Introduction
Lung cancer is a leading cause of cancer-related
mortality worldwide. Non-small cell lung cancer (NSCLC) comprises
~80% of all lung cancers (1).
Cigarette smoking is the most important risk factor of NSCLC.
Nicotine, the principal chemical component in tobacco smoke
associated with addiction, is involved in the progression and
metastasis of NSCLC (2).
Accumulating experimental evidence has demonstrated that nicotine
and its metabolites promotes migration and invasion in lung cancer
cells (3–5). Angiogenesis, the formation of new
blood vessels from a pre-existing vascular network, is essential
for tumor migration, invasion and metastasis (6). Epithelialmesenchymal transition (EMT),
a process by which epithelial cells lose the epithelial phenotype
and gain the mesenchymal phenotype, is largely thought to play a
key role in invasion and metastasis (7). Nicotine has also been found to enhance
angiogenesis (8,9) and EMT (4,5) in
NSCLC cells. Angiogenesis and EMT play a crucial role in the
development and progression of NSCLC (6,10).
Thus the inhibitors of angiogenesis and EMT mediated by nicotine
may be developed into chemopreventive agents of smoking-associated
NSCLC.
Green tea, widely consumed in Asia, contains
polyphenolic compounds that account for 30% of the dry weight of
the leaves. Previous epidemiological studies and meta-analysis have
revealed that green tea consumption reduces the risk of lung cancer
(11,12). The chemopreventive effects of green
tea are mediated by polyphenolic compounds known as catechins
including (−)-epigallocatechin-3-gallate (EGCG),
(−)-epicat-echin-3-gallate (ECG), (−)-epigallocatechin (EGC) and
(−)-epicatechin (EC), with EGCG being the most abundant and
powerful catechin in cancer prevention and treatment (13,14). A
growing body of evidence has demonstrated that EGCG inhibits
migration and invasion in a variety of cancer cells (15–17).
In a previous study, we found that EGCG inhibited angiogenesis by
downregulating hypoxia- and serum-induced hypoxia-inducible factor
1α (HIF-1α) protein accumulation and vascular endothelial growth
factor (VEGF) expression in human cervical carcinoma and hepatoma
cells (18). Previously, we
demonstrated that EGCG inhibits human papillomavirus (HPV)-16
oncoprotein- and IGF-1-induced angiogenesis in NSCLC by targeting
HIF-1α (19,20). Furthermore, nicotine induces HIF-1α
protein expression in NSCLC cells through the nicotinic
acetylcholine receptor-mediated signaling pathway (8). However, the effect of EGCG on
angiogenesis induced by nicotine in NSCLC cells has not been
reported.
EMT promotes the invasion and metastasis of various
types of cancers including NSCLC (7,10).
EGCG has been found to inhibit EMT mediated by a variety of factors
(21–23). Liu et al have reported that
EGCG inhibited transforming growth factor-β (TGF-β)-mediated EMT in
NSCLC cells via inhibition of the Smad2 and Erk1/2 signaling
pathways (21). Ko et al
also found that EGCG negatively affected TGF-β1-mediated EMT by
suppressing the acetylation of Smad2 and Smad3 in A549 NSCLC cells
(22). However, the effect of EGCG
on nicotine-induced EMT in NSCLC cells remains to be
determined.
In the present study, we determined the effect of
EGCG on nicotine-induced migration, invasion, angiogenesis and EMT
in NSCLC cells. To the best of our knowledge, the results of the
present study showed for the first time that, EGCG inhibited
nicotine-induced angiogenesis and EMT in A549 NSCLC cells, leading
to the suppression of migration and invasion. Furthermore, we found
that EGCG suppressed Akt and ERK1/2 activation induced by nicotine.
These findings provide further evidence with regard to the
underlying molecular mechanisms involved in the anticancer effect
of EGCG on smoking-associated NSCLC.
Materials and methods
Drug and reagents
EGCG was purchased from Sigma (St. Louis, MO, USA),
dissolved in distilled water at a concentration of 100 mM and
stored at −80°C as a stock solution. Nicotine was also obtained
from Sigma. Complete protease inhibitor cocktail was from Roche
(Mannheim, Germany). Mouse anti-human HIF-1α monoclonal antibody
was purchased from BD Transduction Laboratories (San Diego, CA,
USA). Vimentin, β-catenin, total and phosphorylated Akt (Ser473) or
ERK1/2 (Thr202/Tyr204) antibodies were purchased from Cell
Signaling Technology (Beverly, MA, USA). Antibody for β-actin was
purchased from Beyotime Biotechnology Corporation (Shanghai,
China). Horseradish peroxidase (HRP)-conjugated secondary
antibodies were obtained from Cell Signaling Technology. The human
VEGF ELISA development kit was purchased from Wuhan Boster
Bio-Engineering Co., Ltd. (Wuhan, China). One-Step SYBR®
PrimeScript® RT-qPCR (no. DRR086A) was purchased from Takara
Biotechnology Co., Ltd. (Dalian, China). Transfection reagent
(Lipofectamine™ 2000) was obtained from Invitrogen Corporation
(Carlsbad, CA, USA). An In Vitro Angiogenesis Assay kit (ECM625)
was obtained from Millipore (Temecula, CA, USA). An In Vivo
Angiogenesis Assay kit (no. 354248) was obtained from BD
Biosciences (Bedford, MA, USA). HiCN Hemoglobin Detection kit was
purchased from Shanghai Rongsheng Biotech Co., Ltd. (Shanghai,
China).
Cell culture and treatment with EGCG
The human A549 NSCLC cell line (adenocarcinoma cell
line) and human umbilical vein endothelial cells (HUVECs) were
obtained from the American Type Culture Collection (ATCC;
Rockville, MD, USA). The cells were cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum (FBS), penicillin (100
U/ml) and streptomycin (100 μg/ml). Cultures were maintained
at 37°C in a humidified atmosphere with 5% CO2.
Exponentially growing cells (~80% confluence) in complete media
were pretreated for 1 h with different concentrations of EGCG,
followed by exposure to nicotine (5 μM) for the indicated
time points according to the purpose of the experiment.
Wound-healing assays
A549 cells were plated and grown to 80% confluence
in a 24-well plate (Corning Inc.). The cells were starved in 1% FBS
for 24 h and then washed with phosphate-buffered saline (PBS). The
cells were scratched with a sterile 200 μl pipette tip in
each well and medium containing 10% FBS and 5 μM nicotine in
the presence or absence of EGCG. After 24 and 48 h, the wounds were
observed and images were captured. The data were representative of
three independent experiments.
Cell invasion assays
The invasive ability of A549 cells was assayed using
24-well invasion chambers (Corning Inc.) with filters coated with
Matrigel (BD Biosciences) on the upper surface with 8.0-μm
pores. Briefly, Matrigel was applied to the upper surface of the
chambers. A549 cells (5×104) in 0.2 ml medium were added
into the upper chamber, and 0.5 ml 10% FBS medium in the presence
or absence of 5 μM nicotine or different concentrations of
EGCG (10, 25, 50 and 100 μM) was added into the lower
chamber. After incubation at 37°C with 5% CO2 for 36 h,
non-migrating A549 cells on the upper surface of the chambers were
removed by wiping with cotton swabs. The filters were stained with
hematoxylin for 30 min. The number of cells migrating on the other
side of the filters was counted. The data are representative of
three independent experiments.
