Introduction
Hepatocellular carcinoma (HCC) is the third leading
cause of cancer-related mortality worldwide (1). Advances are being made in
understanding the mechanisms underlying HCC, which in turn could
lead to novel therapeutics. Signaling pathways have become a major
source of targets for novel therapies for HCC. Multiple signaling
modules are deregulated in HCC, including some related to growth
factor signaling such as IGF, EGF, PDGF, FGF and HGF (2) and cell differentiation such as WNT,
Hedgehog and Notch (3) and
angiogenesis (VEGF) (4).
Intracellular mediators such as RAS and Akt/mTOR may also play a
role in HCC development and progression (5–7).
Different molecular mechanisms have been shown to induce aberrant
pathway activation. These include point mutations, chromosomal
aberrations and epigenetically driven down-regulation.
Among the above-mentioned signaling pathways, the
PI3K/Akt/mTOR pathway has received much attention from researchers
due to its critical role in tumor cell proliferation,
differentiation, survival, motility and invasion (8). Growth factors and hormones activate a
protein known as PI3K, which in turn activates Akt, which then
activates the mammalian target of rapamycin (mTOR) by inactivating
TSC2 to prevent inhibition of mTOR complex 1 (mTORC1). Active
mTORC1 has a number of downstream biological effects including
translation of mRNA via the phosphorylation of downstream targets
(4E-BP1 and p70 S6 kinase), suppression of autophagy and activation
of transcription leading to mitochondrial metabolism or
adipogenesis. Evidence also indicates that the mTORC1 complex plays
a role in HCC progression (9).
Numerous studies have been performed to target mTOR signaling to
impact tumor biology especially for proliferation and survival.
Rapamycin (sirolimus) is an mTOR inhibitor that has been shown to
have antitumor properties (10).
Everolimus is another mTOR inhibitor that has been shown to have
activity against HCC in xenografts (11,12).
Torin-2 is a novel, second-generation
ATP-competitive inhibitor that is potent and selective for mTOR
with a superior pharmacokinetic profile to previous inhibitors
(13,14). Torin-2 inhibits mTORC1-dependent
T389 phosphorylation on S6K (RPS6KB1). Torin-2 also exhibits potent
biochemical and cellular activity against PIKK family kinases
including ATM, ATR and DNA-PK. Torin-2 also displayed marked
anti-proliferative activity across a panel of cancer cell lines
(14,15). Cancer cell treatment with Torin-2
for 24 h resulted in a prolonged blockage of negative feedback and
consequent T308 phosphorylation of Akt. These effects were
associated with strong growth inhibition in vitro. Studies
of Torin-2 suggest that it has potent antitumor activity and
potential efficacy in HCC treatment.
Epigenetic regulation has emerged as an important
mechanism that regulates many chromatin template-based processes,
including transcription, DNA replication and repair (16). An important component of epigenetic
regulation is DNACpG methylation. DNACpG methylation has been shown
to play an important role in tumorigenesis (17). Ubiquitin-like containing PHD and
RING finger domains 1 (UHRF1) has been found to effectively
regulate DNACpG methylation, heterochromatin function and gene
expression (18,19). Overexpression of UHRF1 has been
suggested to contribute to tumorigenesis including HCC (20,21).
However, the relationship between mTORC1 signaling and
UHRF1-regulated DNACpG methylation has not been elucidated.
In the present study, we tested the effect of
Torin-2 on proliferation, cell cycle progression and apoptosis of
different hepatocellular carcinoma cell lines, Hep G2, SNU-182 and
Hep 3B2.1–7. We found higher mTORC1 activity in all three cell
lines, and consequently Torin-2 blocked mTORC1 in a dose-dependent
manner. Torin-2 treatment profoundly inhibited HCC cell
proliferation and survival through autophagy induction.
Unexpectedly, we discovered that Torin-2 also downregulated
expression of UHRF1. UHRF1 transcription was partially decreased by
Torin-2. Collectively, our research provides new clues regarding
the anti-HCC effect of Torin-2 and sheds light on a novel
therapeutic approach for HCC.
