Development. 131:1663–1677. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huelsken J and Birchmeier W: New aspects
of Wnt signaling pathways in higher vertebrates. Curr Opin Genet
Dev. 11:547–553. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li P, Wang K, Zhang J, et al: The 3p21.3
tumor suppressor RBM5 resensitizes cisplatin-resistant human
non-small cell lung cancer cells to cisplatin. Cancer Epidemiol.
36:481–489. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Oh JJ, Taschereau EO, Koegel AK, et al:
RBM5/H37 tumor suppressor, located at the lung cancer hot spot
3p21.3, alters expression of genes involved in metastasis. Lung
Cancer. 70:253–262. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sutherland LC, Wang K and Robinson AG:
RBM5 as a putative tumor suppressor gene for lung cancer. J Thorac
Oncol. 5:294–298. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liang H, Zhang J, Shao C, et al:
Differential expression of RBM5, EGFR and KRAS mRNA and protein in
non-small cell lung cancer tissues. J Exp Clin Cancer Res.
31:362012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lemjabbar-Alaoui H, Dasari V, Sidhu SS, et
al: Wnt and Hedgehog are critical mediators of cigarette
smoke-induced lung cancer. PLoS One. 1:e932006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tian D, Zhu M, Li J, Ma Y and Wu R:
Cigarette smoke extract induces activation of beta-catenin/TCF
signaling through inhibiting GSK3beta in human alveolar epithelial
cell line. Toxicol Lett. 187:58–62. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang X, Xiao T, Cheng S, Tong T and Gao
Y: Cigarette smoke suppresses the ubiquitin-dependent degradation
of OLC1. Biochem Biophys Res Commun. 407:753–757. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Andreeva AV, Kutuzov MA and
Voyno-Yasenetskaya TA: Regulation of surfactant secretion in
alveolar type II cells. Am J Physiol Lung Cell Mol Physiol.
293:L259–L271. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fehrenbach H: Alveolar epithelial type II
cell: defender of the alveolus revisited. Respir Res. 2:33–46.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hoshino Y, Mio T, Nagai S, Miki H, Ito I
and Izumi T: Cytotoxic effects of cigarette smoke extract on an
alveolar type II cell-derived cell line. Am J Physiol Lung Cell Mol
Physiol. 281:L509–L516. 2001.PubMed/NCBI
|
|
20
|
Lannan S, Donaldson K, Brown D and MacNee
W: Effect of cigarette smoke and its condensates on alveolar
epithelial cell injury in vitro. Am J Physiol. 266:L92–L100.
1994.PubMed/NCBI
|
|
21
|
Cavallo D, Ursini CL, Fresegna AM, et al:
Cyto-genotoxic effects of smoke from commercial filter and
non-filter cigarettes on human bronchial and pulmonary cells. Mutat
Res. 750:1–11. 2013. View Article : Google Scholar
|
|
22
|
Pongracz JE and Stockley RA: Wnt
signalling in lung development and diseases. Respir Res. 7:152006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Heijink IH, de Bruin HG, van den Berge M,
et al: Role of aberrant WNT signalling in the airway epithelial
response to cigarette smoke in chronic obstructive pulmonary
disease. Thorax. 68:709–716. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang R, Ahmed J, Wang G, et al:
Down-regulation of the canonical Wnt beta-catenin pathway in the
airway epithelium of healthy smokers and smokers with COPD. PLoS
One. 6:e147932011. View Article : Google Scholar
|
|
25
|
Edamatsu H, Kaziro Y and Itoh H: LUCA15, a
putative tumour suppressor gene encoding an RNA-binding nuclear
protein, is down-regulated in ras-transformed Rat-1 cells. Genes
Cells. 5:849–858. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Welling DB, Lasak JM, Akhmametyeva E,
Ghaheri B and Chang LS: cDNA microarray analysis of vestibular
schwannomas. Otol Neurotol. 23:736–748. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Oh JJ, West AR, Fishbein MC and Slamon DJ:
A candidate tumor suppressor gene, H37, from the human lung cancer
tumor suppressor locus 3p21.3. Cancer Res. 62:3207–3213.
2002.PubMed/NCBI
|
|
28
|
Ji L, Minna JD and Roth JA: 3p21.3 tumor
suppressor cluster: prospects for translational applications.
Future Oncol. 1:79–92. 2005. View Article : Google Scholar
|