Western blot analysis
Nicotine- or EGCG-treated A549 cells were lysed with
cell lysis buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1%
Triton X-100 and sodium pyrophosphate, β-glycerophosphate, EDTA,
Na3VO4, leupeptin phenylmethyl-sulfonyl
fluoride (PMSF), and complete protease inhibitor cocktail (Roche)
followed by incubation at 4°C for 1 h. The lysates were
ultra-sonicated and centrifuged at 12,000 × g for 10 min. Protein
concentrations were determined by BCA methods. Protein (100
μg) was separated on 10% polyacrylamide-SDS gel and
electroblotted onto PVDF membranes (Millipore). After blocking with
5% skim milk, the membrane was incubated overnight at 4°C with
primary antibody against HIF-1α, vimentin, β-catenin, total-Akt
(t-Akt), phosphorylated Akt (p-Akt), total-ERK1/2 (t-ERK1/2), or
phosphorylated-ERK1/2 (p-ERK1/2), followed by incubation with
HRP-conjugated secondary antibodies (1:1,000). As a loading
control, the blots were stripped and re-probed with anti-β-actin
antibody.
RT-qPCR analysis
Total RNA was isolated from cells using TRIzol®
reagent (Invitrogen). RT-qPCR analysis of HIF-1α and VEGF mRNA
levels was performed using One-Step SYBR® PrimeScript® RT-PCR
(Biotechnology Co., Ltd.) according to the manufacturer’s
instructions. The primers designed for RT-qPCR were as follows:
HIF-1α, forward: 5′-TCTGGGTTGAAACTCAAGCAACTG-3′ and reverse:
5′-CAACCGGTTTAAGGACACATTCTG-3′ (GenBank, NM_001243084.1); human
VEGF, forward: 5′-TGCTTCTGAGTTGCCCAGGA-3′ and reverse:
5′-TGGTTTCAATGGTG TGAGGACATAG-3′ (GenBank, NM_003376.5); p53,
forward: 5′-GAGGCCTTGGAACTCAAGGATG-3′ and reverse:
5′-TCAGGCCCTTCTGTCTTGAACA-3′ (GenBank, NM_000546.4); COX-2,
forward: 5′-CTGTAACCAAGATGGATGCAA-AGA-3′ and reverse:
5′-GTCAGTGACAATGAGATGTGGAA-3′ (GenBank, NM_000963.2); β-actin,
forward: 5′-TGGCACCCAGCACAATGAA-3′ and reverse:
5′-CTAAGTCATAGTCCGCCTAGAAGCA-3′ (GenBank, NM_001101.3). The primers
were produced by Takara Biotechnology Co., Ltd. The thermocycling
conditions used were: 42°C for 5 min, 95°C for 10 sec, followed by
40 cycles at 95°C for 5 sec, and 60°C for 31 sec. The size of the
PCR product of HIF-1α, VEGF, p53, COX-2 and β-actin was 150, 176,
137, 195 and 186 bp, respectively. The relative HIF-1α, VEGF, p53
and COX-2 mRNA levels were normalized to β-actin. The experiment
was repeated in triplicate.
Enzyme-linked immunosorbent assay
(ELISA)
The concentration of VEGF protein in the conditioned
media derived from nicotine- or EGCG-treated A549 cells was
determined using a human VEGF ELISA development kit according to
the manufacturer’s instructions. The results were normalized to the
cell number (5×103 cells). The experiments were repeated
in triplicate.
RNA interference
HIF-1α-siRNA (sense strand,
5′-GCCGCUCAAUUUAUGAAUATT-3′) and non-specific (NS)-siRNA (sense
strand, 5′-UUCUCCGAACGUGUCACGUTT-3′) duplexes were generated by
Shanghai GenePharma Co., Ltd. (Shanghai, China). Transfection was
performed using the Lipofectamine™ 2000 transfection reagent
according to the manufacturer’s instructions.
In vitro angiogenesis assay
An in vitro angiogenesis assay kit was
employed according to the manufacturer’s instructions (Millipore).
HUVECs (5×103 cells/well) were seeded onto the surface
of 96-well cell culture plates pre-coated with polymerized
ECMatrix™ and then incubated at 37°C for 6–8 h in the conditioned
media derived from nicotine-treated A549 cells transfection with
HIF-1α-siRNA or NS-siRNA. In parallel studies, HUVECs were cultured
in the conditioned media derived from nicotine-treated A549 cells
pretreated with 100 μM of EGCG. Tubule formation was
observed under a phase-contrast microscope. The total tube length
in three random view-fields/wells was measured by Scion Image
software and the average value was calculated. The experiment was
repeated in triplicate.
In vivo angiogenesis assay
Male nude mice (BALB/C nude) (6–8 weeks old) were
purchased from the Animal Center of Southern Medical University
(Guangzhou, China). Experiments with animals were undertaken in
accordance with the institution guidelines of the Institutional
Animal Care and Use Committee of Southern Medical University and
Guangdong Medical College. Nicotine- or EGCG-treated A549 cells
were re-suspended in serum-free media at a density of
8.0×106 cells/ml and 0.25 ml (2.0×106 cells
in total) of cell suspension was mixed with the same volume of BD
Matrigel Matrix (0.25 ml). The BD Matrigel mixture was
subcutaneously injected into the two flanks of nude mice. On day
11, the mice were euthanized and Matrigel plugs were harvested,
with half of each Matrigel plug fixed with 10% neutral formalin for
immunohistochemical studies and the other half weighed for the
determination of hemoglobin content as previously described
(20).
Immunohistochemistry
The expression of HIF-1α and VEGF proteins in
Matrigel plugs was analyzed by immunohistochemistry as previously
described (20). Briefly,
formalin-fixed Matrigel plugs were paraffin-embedded and sectioned
at 4 μm. The sections were then deparaffinized in xylene,
rehydrated through serial dilutions of alcohol and washed in PBS
(pH 7.2). The sections were heated in a microwave oven twice for 5
min in citrate buffer (pH 6.0) and then incubated overnight at 4°C
with primary antibody, followed by incubation with
streptavidin/HRP-conjugated secondary antibodies. The conventional
streptavidin/peroxidase method (Histostain™-Plus kits, SP0023;
Beijing Biosynthesis Biotechnology, Co., Ltd., Beijing, China) was
used to develop signals, and the cells were counterstained with
hematoxylin. The sections incubated with secondary antibodies in
the absence of primary antibodies served as negative controls.
Statistical analysis
Data are presented as the mean ± SD for three
separate experiments. One-way ANOVA, Bonferroni and Dunnett-T3 were
employed for statistical analysis using SPSS 19.0 for windows
software. P<0.05 was considered to indicate a statistically
significant result.