Materials and methods
Cell culture and treatment
Hep G2 and Hep 3B2.1–7 cells were cultured in
Eagle's minimum Essential medium (DMEM) supplemented with 10% fetal
bovine serum (FBS), 100 µg/ml penicillin, 50 µg/ml
streptomycin and 2 mM L-glutamine. SNU-182 cells were cultured in
RPMI-1640 medium supplemented with 10% FBS, 100 µg/ml
penicillin, 50 µg/ml streptomycin and 2 mM L-glutamine.
Torin-2 was purchased from Selleck Chemicals (Houston, TX, USA) and
was prepared following the manufacturer's instructions. Cells were
treated with Torin-2 at indicated concentrations for indicated
time-periods before they were subject to subsequent analysis. For
growth curve assay, 20,000–30,000 cells were seeded into each well
of 6-well plates. At the indicated time-points, the cells were
trypsinized with 0.25% trypsin-EDTA, and the cell number was
counted on a hemacytometer.
Apoptosis assay
Floating cells were collected after treatment.
Adherent cells were incubated in 2 mM ethylene diamine tetraacetic
acid (EDTA)-PBS. Cells were gently flushed with a pipette to
dissociate cells from the bottom of the wells. Flushed cells were
pooled with floating cells. Cells were then centrifuged and stained
with PI and Annexin V (both from BD Biosciences, Franklin Lakes,
NJ, USA). Briefly, the cells were washed with PBS twice and then
incubated in 100 µl of binding buffer containing Annexin
V-FITC (1:20) and 5 µg/ml PI. Four hundred microliters of
binding buffer was added to each sample. Apoptosis was analyzed by
flow cytometry within 1 h.
Cell proliferation and cell cycle
analysis
For cell proliferation analysis, the cells were
stained with 5 µg/ml FITC-conjugated antibody against Ki67
(Abcam, Cambridge, MA, USA). For cell cycle analysis, the cells
were fixed with 70% ethanol and stained with PI (40 µg/ml;
BD Biosciences), according to the manufacturer's instructions. DNA
content was analyzed on a BD LSRII flow cytometer.
Western blot analysis
The cell lysate was prepared by homogenization in
RIPA buffer [20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM
Na2 EDTA, 1 mM ethylene glycol tetraacetic acid (EGTA),
1% NP-40, 1% sodium deoxycholate] and lysed on ice for 30 min. The
protein amount was quantified using Pierce 660 nm Protein Assay
(Thermo Scientific, Waltham, MA, USA) following the manufacturer's
instructions. Total protein from each sample was loaded onto 10%
SDS-PAGE gel and electrophoresis was performed. For LC3B, proteins
were loaded onto 4–20% Mini-Protean® TGX™ precast gels
(Bio-Rad, Hercules, CA, USA). Proteins were then transferred onto
nitrocellulose membranes and blocked with 5% non-fat dry milk and
PBS for 1 h at room temperature (RT). After 3 washes with 0.05%
Tween-20 PBS (PBST), the membranes were incubated with primary
antibodies overnight at 4°C. Antibodies against LC3B (D11),
Beclin-1 (D40C5) and p62 (D5E2) were purchased from Cell Signaling
Technology (Danvers, MA, USA). Membranes were washed 3 times with
PBST and then incubated with horseradish peroxidase-conjugated
secondary antibodies for 1 h at RT. Membranes were developed with
SuperSignal West Pico Chemiluminescent Substrate (Thermo
Scientific) and were visualized and the optical density of the
identified protein bands on membranes was analyzed using a
Biospectrum 500 Imaging System (UVP LLC, Upland, CA, USA).
Real-time PCR
Total RNAs were extracted using TRIzol and were
reversely transcribed to cDNAs using SuperScript® III
First-Strand Synthesis system according to the manufacturer's
instructions. Real-time PCR was performed using Fast
SYBR®-Green Master Mix on a 7300 Real-Time PCR system.
Reagents and instruments were purchased from Invitrogen Life
Technologies (Carlsbad, CA, USA). Data were analyzed with 7300
System software. GAPDH was used as an internal control for mRNA.
Relative expression was calculated with respect to the control. The
results are expressed as 2−ΔΔCt. Primer sequences for
each gene were: UHRF1 forward, 5′-CCAGCAGAGCAGCCTCATC-3′ and
reverse, 5′-TCCTTGAGTGACGCCAGGA-3′; GAPDH forward,
5′-TGATGACATCAAGAAGGTGGTGAAG-3′ and reverse,
5′-TCCTTGGAGGCCATGTGGGCCAT-3′.
