Introduction
Endometrial carcinoma (EC) is one of the most common
malignancies of the female genital tract. Mortality rates from EC
are increasing in many countries and this may be attributed to an
increased rate of advanced stage cancers and high-risk histologies
(1). Surgery is currently the major
treatment for EC, yet it is critically important to develop new
therapeutic strategies for EC. Research of the molecular mechanisms
of EC may help to identify new diagnostic and therapeutic
targets.
MicroRNAs (miRNAs) are 18–24 nucleotide non-coding
RNAs which play an important role in regulating gene expression by
cleaving or translational repression of mRNAs (2). Recent studies have demonstrated the
dysregulation of specific miRNAs in different types of tumors,
including stomach, lung, breast, ovarian and cervical cancer and
leukemia (3–5). These miRNAs may act as tumor
suppressors or oncogenes by regulating processes such as cell
proliferation, apoptosis, metastasis and invasion (5–8), and
our previous research demonstrated that miR-449a is a suppressor of
EC (9).
The miR-34 family consists of miR-34a (5′-UGGCAG
UGUCUUAGCUGGUUGU-3′), miR-34b (5′-CAAUCACUA ACUCCACUGCCAU-3′) and
miR-34c (5′-AGGCAGUGUAGUUAGCUGAUUGC-3′). Numerous studies have
shown the dysregulation of miR-34 in various types of cancers,
including hepatocellular, mesothelial and colon cancer, melanomas,
neuroblastomas and leukemia (10–12).
Yet, little research has been conducted in endometrial cancer.
E2F3 is a member of the E2F family, and is essential
for tumor growth by regulating cell proliferation and apoptosis
(13,14). Whether E2F3 acts on endometrial
cancer cells remains unclear.
In the present study, we investigated the
relationship between miR-34c and EC. It has been reported that
miR-34c is significantly reduced in EC (15,16),
and numerous studies have shown that decreased expression of
miR-34c in cancer cells correlates with aberrant DNA methylation
(17,18). To further study the function of
miR-34c in EC, we upregulated the expression of miR-34c in the EC
cell line, HEC-1-B, and then sought to identify the effect of
miR-34c on cell proliferation, colony formation, cell cycle arrest,
apoptosis, migration and invasion. Finally, we determined the
expression of E2F3 in the HEC-1-B cells using western blot
analysis.
Materials and methods
Cell line and culture
EC cell line (HEC-1-B) was obtained from the
Shanghai Institute of Cell Biology (Shanghai, China). The cells
were maintained in Dulbecco’s modified Eagle’s medium (DMEM)
supplemented with 10% fetal bovine serum (FBS) (both from Gibco),
and incubated under a condition of a humidified atmosphere of 5%
CO2 and 95% air at 37°C.
Cell transfection
We purchased hsa-miR-34c-5p mimics and a negative
control (NC) from GenePharma (Shanghai, China). The sequence of
hsa-miR-34c-5p mimics was 5′-AGGCAGUGUAGUUAGCUGAUUGC-3′ and the
sequence of NC was 5′-UUCUCCGAACGUGUCACGUTT-3′. One day before
transfection, HEC-1-B cells were seeded in 96-well plates at
2.5×103 or 2×105 cells/well in 6-well plates
in growth medium (DMEM supplemented with 10% FBS) without
antibiotics, such that they were 30–50% confluent at the time of
transfection. On the following day, the cells were transfected with
hsa-miR-34c-5p mimics or NC at a final concentration of 50 nmol/l
using Lipofectamine 2000 transfection reagent (Invitrogen)
according to the manufacturer’s instructions. There were three
groups: the miR-34c group, the NC group, and the untransfected
group.
RNA isolation and RT-qPCR
Reverse transcription quantitative PCR (RT-qPCR) was
applied to ensure that the HEC-1-B cells were successfully
transfected. Forty-eight hours after transfection, total RNA was
isolated from the HEC-1-B cells with TRIzol reagent (Invitrogen)
according to the manufacturer’s instructions. Reverse transcription
PCR (RT-PCR) and real-time PCR (qPCR) were performed according to
the manufacturer’s instructions. Briefly, miRNAs (contained in 500
ng total RNA each group) were polyadenylated and further reverse
transcribed into cDNA using miRNA First-Strand cDNA Synthesis and
qRT-PCR kits (Invitrogen). Next, we diluted the cDNA 1:10 in
DEPC-treated water and the diluent cDNA served as the template for
qPCR.
We used Applied Biosystems 7500 detection system to
perform the qPCR. U6 snRNA was used as an endogenous control for
normalization. The forward primers of hsa-miR-34c-5p and U6 used in
our study were synthesized by Invitrogen. The primer sequence of
hsa-miR-34c-5p was 5′-AGGCAGTGTAGTTAGCTGATTGC-3′ and the primer
sequence of U6 was 5′-CGCAAGGATGACACGCAAA TTC-3′. The universal
reverse primer was offered in the kit. After mixing 10 μl of
the PCR system, which included 5 μl of SYBR-Green Master Mix
(Toyobo, Japan), 1 μl of the specific forward primer (1
μmol/l), and 1 μl of the reverse primer (1
μmol/l), 1 μl plus solution, 1 μl template and
1 μl DEPC-treated water, we placed the reactions in a
real-time instrument at 95°C for 3 min, and 40 cycles (95°C, 15 sec
and 60°C, 1 min for each cycle). Then, we obtained Ct values for
each sample, and the relative expression of miR-34c was calculated
by the 2−ΔΔCt method.