Results
EGCG inhibits nicotine-induced migration
and invasion in A549 cells
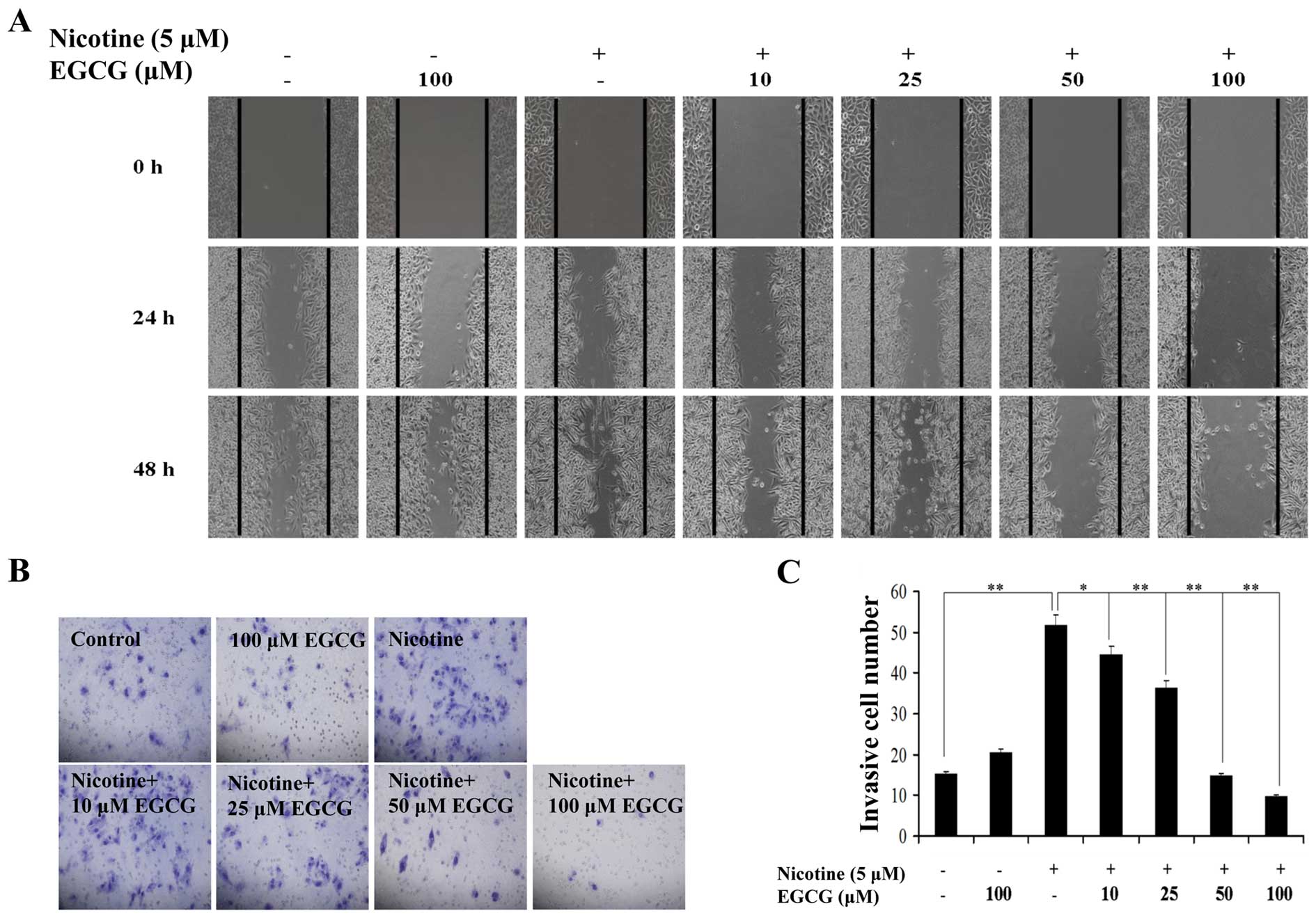
A wound-healing assay was carried out to assess
whether EGCG affects the migratory properties of A549 cells induced
by nicotine. Similar to recent results (3), we found that nicotine enhanced A549
cell migration (Fig. 1A). However,
EGCG significantly inhibited the migration of A549 cells induced by
nicotine in a dose-dependent manner, with the maximal efficiency
being observed at 100 μM EGCG treatment (Fig. 1A). The effect of EGCG on invasion
induced by nicotine in A549 cells was examined. As shown in
Fig. 1B and C, treatment of cells
with nicotine promoted cell invasive property. However, EGCG
inhibited the invasive property of A549 cells induced by nicotine
in a dose-dependent manner, and the maximum effect was observed at
100 μM EGCG treatment. Taken together, the results indicated
that EGCG inhibited nicotine-induced migration and invasion in A549
cells.
EGCG reverses the upregulation of HIF-1α,
VEGF, COX-2, p-Akt, p-ERK and vimentin protein levels and the
down-regulation of p53 and β-catenin protein levels mediated by
nicotine in A549 cells
To determine the effects of EGCG on angiogenesis and
EMT mediated by nicotine in A549 cells, the expression of
angiogenesis- and EMT-associated proteins was analyzed. Western
blot analysis was carried out to assess the expression of HIF-1α,
p53, COX-2, vimentin, β-catenin, p-Akt and p-ERK, and ELISA was
performed to determine VEGF concentrations in the conditioned media
derived from differentially treated cells. The results showed that
nicotine significantly upregulated the expression of HIF-1α, VEGF,
COX-2 and vimentin proteins in A549 cells, but downregulated the
expression of β-catenin and p53 proteins in A549 cells (Fig. 2A, B and D). However, EGCG
significantly reversed the effects of nicotine on the expression of
these proteins in A549 cells (Fig. 2A,
B and D). We also assessed whether EGCG affects the activation
of Akt and ERK induced by nicotine in A549 cells. The results
showed that EGCG inhibited the expression of p-Akt and p-ERK
induced by nicotine in A549 cells (Fig.
2C). Taken together, our results suggested that EGCG inhibited
angiogenesis and EMT mediated by nicotine in A549 cells.
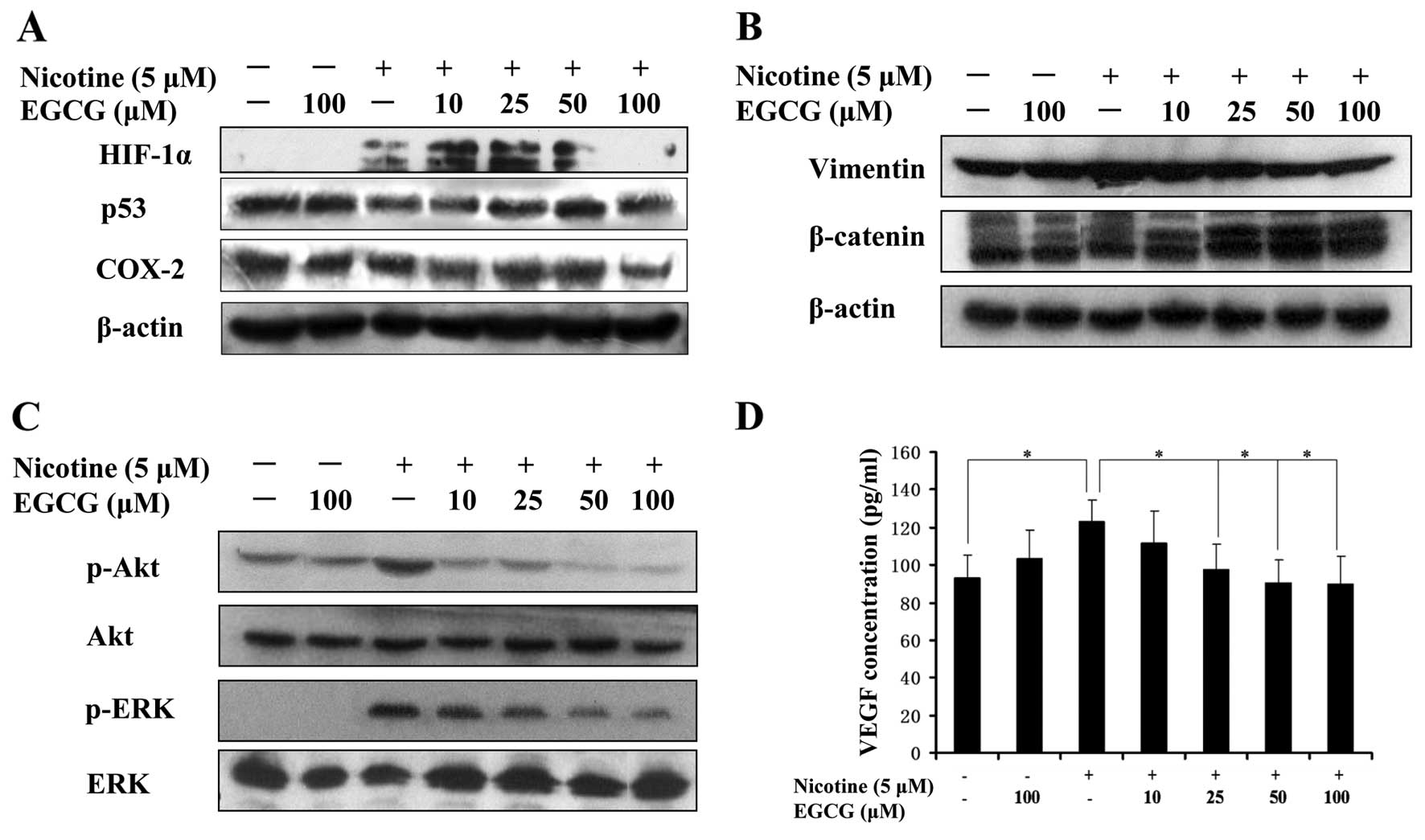 | Figure 2EGCG reverses the upregulation of
HIF-1α, VEGF, COX-2, p-Akt, p-ERK and vimentin protein levels and
the downregulation of p53 and β-catenin protein levels mediated by
nicotine in A549 cells. Exponentially growing cells in complete
media were pretreated for 1 h with different concentrations of
EGCG, followed by exposure to nicotine (5 μM) for 16 h.