Statistical analysis
Data were analyzed and the results are presented as
the mean ± standard deviation. Student's t-test or one-way ANOVA
was used for comparison of the mean between the groups and
P<0.05 was considered to indicate a statistically significant
result.
Results
Torin-2 inhibits the proliferation of HCC
cell lines
To determine whether Torin-2 impacts cancer cell
biology, we firstly measured the growth curves of three HCC cell
lines Hep G2, SNU-182 and Hep 3B2.1–7. As shown in Fig. 1A–C, Torin-2 reduced HCC cell growth
in a dose-dependent manner and 1 µM Torin-2 significantly
inhibited the cell growth of all three cell lines tested. Torin-2
(0.5 µM) exhibited similar but relatively weaker inhibitory
effects. The inhibitory effect lasted for 4 to 5 days. Cell cycle
analysis with PI staining indicated that on day 3 post-treatment, 1
µM Torin-2 induced significant G0/G1
phase arrest of the three cell lines, while the percentage of cells
in the S phase were markedly reduced, suggesting that cancer cell
proliferation was decreased by Torin-2 (Fig. 1D–F). Ki67 staining, which is a
marker of cell cycle progression, also demonstrated that a lower
percentage of cancer cells entered the cell cycle on day 3 after
Torin-2 treatment (Fig. 1G and H).
Collectively, our data indicated that Torin-2 effectively decreased
cancer cell growth in our experimental settings.
 | Figure 1Torin-2 inhibits the proliferation of
HCC cell lines. (A–C) Growth curves for the HCC cell lines after
Torin-2 treatment. (D–F) Cell cycle analysis with PI staining for
HCC cell lines on day 3 after Torin-2 treatment. (G) Representative
histograms of Ki67 staining for HCC cell lines. Dotted line,
isotype control. Red line, vehicle-treated cells. Blue line, 1
µM Torin-2-treated cells. (H) Quantification of Ki67
staining. All experiments were repeated 4 times independently.
n=8/group. SNU, SNU-182; Hep 3B, Hep 3B2.1–7; C, vehicle control;
T, 1 µM Torin-2-treated cells. *P<0.05,
**P<0.01 and ***P<0.001, in comparison
with the vehicle control; HCC, hepatocellular carcinoma; PI,
propidium iodide. |
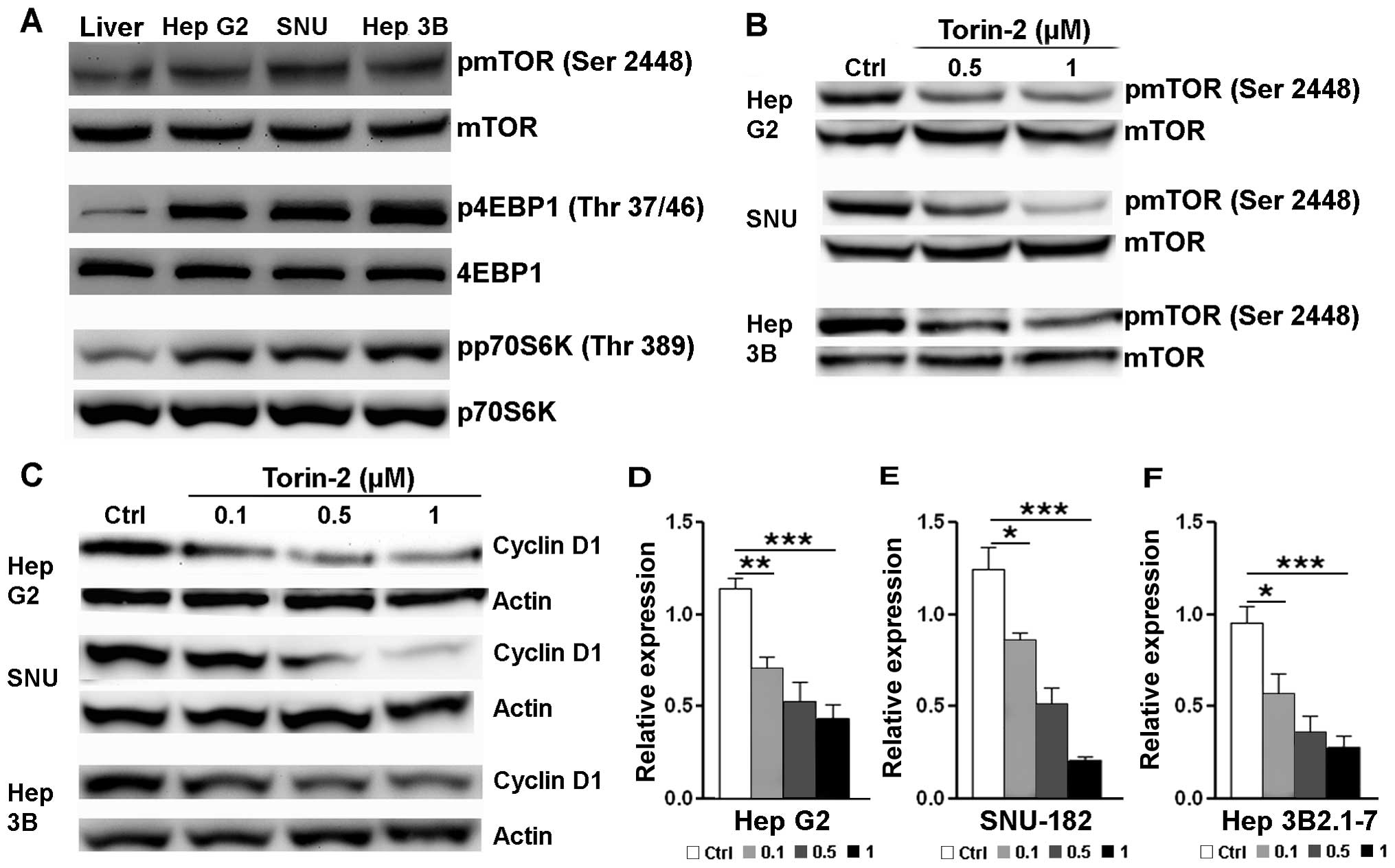
Torin-2 blocks mTORC1 signaling to
inhibit cancer cell proliferation
It has been reported that Torin-2 is an mTORC1