Cell proliferation assay
Cells were transfected 24 h after seeding in 96-well
plates at 2,500 cells/well. Then we evaluated cell proliferation
using CCK-8 reagent (Beyotime Institute of Biotechnology, China) at
24, 48, 72, and 96 h after transfection, respectively. Briefly, we
replaced the DMEM in the well, then added 10 μl of CCK-8
solution into each well and incubation was carried out at 37°C for
an additional 2 h. We then determined the absorbance at a
wavelength of 450 nm.
Colony formation assay
Seventy-two hours after transfection, the cells were
collected and resuspended in DMEM with 10% FBS. Then the cells were
seeded in 6-well plates at an amount of 800 cells/well, and
incubated at 37°C in a humidified incubator with 5% CO2
for 14 days until visible colonies (containing 40 or more cells)
formed. The cell colonies were stained with crystal violet for 30
min. We counted the numbers of colonies in each group.
Cell apoptosis assay
The cell apoptosis assay was performed using the
Alexa Fluor 488 Annexin V apoptosis kit (Invitrogen). Briefly, the
cells were collected 72 h after transfection, washed twice with
cold PBS and resuspended in Annexin-binding buffer supplied in the
kit. Subsequently, the cells were stained using Annexin V and PI
according to the manufacturer’s instructions. Cell apoptosis was
analyzed by flow cytometry.
Cell cycle assay
Approximately 2×105–1×106
cells were collected 48 h after transfection, and washed once with
PBS. Then the cells were resuspended in 1 ml DNA staining solution
(MultiSciences Biotech Co., Ltd.), incubated for 30 min at room
temperature and analyzed by flow cytometry in the presence of the
dye.
Transwell migration and invasion
assays
For the Transwell migration assay, the cells were
harvested 72 h after transfection and resuspended in serum-free
medium at a concentration of 1.5×105 cells/ml. Two
hundred microliters of the cell suspension was transferred into the
top chamber with a non-coated membrane (8-μm pore; BD
Biosciences), and the chamber was inserted in a 24-well plate. For
the Transwell invasion assay, 200 μl of the cell suspension
at a concentration of 3.0×105 cells/ml was transferred
into the top chamber with a Matrigel-coated membrane (8-μm
pore; BD Biosciences). In both assays, 500 μl DMEM
containing 10% FBS was added to the bottom chamber in a 24-well
plate. The plate was placed in an incubator for 24 h. After that,
we wiped away the cells on the upper surface of the membrane with a