(A–C) Western blot analysis was performed to determine the
expression of HIF-1α, p53, COX-2, vimentin, β-catenin, p-Akt and
p-ERK proteins. (D) VEGF concentration in the conditioned media
derived from A549-treated cells was determined by ELISA. The data
are presented as the mean ± SD from three replicate experiments.
*P<0.05. EGCG, epigallocatechin-3-gallate; VEGF,
vascular endothelial growth factor. |
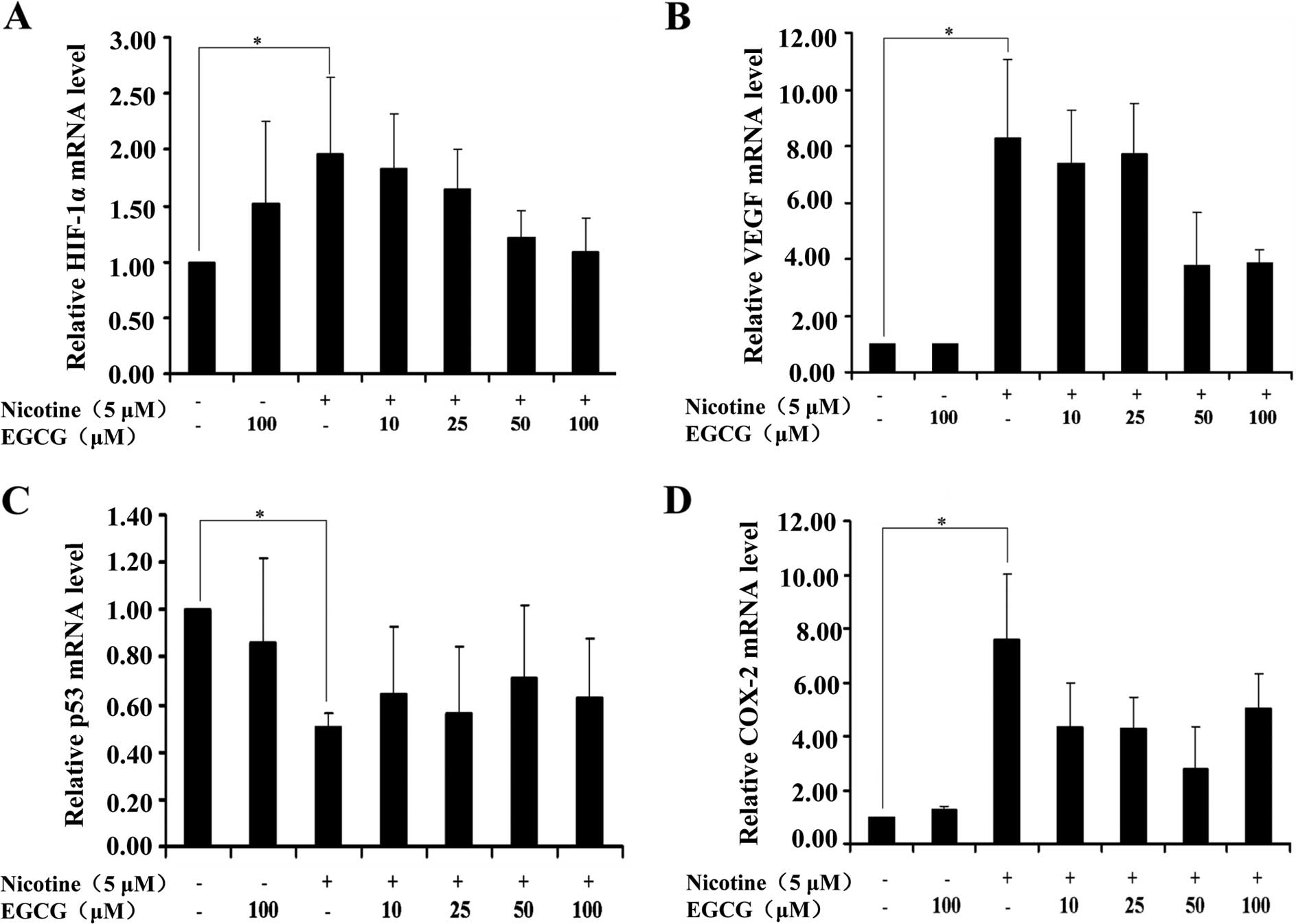
EGCG has no significant effect on the
expression of HIF-1α, VEGF, COX-2 and p53 mRNA mediated by nicotine
in A549 cells
Since EGCG affected the expression of HIF-1α, VEGF,
p53 and COX-2 proteins induced by nicotine in A549 cells, to assess
whether the alteration of HIF-1α, VEGF, p53 and COX-2 expression
can be regulated at the transcriptional levels, we measured mRNA
levels by RT-qPCR. As shown in Fig.
3, treatment of cells with nicotine upregulated HIF-1α, VEGF
and COX-2 mRNA levels and downregulated the p53 mRNA level as
compared to the controls, whereas EGCG treatment had no obvious
effects on HIF-1α, VEGF, p53 and COX-2 mRNA levels mediated by
nicotine. These results suggested that EGCG affected the expression
of HIF-1α, VEGF, p53 and COX-2 proteins mediated by nicotine in
A549 cells possibly through translational or post-translational
mechanisms.