signaling inhibitor (14) and
mTORC1 signaling has been shown to be important for tumor cell
growth and metabolism (8). To
ascertain whether the above anti-proliferative effect of Torin-2 is
related to the blockage of mTORC1 signal transduction, we assessed
activated phosphorylation of mTORC1 components using western blot
analysis. Firstly, we confirmed that mTORC1 signaling was
significantly activated in all three cancer cell lines in
comparison with normal liver tissues, demonstrated by a higher
phosphorylation level of mTOR and mTORC1 target proteins 4EBP1 and
p70S6K (Fig. 2A). Torin-2 decreased
activating phosphorylation of mTOR dose-dependently, suggesting
that, similar to leukemic cells, Torin-2 effectively blocks mTORC1
signaling (Fig. 2B). In association
with less mTORC1 activation, cyclin D1 expression was profoundly
reduced in the three cell lines by Torin-2 dose-dependently
(Fig. 2C–F). Cyclin D is critical
for G1/S transition in cell cycle progression (22). Thus, this reduction was consistent
with our previous data showing G0/G1 arrest
and a decreased S phase population of cancer cells. Furthermore,
cyclin D translation is regulated by the mTORC1 signaling pathway
(23). Hence, a reduced cyclin D1
level was possibly due to the mTORC1 blockage by Torin-2.
Collectively, our data indicated that Torin-2 indeed inhibits
mTORC1 signaling in HCC cells.
Torin-2 induces cancer cell
apoptosis
An initial reduction in cell number within 3 days
after high-dose Torin-2 treatment (Fig.
1A–C) indicated that Torin-2 is cytotoxic to cancer cells. To
test this hypothesis, we determined the extent of cell apoptosis
with Annexin V and PI staining. As shown in Fig. 3, the percentage of
Annexin-V-positive cells was markedly increased with increasing
Torin-2 concentrations, suggesting that Torin-2 induced cancer cell
apoptosis in a dose-dependent manner. Surprisingly, necrosis was
not profound even in the treated HCC cells. Less than 6% of cells
underwent necrosis in the Torin-2-treated groups, although the
moderate increase in necrosis was statistically significant. The
weak necrosis induction by Torin-2 may be beneficial for future
cancer therapy, since necrosis would trigger a severe inflammatory
response, while apoptosis would not.