cotton swab, then fixed the cells on the lower surface of the
membrane in 4% paraformaldehyde for 30 min, stained them with
crystal violet for 30 min, and evaluated the migration or invasive
ability by counting the numbers of stained cells under a
microscope.
Western blot analysis
Total proteins in each group of cells were extracted
72 h after transfection, and resolved by 10% SDS-PAGE. The proteins
were transferred onto PVDF membranes (Solarbio) and blocked in TBST
containing 5% skim milk for 1 h. Next, the membranes were incubated
with primary antibodies for E2F3 (rabbit anti-human polyclonal
antibody, diluted as 1:1,000, ab50917; Abcam) and β-actin
(1:10,000, AP0060; BioWorld) for 1 h, respectively. After washing 3
times with TBST (each time 15 min), the membranes were incubated
with secondary antibody (peroxidase conjugated goat anti-rabbit
IgG, 1:5,000, GAR007; MultiSciences Biotech Co., Ltd.) for 1 h.
After washing the membranes 3 times with TBST (each time 15 min),
we detected the blots using SuperSignal West Femto Maximum
Sensitivity Substrate kit (Thermo Fisher Scientific) and protein
bands were exposed to Kodak film (Sigma-Aldrich). Relative
expression of E2F3 protein was normalized to β-actin.
Statistical analysis
Statistical analysis of all data was performed using
SPSS 19.0 software package. Each experiment was performed at least
3 times, and differences between each group were evaluated by
one-way ANOVA. The results are shown as the mean ± SD. P<0.05
was considered to indicate a statistically significant result.
Results
Expression of miR-34c after
transfection
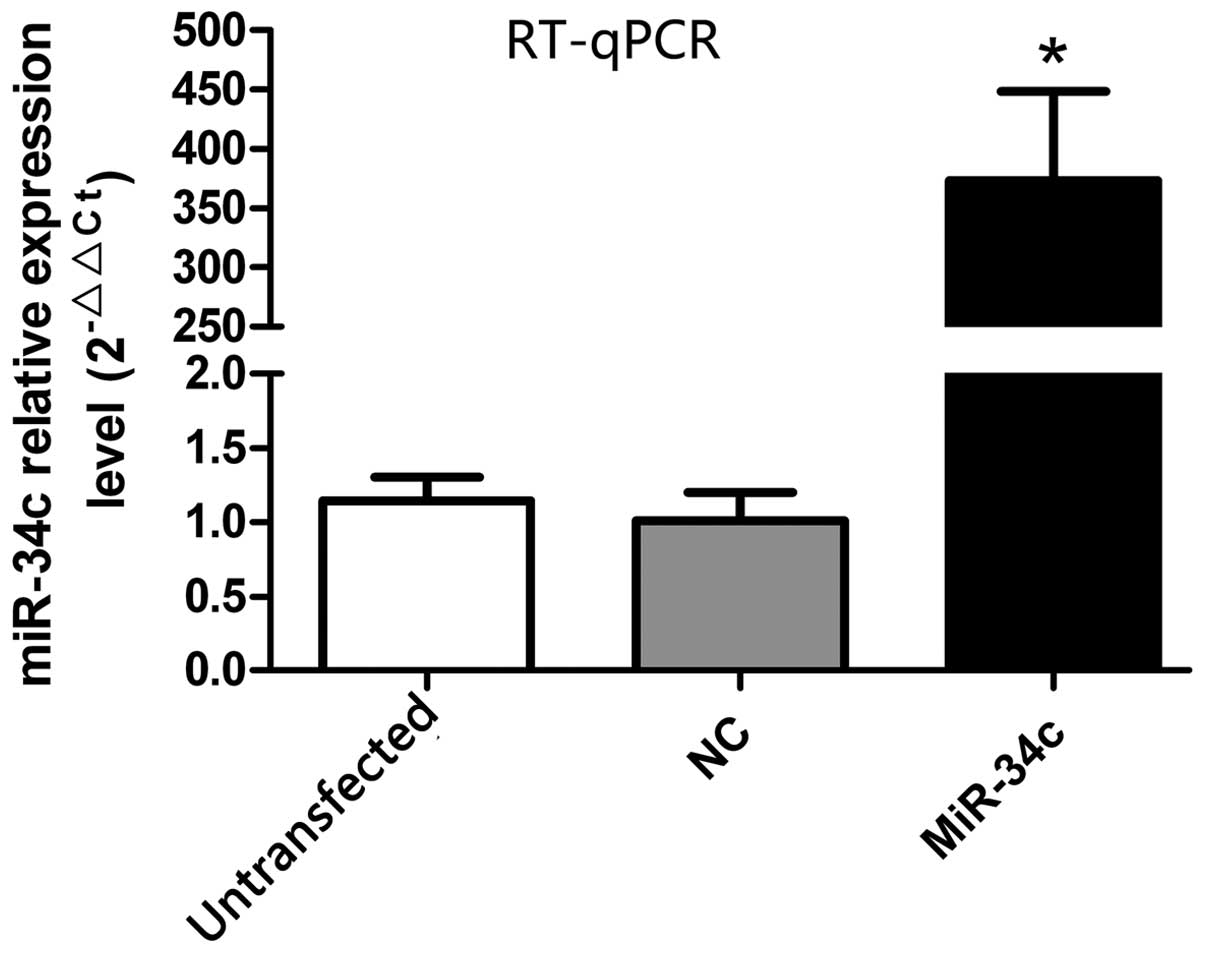
The expression of miR-34c was detected by RT-qPCR 48
h after transfection (Fig. 1).
There was increased expression of miR-34c in the miR-34c group
compared with the NC and untransfected group (miR-34c group,
373.43±74.95; NC group, 1.01±0.19; untransfected group, 1.14±0.16,
P<0.01).
miR-34c inhibits the growth of HEC-1-B
cells
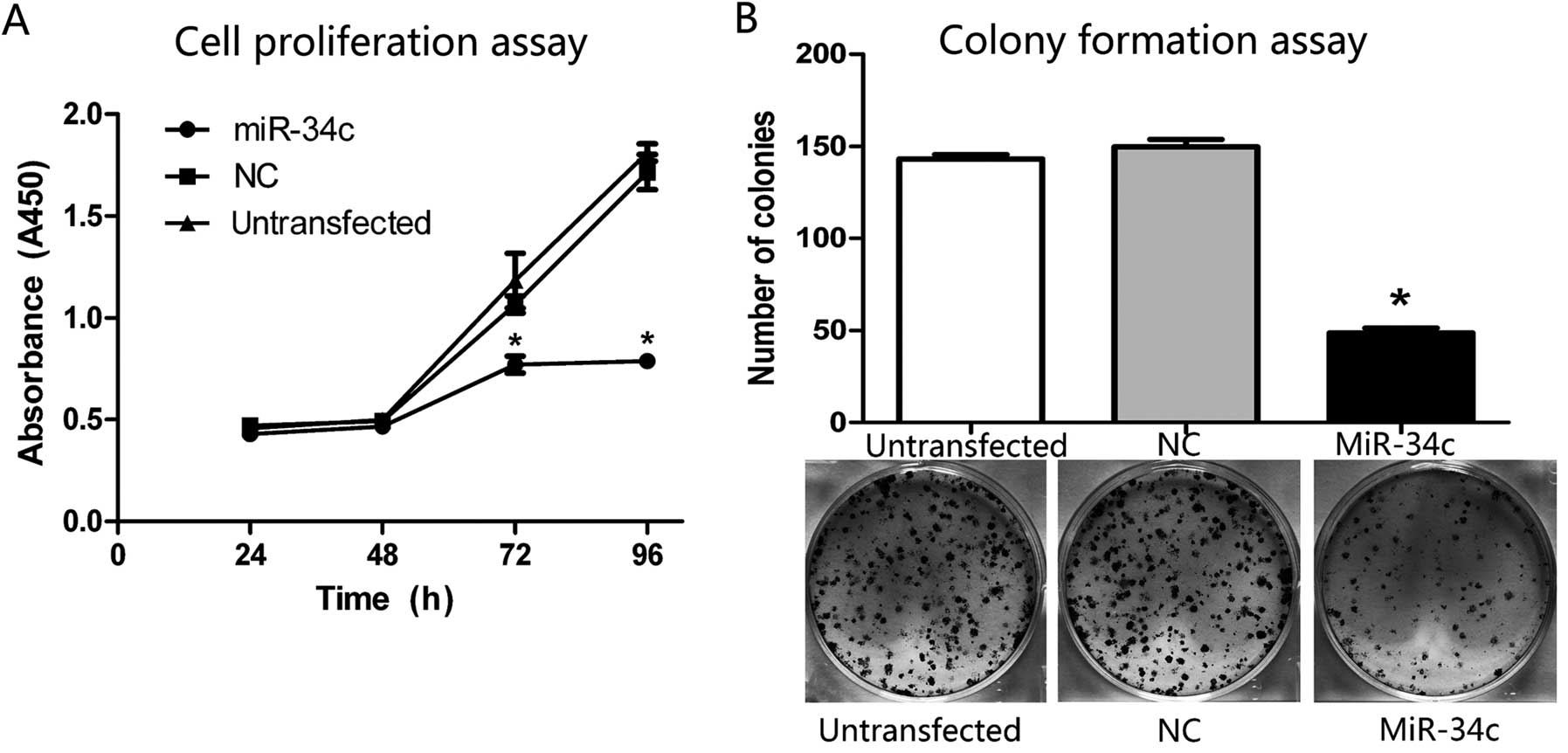
To confirm a proliferation-suppressive effect of
miR-34c in the HEC-1-B cells, the absorbance at the wavelength of
450 nm was measured 2 h after adding CCK-8 to reflect the amount of
cells indirectly. The absorbance in the miR-34c group was
significantly decreased by ~28.0% (P<0.01) at 72 h and 54.1%
(P<0.01) at 96 h compared with the NC and untransfected group
(Fig. 2A), indicating that the cell
proliferation was suppressed followed by increased expression of
miR-34c.