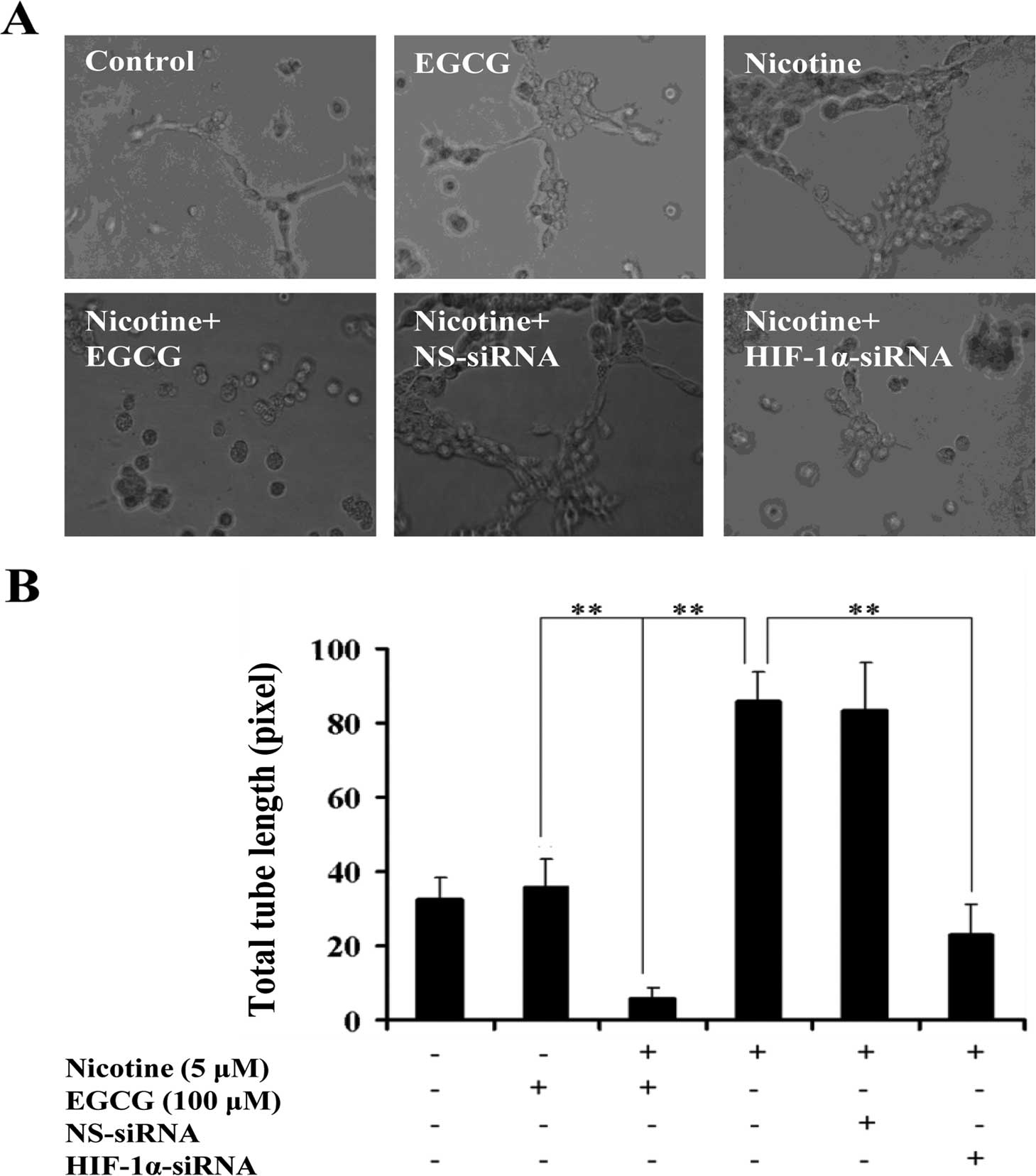
EGCG inhibits HIF-1α-dependent
angiogenesis induced by nicotine in vitro and in vivo, and
suppressed HIF-1α and VEGF protein expression induced by nicotine
in A549 xenografts of nude mice
To determine the effects of EGCG on nicotine-induced
angiogenesis in A549 cells, an in vitro angiogenesis model
was employed to evaluate the capillary tube formation of HUVECs
stimulated by the conditioned media derived from A549-treated
cells. Since VEGF is one of the downstream genes of HIF-1α
(24,25), to investigate whether the increased
expression of VEGF and angiogenesis induced by nicotine in A549
cells was mediated by HIF-1α, A549 cells were transfected with
HIF-1α-specific siRNA (HIF-1α-siRNA) or HIF-1α non-specific siRNA
(NS-siRNA). The results showed that A549 cells treated with
nicotine enhanced the ability to stimulate the formation of
capillary tube-like structures by HUVECs as compared to the
controls. Furthermore, A549 cells treated with EGCG or transfection
with HIF-1α-siRNA inhibited the nicotine-induced ability to
stimulate the formation of capillary tube-like structures by HUVECs
as compared with nicotine-treated cells (Fig. 4A), which was further confirmed by
quantification of the total tube length pixel values (P<0.05;
Fig. 4B). However, such inhibition
of angiogenic abilities was not observed in NS-siRNA-transfected
cells (P>0.05; Fig. 4A and
B).
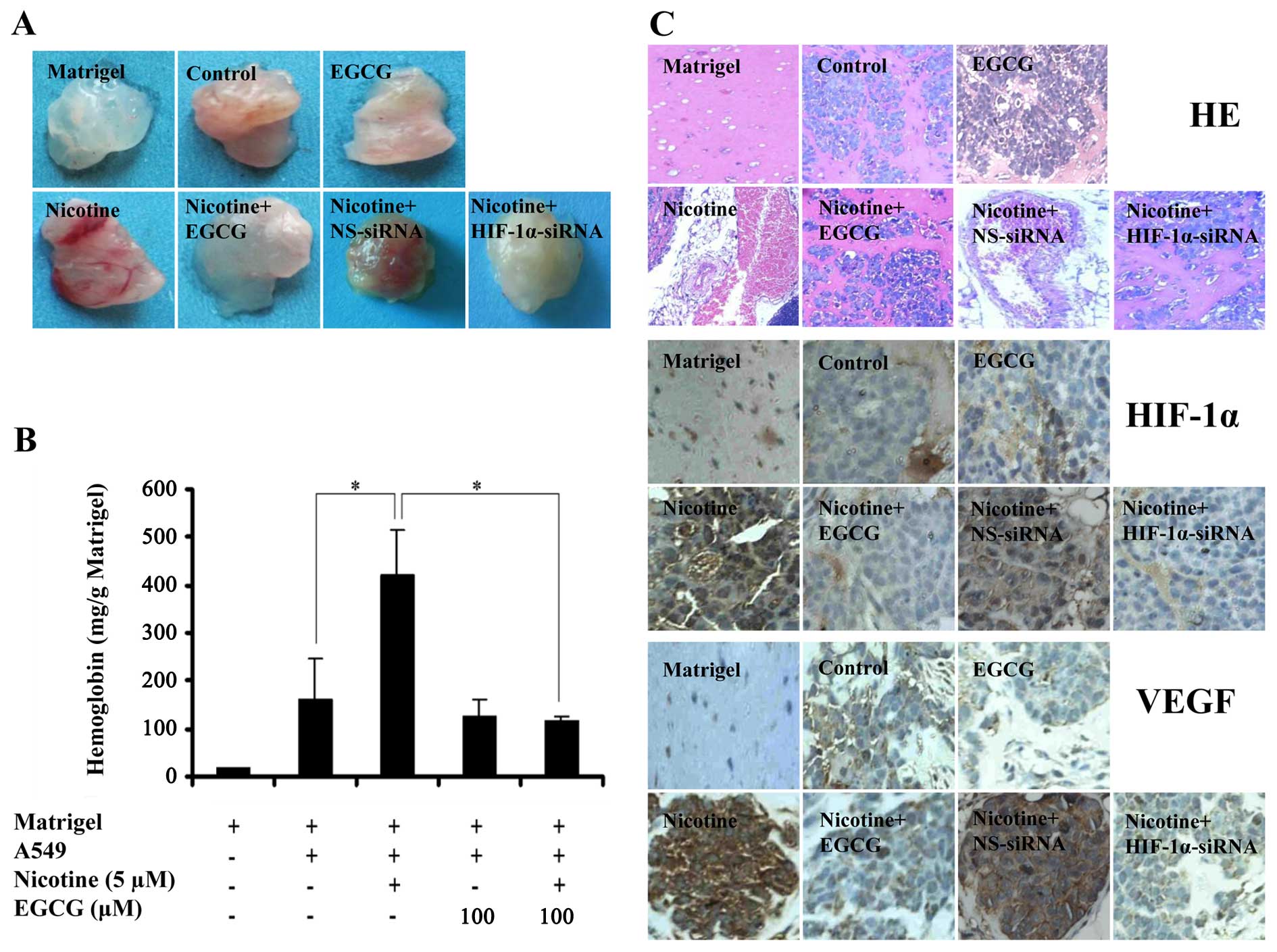
We performed a Matrigel plug angiogenesis assay
using BALB/C nude mice to observe the effect of EGCG on
nicotine-induced tumor angiogenesis in vivo. The results
showed that Matrigel plugs mixed with nicotine-treated cells
obviously induced angiogenesis in vivo (Fig. 5A), showing much higher hemoglobin
levels than those in the controls (P<0.05; Fig. 5B). Moreover, Matrigel plugs mixed
with EGCG or HIF-1α-siRNA-transfected cells significantly inhibited
angiogenesis in vivo stimulated by nicotine (Fig. 5A), showing barely detectable or low
hemoglobin levels (P<0.05; Fig.