Torin-2 upregulates autophagy in liver
cancer cells
A previous study showed that Torin-2 promotes
autophagy in leukemic cell lines (24). Since autophagy is closely related to
cancer cell survival, we ascertained whether Torin-2 induced
autophagy in liver cancer cells by detecting autophagic marker
expression. As shown in Fig. 4, in
all three cell lines, Torin-2 significantly elevated LC3B
conversion from LC3B-I to LC3B-II, demonstrated by higher LC3B-II
levels. Beclin-1, which initiates autophagy, was also upregulated
by Torin-2. p62, which is degraded by autophagosomes, reflects the
extent of autophagic protein degradation. The p62 level was
markedly reduced following Torin-2 treatment. Collectively, our
data indicated that Torin-2 effectively promoted autophagy in HCC
cells.
Torin-2-induced autophagy inhibits the
proliferation and survival of liver cancer cells
To confirm that Torin-2-induced autophagy is the
cause of the inhibition of proliferation and increased apoptosis of
liver cancer cells, we blocked autophagy with the well-known
autophagy inhibitor chloroquine, which is a lysosomotropic agent
preventing endosomal acidification (25). Chloroquine significantly upregulated
the LC3B-II level due to inhibition of autophagic protein
degradation. The p62 level was also enhanced for the same reason
(Fig. 5A), suggesting that
autophagy was indeed inhibited. Cell proliferation assay with Ki67
staining showed that chloroquine partially restored cell
proliferation in the presence of Torin-2 in all three cell lines
(Fig. 5B), suggesting that
Torin-2-induced autophagy was at least one of the causes of cell
proliferation inhibition. Apoptosis assay with Annexin-V staining
indicated that chloroquine significantly reduced the percentage of
apoptotic cells in the presence of Torin-2 (Fig. 5C), suggesting that Torin-2-induced
autophagy indeed triggers apoptosis.
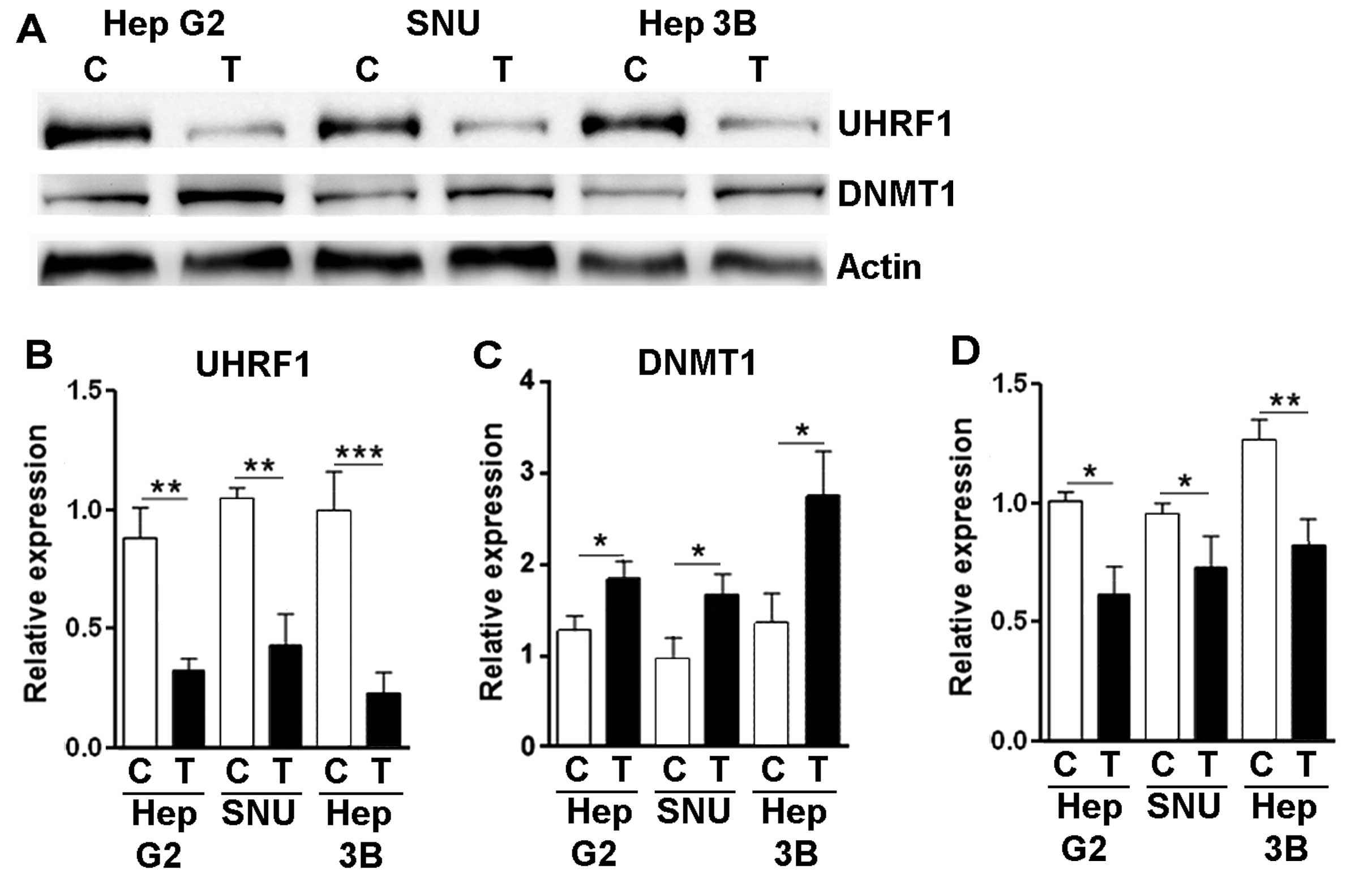