To further prove the function of miR-34c on growth
inhibition of the HEC-1-B cells, we examined the role of miR-34c on
cell colony formation. As shown in Fig.
2B, the colony formation capacity was markedly decreased in the
miR-34c group. We counted the numbers of colonies in each group,
and the numbers of colonies in the NC and untransfected group
reached 149.7±4.0 and 143.0±2.6, respectively, while it was only
49.0±2.5 in the miR-34c group (P<0.01).
miR-34c promotes the apoptosis of HEC-1-B
cells
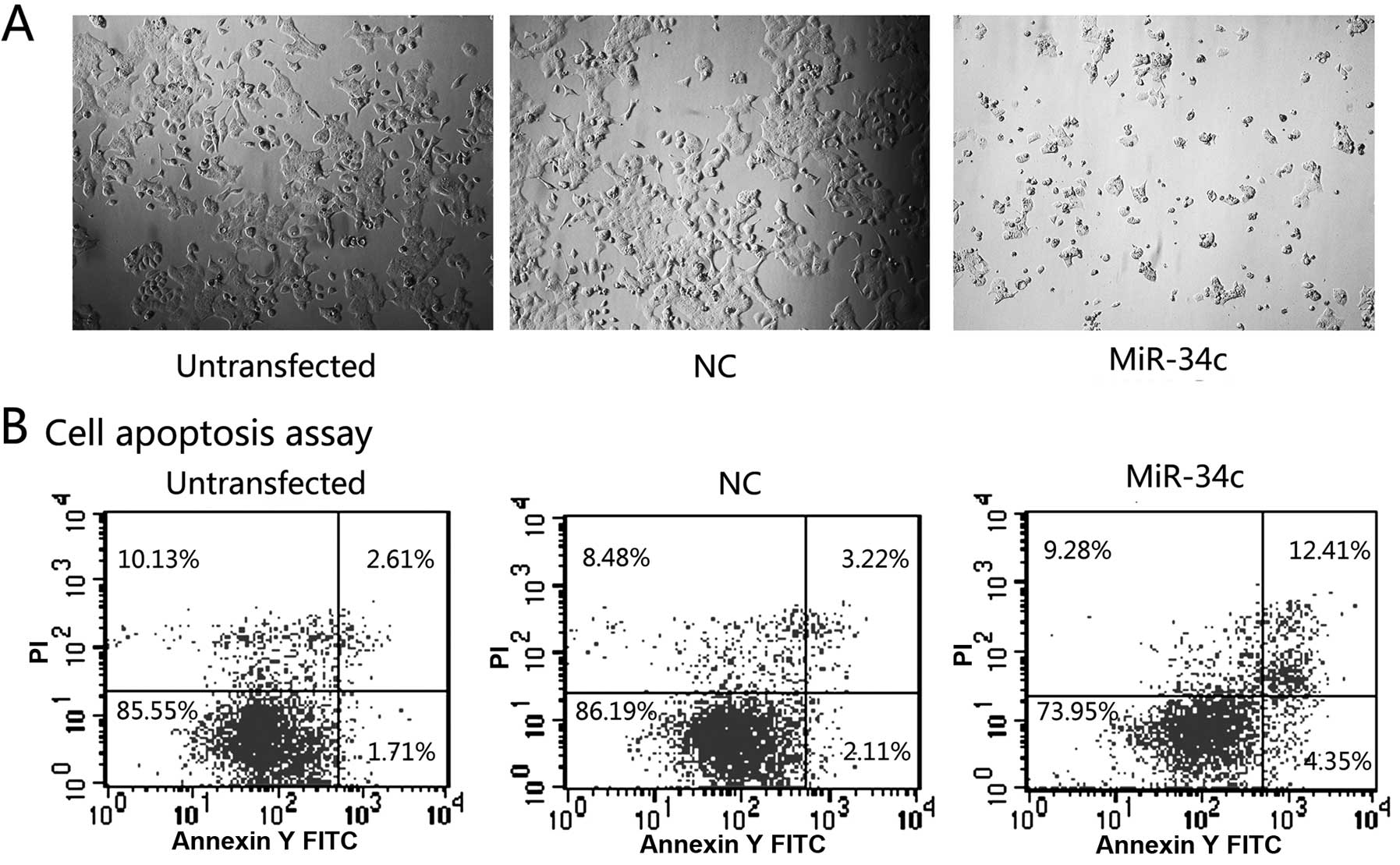
Seventy-two hours after transfection, the cells in
the miR-34c group were in an obviously poorer status when compared
to the NC and untransfected group (Fig.