5B). However, Matrigel plugs mixed with NS-siRNA-transfected
cells did not inhibit angiogenesis in vivo induced by
nicotine (Fig. 5A and B).
In addition, immunohistochemical results showed that
xenografts of A549 cells treated with nicotine exhibited a
significantly enhanced expression of HIF-1α and VEGF proteins as
compared to the controls (Fig. 5C).
The enhanced expression of HIF-1α protein was inhibited by
transfection with HIF-1α-siRNA, but not by transfection with
NS-siRNA (Fig. 5C). The increase of
VEGF expression induced by nicotine was abrogated by transfection
with HIF-1α-siRNA, but not by transfection with NS-siRNA. These
results indicated that the upregulation of VEGF protein expression
induced by nicotine was HIF-1α-dependent in the xenografts of A549
cells. Taken together, these results indicated EGCG markedly
inhibited HIF-1α-dependent angiogenesis induced by nicotine in
vitro and in vivo, and downregulated HIF-1α and VEGF
protein expression in A549 xenografts of nude mice.
Discussion
Cancer progression is associated with the abrogation
of normal biological characteristics that limit cell proliferation,
migration and invasion, eventually leading to metastasis (26–28).
Since metastases are a major cause of recurrence and mortality in
cancer patients, the development of new drugs that reduce invasion
and metastasis is extremely important for the prevention and
treatment of cancer. In the present study, we demonstrate that EGCG
obviously inhibited the migration and invasion induced by nicotine
in A549 cells, suggesting that EGCG holds great potential as a
chemopreventive agent in nicotine-associated NSCLC.
EMT is a critical normal process during development
and wound healing. However, the properties of EMT were shown to be
involved in human pathology, including fibrosis and cancer
metastasis (29,30). EMT facilitates tumor cell migration,
invasion and metastasis, and therefore may also be a major
mechanism of tumor progression (26–28).
The results presented in this study showed that nicotine
upregulated the expression of vimentin but downregulated the
expression level of β-catenin, an extracellular matrix protein
marker. However, treatment with EGCG reversed the expression of
vimentin and β-catenin mediated by nicotine in A549 cells. Previous
findings have demonstrated that the decrease in E-cadherin and
β-catenin levels with a concurrent increase in the vimentin level
is one of the hallmarks of EMT in lung cancer cells (31–33).
Therefore, EGCG may be important in suppression of EMT mediated by
nicotine in A549 NSCLC cells. Angiogenesis is required for tumor
migration, invasion and metastasis (6). In the present study, we found that
EGCG also inhibited nicotine-induced angiogenesis in vitro
and in vivo. Thus, the results suggest that EGCG-inhibited
nicotine-induced migration and invasion may at least occur in part,
through the suppression of EMT and angiogenesis.
HIF-1α and VEGF play an important role in tumor
invasion and metastasis, particularly in tumor angiogenesis
(34). It has been reported that
the expression of HIF-1α and VEGF was related to a poor prognosis
and a worse overall survival (34–36).
It has been previously demonstrated that hypoxia-stabilized HIF-1α
promoted EMT through increasing SNAI1 transcription in
hepatocellular carcinoma cells (37), and that vascular endothelial growth
factor receptor-1 (VEGFR-1) activation enhanced migration and
invasion of breast cancer cells through EMT (38). To identify the mechanisms by which
EGCG inhibited nicotine-induced angiogenesis and EMT, we determined
the expression of HIF-1α and VEGF. Our results show that EGCG
significantly inhibited nicotine-induced HIF-1α and VEGF protein
expression in A549 cells and in A549 xenografts of the nude mice,
indicating that EGCG-inhibited nicotine-induced angiogenesis and
EMT may be associated with the inhibition of HIF-1α and VEGF
protein expression.
HIF-1α, a key transcriptional factor, governs the
expression of a large array of hypoxia-responsive genes, including
VEGF, COX-2 and p53 (39–42)
COX-2 a key regulator of inflammation-producing prostaglandins, can
promote tumor cell proliferation and growth. p53, an important
tumor suppressor, holds a crosstalk with HIF-1 (42). In the present study, we found that
nicotine-induced angiogenesis in vitro and in vivo
and VEGF protein expression in A549 cells were HIF-1α-dependent.
Additionally, we analyzed the expression of other downstream
protein of HIF-1α, including COX-2 and p53. We found that EGCG
inhibited nicotine-induced COX-2 protein expression, and
concomitantly upregulated the p53 protein level. However, EGCG had
no obvious effects on the expression of HIF-1α, VEGF, COX-2 and p53
mRNA mediated by nicotine in A549 cells. These results suggest that
EGCG altered the expression of HIF-1α, VEGF, COX-2 and p53 proteins
mediated by nicotine through a post-transcriptional mechanism.
The PI3K/Akt and ERK1/2 signaling pathways have been
found to be involved in tumor angiogenesis and EMT stimulated by
various factors (43,44). These signaling pathways are
important in the development and progression of NSCLC (45,46).
Therefore, targeting PI3KAKT and ERK1/2 signaling pathways may be
an emerging treatment strategy for NSCLC (47,48).
In the present study, to the best of our knowledge we have for the
first time demonstrated that, EGCG suppressed nicotine-induced Akt
and ERK1/2 activation, providing the new evidence of a novel
molecular mechanism for the anticancer action of EGCG on
smoking-associated NSCLC.
In conclusion, to the best of our knowledge, we have
demonstrated for the first time that, EGCG significantly inhibited
nicotine-induced EMT and angiogenesis in vitro and in
vivo, leading to the suppression of migration and invasion in
A549 cells. Our findings suggest that EGCG is a potential agent for
the prevention and treatment of smoking-associated NSCLC.