Torin-2 downregulates UHRF1 expression in
liver cancer cells
UHRF1 has been identified as an important regulator
for DNA methylation during DNA replication. Recently it was
reported that UHRF1 is responsible for hepatocellular
carcinogenesis (20). The
relationship between mTORC1 signaling and UHRF1 expression has not
yet been elucidated. We detected the UHRF1 protein level using
western blot analysis and found that Torin-2 treatment
significantly decreased UHRF1 expression in the HCC cell lines
(Fig. 6A and B). Consistently, the
expression of DNMT1, which is destabilized by URHF1, was
upregulated in the presence of Torin-2 (Fig. 6A and C). We further determined the
mRNA level of URHF1 using real-time PCR. Our data showed that
Torin-2 treatment moderately decreased URHF1 mRNA in these cells
(Fig. 6D), suggesting that Torin-2
inhibited URHF1 transcription, or destabilized URHF1 mRNA. However,
it is still not clear whether Torin-2 decreased URHF1 mRNA through
mTORC1 signaling, since few studies have reported that mTORC1
signaling can regulate gene transcription. Further studies are
needed to dissect the mechanisms responsible for the reduction in
URHF1 mRNA.
Discussion
HCC is the third leading cause of cancer-related
mortality worldwide. The incidence of this disease is increasing
and it is one of the key indications for liver transplantation. It
is the most common primary liver tumor, which is notoriously
resistant to systemic therapies and often recurs even after
aggressive local therapies. HCC initiation and progression are
complicated, involving alterations in multiple signal transduction
pathways and subsequent cellular behavior change.
In the present study, we tested the effect of
Torin-2 on the proliferation and survival of three HCC cell lines.
Torin-2 is a novel ATP-competitive inhibitor of mTOR. mTOR is a
highly conserved serine/threonine protein kinase that serves as a
central regulator of cell growth, survival and autophagy.