3A). Following analysis by flow cytometry (Fig. 3B), the percentages of early and late
apoptotic cells were significantly increased in the miR-34c group
(16.76% in total) when compared with these percentages in the other
2 groups (4.32 and 5.33% in total, respectively).
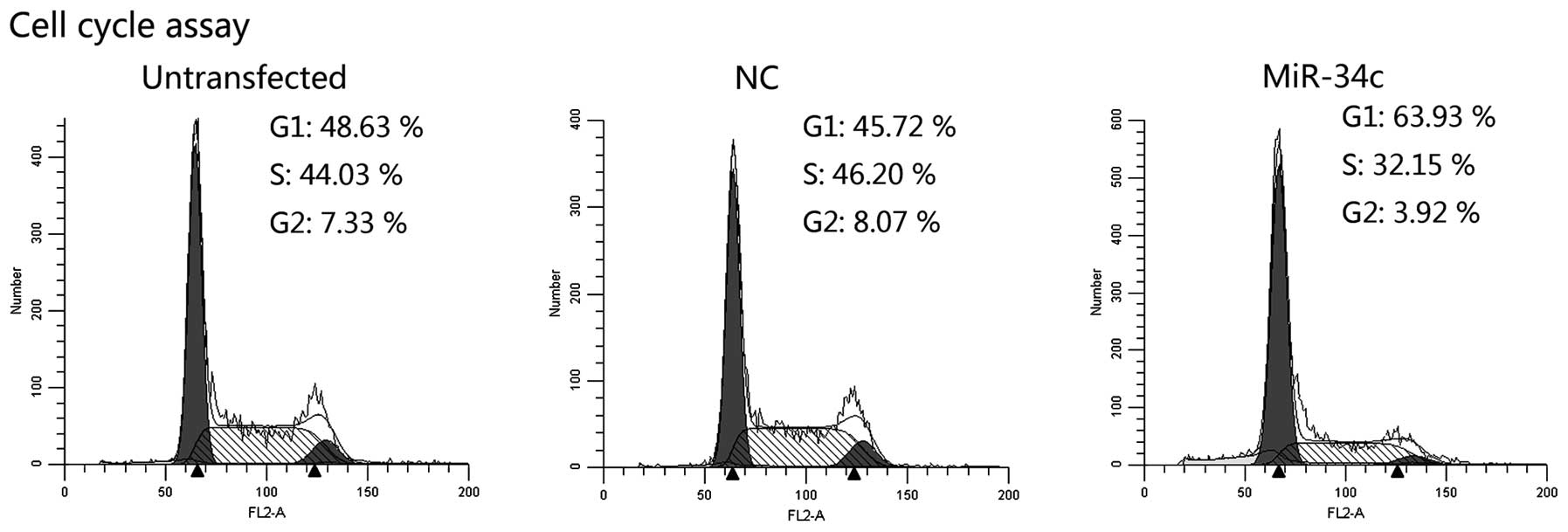
miR-34c induces cell cycle arrest
Cell cycle distribution was evaluated by flow
cytometry. Overexpression of miR-34c led to the accumulation of
cells in the G1 phase and the reduction of cells in the
S and G2 phase (Fig. 4).
These results suggest that miR-34c influenced cell cycle transition
from G1 to the S phase and induced cell cycle arrest at
the G1 phase.
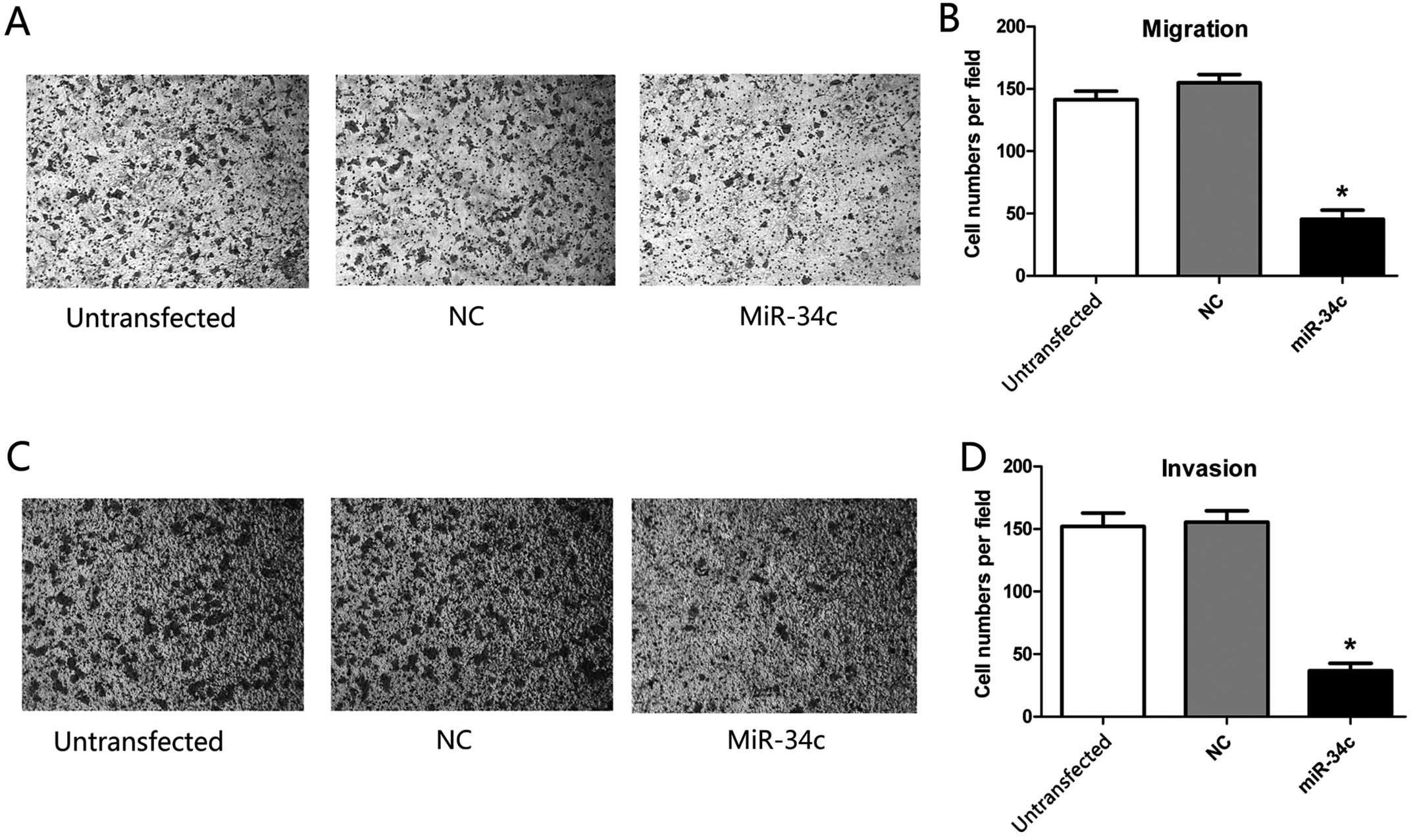
miR-34c inhibits the migration and
invasion of HEC-1-B cells