Acknowledgments
We thank Dr Anh D. Le and Dr Qunzhou Zhang
(Department of Oral Surgery and Pharmacology, University of
Pennsylvania School of Dental Medicine, Philadelphia, USA) for
their kind assistance and guidance. The present study was supported
by grants from the National Natural Science Foundation of China,
nos. 81372511 and 81073103 (to X.T.), the Guangdong Natural Science
Foundation S2012010008232 (to X.T.), the Science and Technology of
Guangdong Province 2013B031100002 (to X.T.), the Specialized
Foundation for Introduced Talents of Guangdong Province Higher
Education (Foundation for High-Level Talent) 2050205 (to X.T.), the
Zhanjiang Municipal Governmental Specific Financial Fund Allocated
for Competitive Scientific and Technological Projects 2012C0303-56
(to X.T.), and the China Scholarship Council and the State
Scholarship Fund 201308440331 (to X.T.).
Abbreviations:
|
EGCG
|
epigallocatechin-3-gallate
|
|
NSCLC
|
non-small cell lung cancer
|
|
EMT
|
epithelial-mesenchymal transition
|
References
|
1
|
Pesch B, Kendzia B, Gustavsson P, Jöckel
KH, Johnen G, Pohlabeln H, Olsson A, Ahrens W, Gross IM, Brüske I,
et al: Cigarette smoking and lung cancer - relative risk estimates
for the major histological types from a pooled analysis of
case-control studies. Int J Cancer. 131:1210–1219. 2012. View Article : Google Scholar :
|
|
2
|
Schaal C and Chellappan SP:
Nicotine-mediated cell proliferation and tumor progression in
smoking-related cancers. Mol Cancer Res. 12:14–23. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nair S, Bora-Singhal N, Perumal D and
Chellappan S: Nicotine-mediated invasion and migration of non-small
cell lung carcinoma cells by modulating STMN3 and GSPT1 genes in an
ID1-dependent manner. Mol Cancer. 13:1732014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dasgupta P, Rizwani W, Pillai S, Kinkade
R, Kovacs M, Rastogi S, Banerjee S, Carless M, Kim E, Coppola D, et
al: Nicotine induces cell proliferation, invasion and
epithelial-mesenchymal transition in a variety of human cancer cell
lines. Int J Cancer. 124:36–45. 2009. View Article : Google Scholar
|
|
5
|
Wu SQ, Lv YE, Lin BH, Luo LM, Lv SL, Bi AH
and Jia YS: Silencing of periostin inhibits nicotine-mediated tumor
cell growth and epithelial-mesenchymal transition in lung cancer
cells. Mol Med Rep. 7:875–880. 2013.PubMed/NCBI
|
|
6
|
Jackson AL, Zhou B and Kim WY: HIF,
hypoxia and the role of angiogenesis in non-small cell lung cancer.
Expert Opin Ther Targets. 14:1047–1057. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Creighton CJ, Gibbons DL and Kurie JM: The
role of epithelial-mesenchymal transition programming in invasion
and metastasis: A clinical perspective. Cancer Manag Res.
5:187–195. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Q, Tang X, Zhang ZF, Velikina R, Shi
S and Le AD: Nicotine induces hypoxia-inducible factor-1alpha
expression in human lung cancer cells via nicotinic acetylcholine
receptor-mediated signaling pathways. Clin Cancer Res.
13:4686–4694. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma X, Jia Y, Zu S, Li R, Jia Y, Zhao Y,
Xiao D, Dang N and Wang Y: α5 nicotinic acetylcholine receptor
mediates nicotineinduced HIF-1α and VEGF expression in non-small
cell lung cancer. Toxicol Appl Pharmacol. 278:172–179. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nurwidya F, Takahashi F, Murakami A and
Takahashi K: Epithelial mesenchymal transition in drug resistance
and metastasis of lung cancer. Cancer Res Treat. 44:151–156. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang L, Zhang X, Liu J, Shen L and Li Z:
Tea consumption and lung cancer risk: A meta-analysis of
case-control and cohort studies. Nutrition. 30:1122–1127. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin IH, Ho ML, Chen HY, Lee HS, Huang CC,
Chu YH, Lin SY, Deng YR, He YH, Lien YH, et al: Smoking, green tea
consumption, genetic polymorphisms in the insulin-like growth
factors and lung cancer risk. PLoS One. 7:e309512012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ravindranath MH, Ramasamy V, Moon S, Ruiz
C and Muthugounder S: differential growth suppression of human
melanoma cells by tea (Camellia sinensis) epicatechins (ECG, EGC
and EGCG). Evid Based Complement Alternat Med. 6:523–530. 2009.
View Article : Google Scholar :
|
|
14
|
Du GJ, Zhang Z, Wen XD, Yu C, Calway T,
Yuan CS and Wang CZ: Epigallocatechin gallate (EGCG) is the most
effective cancer chemopreventive polyphenol in green tea.
Nutrients. 4:1679–1691. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang F, Chang Z, Fan Q and Wang L:
Epigallocatechin-3-gallate inhibits the proliferation and migration
of human ovarian carcinoma cells by modulating p38 kinase and
matrix metallo-proteinase-2. Mol Med Rep. 9:1085–1089.
2014.PubMed/NCBI
|
|
16
|
Zhang W, Yang P, Gao F, Yang J and Yao K:
Effects of epigallocatechin gallate on the proliferation and
apoptosis of the nasopharyngeal carcinoma cell line CNE2. Exp Ther
Med. 8:1783–1788. 2014.PubMed/NCBI
|
|
17
|
Li H, Li Z, Xu YM, Wu Y, Yu KK, Zhang C,
Ji YH, Ding G and Chen FX: Epigallocatechin-3-gallate induces
apoptosis, inhibits proliferation and decreases invasion of glioma
cell. Neurosci Bull. 30:67–73. 2014. View Article : Google Scholar
|
|
18
|
Li X, Feng Y, Liu J, Feng X, Zhou K and
Tang X: Epigallocatechin-3-gallate inhibits IGF-I-stimulated lung
cancer angiogenesis through downregulation of HIF-1α and VEGF
expression. J Nutrigenet Nutrigenomics. 6:169–178. 2013. View Article : Google Scholar
|
|
19
|
Zhang Q, Tang X, Lu Q, Zhang Z, Rao J and
Le AD: Green tea extract and (−)-epigallocatechin-3-gallate inhibit
hypoxia- and serum-induced HIF-1alpha protein accumulation and VEGF
expression in human cervical carcinoma and hepatoma cells. Mol
Cancer Ther. 5:1227–1238. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
He L, Zhang E, Shi J, Li X, Zhou K, Zhang
Q, Le AD and Tang X: (−)-Epigallocatechin-3-gallate inhibits human
papillomavirus (HPV)-16 oncoprotein-induced angiogenesis in
non-small cell lung cancer cells by targeting HIF-1α. Cancer
Chemother Pharmacol. 71:713–725. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu LC, Tsao TC, Hsu SR, Wang HC, Tsai TC,
Kao JY and Way TD: EGCG inhibits transforming growth
factor-β-mediated epithelial-to-mesenchymal transition via the
inhibition of Smad2 and Erk1/2 signaling pathways in nonsmall cell
lung cancer cells. J Agric Food Chem. 60:9863–9873. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ko H, So Y, Jeon H, Jeong MH, Choi HK, Ryu
SH, Lee SW, Yoon HG and Choi KC: TGF-β1-induced
epithelial-mesenchymal transition and acetylation of Smad2 and
Smad3 are negatively regulated by EGCG in human A549 lung cancer
cells. Cancer Lett. 335:205–213. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takahashi A, Watanabe T, Mondal A, Suzuki
K, Kurusu-Kanno M, Li Z, Yamazaki T, Fujiki H and Suganuma M:
Mechanism-based inhibition of cancer metastasis with
(−)-epigal-locatechin gallate. Biochem Biophys Res Commun. 443:1–6.