We identified the effect of miR-34c on the migration
(Fig. 5A) and invasion (Fig. 5C) of the HEC-1-B cells by Transwell
assay. In the Transwell migration assay, a 70.5% decrease in
HEC-1-B cells (P<0.01) was detected 72 h after transfection of
the hsa-miR-34c-5p mimics (Fig.
5B). In the Transwell invasion assay, a 75.8% decrease in
HEC-1-B cells was noted after transfection of the hsa-miR-34c-5p
mimics (P<0.01, Fig. 5D).
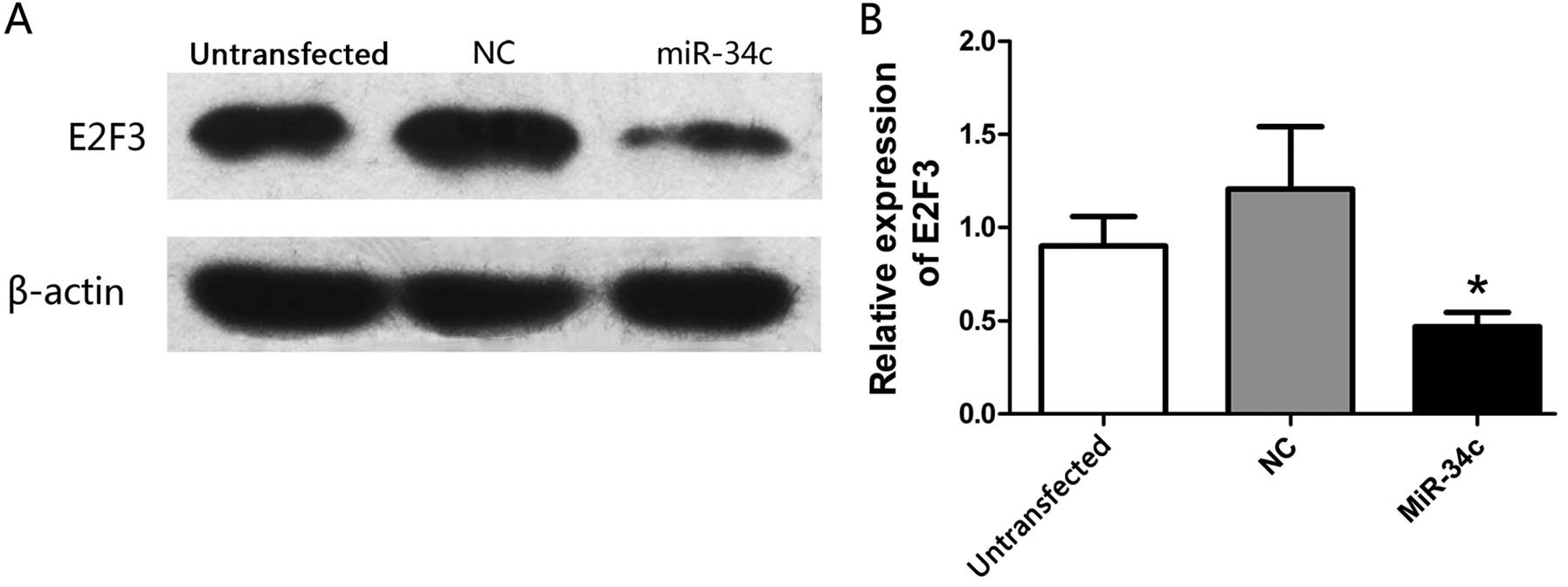
miR-34c suppresses the expression of E2F3
protein
To determine whether miR-34c regulates the
expression of E2F3 protein, the expression of E2F3 was detected by
western blot analysis 72 h after transfection. As expected, the
expression of E2F3 protein was apparently inhibited after the
transfection of the hsa-miR-34c-5p mimics (Fig. 6A and B).
Discussion
Our research together with a study conducted by
Hiroki et al (15) showed
that miR-34c is downregulated in EC, and this transformation may be
an important factor in the poor prognosis of EC. To further study
the function of miR-34c, we upregulated the expression of miR-34c
in HEC-1-B cells by transfection with the hsa-miR-34c-5p mimics,
and we found that the overexpression of miR-34c inhibited cell
proliferation, colony formation, migration and invasion, promoted
apoptosis, and induced cell cycle arrest, demonstrating that
miR-34c is a suppressor of HEC-1-B cells, and western blot analysis
demonstrated that E2F3 protein may be a target of miR-34c. To our
knowledge, the present study is the first to confirm that miR-34c
suppresses HEC-1-B cells by targeting E2F3.
In the present study, we found that the decrease in
the number of cells may be partly related to cell apoptosis.
Inhibition of cell proliferation together with apoptosis ultimately
inhibited the formation of cell colonies. A previous study showed
that inhibition of cell proliferation and apoptosis may be partly
caused by cell cycle arrest after transfection with miR-34s
(19). Notably, our results found
that both inhibition of cell proliferation and apoptosis appeared
72 h after transfection with the hsa-miR-34c-5p mimics, later than
cell cycle arrest (48 h after transfection). Cell cycle arrest at
the G1 phase is likely to be the key function of miR-34c
in HEC-1-B cells. We also found that miR-34c significantly
inhibited the migration and invasion of the HEC-1-B cells, which
may also be clinically significant.
TargetScan analysis indicates that E2F3 may be a
target of miR-34c. E2F3 is a member of the E2F family, whose
potential oncogenic capacity was reported (20), and studies show that E2F3 regulates
E2F-mediated DNA replication and cell proliferation by a
p53-dependent negative feedback loop (21). Lax et al (22) reported that p53 mutations were found
in 93% of uterine serous carcinoma and 17% of uterine endometrioid
carcinoma, and that this may be responsible for their morphology
and biologic behavior. In addition, it has been demonstrated that
members of the miR-34 family are direct transcriptional targets of
p53, playing a role as tumor suppressors (19,23).
Based on the reports mentioned above, we proposed that the effects
of miR-34c on HEC-1-B cells operate through E2F3 protein. To test
this hypothesis, we used western blot analysis to demonstrate that
E2F3 was markedly downregulated after transfection with the
hsa-miR-34c-5p mimics. These studies, as well as our results,
suggest that E2F3 protein is a target protein of miR-34c. It has
been reported that overexpression of E2F3 protein is an important
marker for poor clinical outcome in prostate cancer (24). These results suggest that E2F3 may
exert a significant influence on tumor progression.
Leone et al (25) demonstrated that E2F3 influences cell
cycle arrest in the induction of S phase during the transition from
quiescence to proliferation by regulating Cdc6, cyclin E, Cdk2 and
Mcm proteins. Similarly, our data showed reduced E2F3, and cell
cycle arrest at G1 phase accompanied by reduction in the
percentage of cells in the S phase after transfection of
hsa-miR-34c-5p mimics, suggesting that miR-34c acts as a tumor
suppressor by inducing cell cycle arrest at the G1 phase
via E2F3.
Invasive growth in many human tumors is associated
with overexpression of MET (26),
and recent studies show that knockdown of cyclophilin A leads to a
significant inhibition of cell migration and invasion in HEC-1-B
cells by down-regulating focal adhesion signaling (27). Our data revealed that miR-34c
significantly inhibited migration and invasion of HEC-1-B cells;
however, futher studies are necessary to confirm the effects of
miR-34c on the migration and invasion of HEC-1-B cells and whether
this is also associated with MET and cyclophilin A.
In conclusion, miR-34c acts as a direct target of
p53, its overexpression suppresses the expression of E2F3 protein,
and a low level of E2F3 causes cell cycle arrest at the
G1 phase by acting on a series of cell cycle
arrest-related proteins. Then, cell cycle arrest induces cell
proliferation inhibition and apoptosis partly and further inhibits
cell colony formation. miR-34c can also inhibit cell migration and
invasion.
Although therapeutic strategies for EC have improved
greatly in the last few decades, EC still has a significantly poor
prognosis. Understanding the pathogenesis at the molecular level is
essential for the development of new targeted therapies. In the
present study, we successfully identified miR-34c as a suppressor
of the EC cell line (HEC-1-B), and demonstrated E2F3 as a target of
miR-34c in HEC-1-B cells. Further studies to ascertain whether
overexpression of miR-34c has a therapeutic role in EC are
necessary. We believe that miR-34c has potential value in the early
diagnosis and molecular therapy of EC.
Acknowledgments
This study was supported by the Zhejiang Provincial
Natural Science Foundation of China (no. Y2090699).
References
|
1
|
Ueda SM, Kapp DS, Cheung MK, Shin JY,
Osann K, Husain A, Teng NN, Berek JS and Chan JK: Trends in
demographic and clinical characteristics in women diagnosed with
corpus cancer and their potential impact on the increasing number
of deaths. Am J Obstet Gynecol. 198:218.e1–218.e6. 2008. View Article : Google Scholar
|
|
2
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Takamizawa J, Konishi H, Yanagisawa K,
Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y,
et al: Reduced expression of the let-7 microRNAs in human lung
cancers in association with shortened postoperative survival.
Cancer Res. 64:3753–3756. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bou Kheir T, Futoma-Kazmierczak E,
Jacobsen A, Krogh A, Bardram L, Hother C, Grønbæk K, Federspiel B,
Lund AH and Friis-Hansen L: miR-449 inhibits cell proliferation and
is down-regulated in gastric cancer. Mol Cancer. 10:292011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev Cancer. 6:259–269. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jiang F, Liu T, He Y, Yan Q, Chen X, Wang
H and Wan X: MiR-125b promotes proliferation and migration of type
II endometrial carcinoma cells through targeting TP53INP1 tumor
suppressor in vitro and in vivo. BMC Cancer. 11:4252011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tsuruta T, Kozaki K, Uesugi A, Furuta M,
Hirasawa A, Imoto I, Susumu N, Aoki D and Inazawa J: miR-152 is a
tumor suppressor microRNA that is silenced by DNA hypermethylation
in endometrial cancer. Cancer Res. 71:6450–6462. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Le XF, Merchant O, Bast RC and Calin GA:
The roles of microRNAs in the cancer invasion-metastasis cascade.