2014. View Article : Google Scholar
|
|
24
|
Nilsson I, Shibuya M and Wennström S:
differential activation of vascular genes by hypoxia in primary
endothelial cells. Exp Cell Res. 299:476–485. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li G, He L, Zhang E, Shi J, Zhang Q, Le
AD, Zhou K and Tang X: Overexpression of human papillomavirus (HPV)
type 16 oncoproteins promotes angiogenesis via enhancing HIF-1α and
VEGF expression in non-small cell lung cancer cells. Cancer Lett.
311:160–170. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Skinner HD, Zheng JZ, Fang J, Agani F and
Jiang BH: Vascular endothelial growth factor transcriptional
activation is mediated by hypoxia-inducible factor 1alpha, HDM2,
and p70S6K1 in response to phosphatidylinositol 3-kinase/AKT
signaling. J Biol Chem. 279:45643–45651. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang L, Gu F, Ma N, Zhang L, Bian JM and
Cao HY: CIP2A expression is associated with altered expression of
epithelial-mesenchymal transition markers and predictive of poor
prognosis in pancreatic ductal adenocarcinoma. Tumour Biol.
34:2309–2313. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao D, Yang K, Tang XF, Lin NN and Liu
JY: Expression of integrin-linked kinase in adenoid cystic
carcinoma of salivary glands correlates with epithelial-mesenchymal
transition markers and tumor progression. Med Oncol. 30:6192013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Balanis N, Wendt MK, Schiemann BJ, Wang Z,
Schiemann WP and Carlin CR: Epithelial to mesenchymal transition
promotes breast cancer progression via a fibronectin-dependent
STAT3 signaling pathway. J Biol Chem. 288:17954–17967. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xing Y, Qi J, Deng S, Wang C, Zhang L and
Chen J: Small interfering RNA targeting ILK inhibits metastasis in
human tongue cancer cells through repression of
epithelial-to-mesenchymal transition. Exp Cell Res. 319:2058–2072.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
López-Novoa JM and Nieto MA: Inflammation
and EMT: An alliance towards organ fibrosis and cancer progression.
EMBO Mol Med. 1:303–314. 2009. View Article : Google Scholar
|
|
32
|
Pirozzi G, Tirino V, Camerlingo R, Franco
R, La Rocca A, Liguori E, Martucci N, Paino F, Normanno N and Rocco
G: Epithelial to mesenchymal transition by TGFβ-1 induction
increases stemness characteristics in primary non small cell lung
cancer cell line. PLoS One. 6:e215482011. View Article : Google Scholar
|
|
33
|
Kim H, Yoo SB, Sun P, Jin Y, Jheon S, Lee
CT and Chung JH: Alteration of the E-cadherin/β-catenin complex is
an independent poor prognostic factor in lung adenocarcinoma.
Korean J Pathol. 47:44–51. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhan H, Liang H, Liu X, Deng J, Wang B and
Hao X: Expression of Rac1, HIF-1α, and VEGF in gastric carcinoma:
Correlation with angiogenesis and prognosis. Onkologie. 36:102–107.
2013. View Article : Google Scholar
|
|
35
|
Simonetti O, Lucarini G, Rubini C, Goteri
G, Zizzi A, Staibano S, Campanati A, Ganzetti G, Di Primio R and
Offidani A: Microvessel density and VEGF, HIF-1α expression in
primary oral melanoma: Correlation with prognosis. Oral dis.
19:620–627. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kang FW, Gao Y, Que L, Sun J and Wang ZL:
Hypoxia-inducible factor-1α overexpression indicates poor clinical
outcomes in tongue squamous cell carcinoma. Exp Ther Med.
5:112–118. 2013.
|
|
37
|
Zhang L, Huang G, Li X, Zhang Y, Jiang Y,
Shen J, Liu J, Wang Q, Zhu J, Feng X, et al: Hypoxia induces
epithelial-mesenchymal transition via activation of SNAI1 by
hypoxia-inducible factor -1α in hepatocellular carcinoma. BMC
Cancer. 13:108–117. 2013. View Article : Google Scholar
|
|
38
|
Ning Q, Liu C, Hou L, Meng M, Zhang X, Luo
M, Shao S, Zuo X and Zhao X: Vascular endothelial growth factor
receptor-1 activation promotes migration and invasion of breast
cancer cells through epithelial-mesenchymal transition. PLoS One.
8:e652172013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Semenza GL: Defining the role of
hypoxia-inducible factor 1 in cancer biology and therapeutics.
Oncogene. 29:625–634. 2010. View Article : Google Scholar :
|
|
40
|
Jiang Y, Cukic B, Adjeroh DA, Skinner HD,
Lin J, Shen QJ and Jiang BH: An algorithm for identifying novel
targets of transcription factor families: Application to
hypoxia-inducible factor 1 targets. Cancer Inform. 7:75–89.
2009.PubMed/NCBI
|
|
41
|
Kaidi A, Qualtrough D, Williams AC and
Paraskeva C: Direct transcriptional up-regulation of
cyclooxygenase-2 by hypoxia-inducible factor (HIF)-1 promotes
colorectal tumor cell survival and enhances HIF-1 transcriptional
activity during hypoxia. Cancer Res. 66:6683–6691. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Obacz J, Pastorekova S, Vojtesek B and
Hrstka R: Cross-talk between HIF and p53 as mediators of molecular
responses to physiological and genotoxic stresses. Mol Cancer.
12:932013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang E, Feng X, Liu F, Zhang P, Liang J
and Tang X: Roles of PI3K/Akt and c-Jun signaling pathways in human
papillomavirus type 16 oncoprotein-induced HIF-1α, VEGF, and IL-8
expression and in vitro angiogenesis in non-small cell lung cancer
cells. PLoS One. 9:e1034402014. View Article : Google Scholar
|
|
44
|
Yu H, Zhang L and Liu P: CXCR7 signaling
induced epithelial-mesenchymal transition by AKT and ERK pathways
in epithelial ovarian carcinomas. Tumour Biol. Oct 31–2014.Epub
ahead of print.
|
|
45
|
Wojtalla A and Arcaro A: Targeting
phosphoinositide 3-kinase signalling in lung cancer. Crit Rev Oncol
Hematol. 80:278–290. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang J, Cao J, Ma S, et al: Tumor hypoxia
enhances non-small cell lung cancer metastasis by selectively
promoting macrophage M2 polarization through the activation of ERK
signaling. Oncotarget Oct. 5:9664–9677. 2014.
|
|
47
|
Beck JT, Ismail A and Tolomeo C: Targeting
the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of
rapamycin (mTOR) pathway: An emerging treatment strategy for
squamous cell lung carcinoma. Cancer Treat Rev. 40:980–989. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Stinchcombe TE and Johnson GL: MEK
inhibition in non-small cell lung cancer. Lung Cancer. 86:121–125.
2014. View Article : Google Scholar : PubMed/NCBI
|