Cancer Microenviron. 3:137–147. 2010. View Article : Google Scholar
|
|
9
|
Ye W, Xue J, Zhang Q, Li F, Zhang W, Chen
H, Huang Y and Zheng F: miR-449a functions as a tumor suppressor in
endometrial cancer by targeting CDC25A. Oncol Rep. 32:1193–1199.
2014.PubMed/NCBI
|
|
10
|
Wong MY, Yu Y, Walsh WR and Yang JL:
microRNA-34 family and treatment of cancers with mutant or
wild-type p53 (Review). Int J Oncol. 38:1189–1195. 2011.PubMed/NCBI
|
|
11
|
Son MS, Jang MJ, Jeon YJ, Kim WH, Kwon CI,
Ko KH, Park PW, Hong SP, Rim KS, Kwon SW, et al: Promoter
polymorphisms of pri-miR-34b/c are associated with hepatocellular
carcinoma. Gene. 524:156–160. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Roy S, Levi E, Majumdar AP and Sarkar FH:
Expression of miR-34 is lost in colon cancer which can be
re-expressed by a novel agent CDF. J Hematol Oncol. 5:582012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Oeggerli M, Tomovska S, Schraml P,
Calvano-Forte D, Schafroth S, Simon R, Gasser T, Mihatsch MJ and
Sauter G: E2F3 amplification and overexpression is associated with
invasive tumor growth and rapid tumor cell proliferation in urinary
bladder cancer. Oncogene. 23:5616–5623. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ziebold U, Reza T, Caron A and Lees JA:
E2F3 contributes both to the inappropriate proliferation and to the
apoptosis arising in Rb mutant embryos. Genes Dev. 15:386–391.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hiroki E, Suzuki F, Akahira J, Nagase S,
Ito K, Sugawara J, Miki Y, Suzuki T, Sasano H and Yaegashi N:
MicroRNA-34b functions as a potential tumor suppressor in
endometrial serous adenocarcinoma. Int J Cancer. 131:E395–E404.
2012. View Article : Google Scholar
|
|
16
|
Jiang L, Meng W, Zeng J, Hu H and Lu L:
MiR-34c oligonucleotide enhances chemosensitivity of Ishikawa cell
to cisplatin by inducing apoptosis. Cell Biol Int. 37:577–583.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kubo T, Toyooka S, Tsukuda K, Sakaguchi M,
Fukazawa T, Soh J, Asano H, Ueno T, Muraoka T, Yamamoto H, et al:
Epigenetic silencing of microRNA-34b/c plays an important role in
the pathogenesis of malignant pleural mesothelioma. Clin Cancer
Res. 17:4965–4974. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Toyota M, Suzuki H, Sasaki Y, Maruyama R,
Imai K, Shinomura Y and Tokino T: Epigenetic silencing of
microRNA-34b/c and B-cell translocation gene 4 is associated with
CpG island methylation in colorectal cancer. Cancer Res.
68:4123–4132. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
He L, He X, Lim LP, de Stanchina E, Xuan
Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, et al: A microRNA
component of the p53 tumour suppressor network. Nature.
447:1130–1134. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen HZ, Tsai SY and Leone G: Emerging
roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev
Cancer. 9:785–797. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Timmers C, Sharma N, Opavsky R, Maiti B,
Wu L, Wu J, Orringer D, Trikha P, Saavedra HI and Leone G: E2f1,
E2f2, and E2f3 control E2F target expression and cellular
proliferation via a p53-dependent negative feedback loop. Mol Cell
Biol. 27:65–78. 2007. View Article : Google Scholar :
|
|
22
|
Lax SF, Kendall B, Tashiro H, Slebos RJ
and Hedrick L: The frequency of p53, K-ras mutations, and
microsatellite instability differs in uterine endometrioid and
serous carcinoma: evidence of distinct molecular genetic pathways.
Cancer. 88:814–824. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Corney DC, Flesken-Nikitin A, Godwin AK,
Wang W and Nikitin AY: MicroRNA-34b and microRNA-34c are targets of
p53 and cooperate in control of cell proliferation and
adhesion-independent growth. Cancer Res. 67:8433–8438. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Foster CS, Falconer A, Dodson AR, Norman
AR, Dennis N, Fletcher A, Southgate C, Dowe A, Dearnaley D, Jhavar
S, et al: Transcription factor E2F3 overexpressed in prostate
cancer independently predicts clinical outcome. Oncogene.
23:5871–5879. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Leone G, DeGregori J, Yan Z, Jakoi L,
Ishida S, Williams RS and Nevins JR: E2F3 activity is regulated
during the cell cycle and is required for the induction of S phase.
Genes Dev. 12:2120–2130. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Migliore C, Petrelli A, Ghiso E, Corso S,
Capparuccia L, Eramo A, Comoglio PM and Giordano S: MicroRNAs
impair MET-mediated invasive growth. Cancer Res. 68:10128–10136.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li Z, Gou J and Xu J: Down-regulation of
focal adhesion signaling in response to cyclophilin A knockdown in
human endometrial cancer cells, implicated by cDNA microarray
analysis. Gynecol Oncol. 131:191–197. 2013. View Article : Google Scholar : PubMed/NCBI
|