Deregulation of the PI3K/Akt/mTOR signaling pathway commonly occurs
in cancer (26). Given its
importance in cell growth and metabolism, mTOR also plays a pivotal
role in HCC (6). mTORC1 and mTORC2
pathways, including pRPS6, p-AKT, IGF-1R and RICTOR are upregulated
in 40–50% of HCCs (27). Moreover,
an upregulation of mTOR signaling is frequently observed in
cholangiocarcinoma, the second most common primary cancer of the
liver (5). Activation of the mTOR
pathway in HCC is associated with less differentiated tumors, poor
prognosis and earlier recurrence independently of the underlying
etiology of liver cancer. We tested the mTORC1 activation status in
these HCC cell lines and found that all had upregulated mTORC1
signaling, in comparison with the normal mouse liver tissue. Thus,
it is highly possible that mTORC1 activation is critical for the
initiation and/or maintenance of carcinogenic features in these HCC
cells. Therefore, blocking this pathway is an attractive strategy
for HCC treatment. Until recently, several inhibitors targeting
mTOR have been studied for their efficacy in tumor therapy,
including rapamycin (28) and its
derivatives everolimus (11),
temsirolimus (29–31) and deforolimus (32). In contrast to these first generation
mTOR inhibitors, Torin-2 inhibits mTOR kinase and mTORC1 signaling
activities in a sustained manner suggestive of a slow dissociation
from the kinase (14). It has been
reported that cancer cell treatment with Torin-2 for 24 h resulted
in a prolonged block in negative feedback and consequent T308
phosphorylation on Akt (14). These
effects were associated with strong growth inhibition in
vitro. To our knowledge, however, Torin-2 has not been assessed
for treating HCC in vitro or in vivo.
Our in vitro study indicated that Torin-2
profoundly reduced HCC cell growth in a dose-dependent manner. This
growth inhibition was attributed to G0/G1
phase arrest in the cell cycle. Consistently, cyclin D expression
was downregulated by Torin-2. Cyclin D is the partner of
cyclin-dependent kinases CDK4 and CDK6. CDK4 and CDK6 drive the
transition of G1 to S phase in the cell cycle. Activity
of the cyclin-dependent CDK4 and CDK6 is regulated by T-loop
phosphorylation, by the abundance of cyclin D (33,34).
Thus, the downregulation of cyclin D could be the reason for
G0/G1 arrest, preventing cells from entering
the DNA synthesis phase. Moreover, the translation of cyclin D
protein is tightly regulated by mTORC1 signaling, since mTORC1
activates 4EBP1 and p70S6K, which both are important participants
in the protein synthesis machinery (35). We indeed confirmed that Torin-2
inhibited the activating phosphorylation of mTOR. Therefore, it is
highly possible that the blockage of mTORC1 activation by Torin-2
decreased the translation of cyclin D, consequently reducing the
activity of CDK4 and CDK6 to prevent cell entry to the S phase.
However, we did not test whether other cell cycle regulators, such
as CDK inhibitors and cdc2 are influenced by Torin-2. In addition,
it is still not clear whether Torin-2 impacts autocrine/paracrine
factors which profoundly modulate HCC initiation and progression,
such as IGF-1, IGF-2, IGFBP-3, TGF-α, TGF-β, EGF and FGF. Further
studies are needed to include or exclude the effects of Torin-2 on
these regulatory factors.
In addition, our research revealed that Torin-2 can
induce HCC cell apoptosis, which could also contribute to the
inhibition of HCC cell growth. Furthermore, Torin-2-induced
apoptosis is closely related to autophagy, which is a crucial
mechanism underlying tumor cell metabolism and survival. Autophagy
is a homeostatic, catabolic degradation process whereby cellular
proteins and organelles are engulfed by autophagosomes, digested in
lysosomes and recycled to sustain cellular metabolism (36). Autophagy genes are frequently
mono-allelically deleted, silenced or mutated in human tumors,
resulting in an environment of increased oxidative stress that is
conducive to DNA damage, genomic instability and tumor progression.
It has been known that autophagy can lead to autophagic cell death
in HCCs (37–40). However, previous research has also
revealed that autophagy induction could also be beneficial for
tumor cell growth (41–44). Thus, the extent of autophagy may be
very important to decide the fate of HCC cells. To our knowledge,
how much autophagy induction is needed for tumor growth or death is
still poorly understood. Previous studies on rapamycin found that
rapamycin-induced autophagy could be either pro-apoptotic (45) or anti-apoptotic (46). The Torin-2 concentration we used
could have been high enough to cause autophagic cell death of HCC
cells. It will be interesting to test whether lower doses of
Torin-2 induce weaker autophagy and promote HCC cell survival. If
this was the case, Torin-2 dosage must be taken into account in
future therapeutic applications for tumors.
What was unexpected in our research was the
downregulation of UHRF1 by Torin-2. UHRF1 plays an essential role
in DNA methylation by recognizing hemimethylated DNA generated
during DNA replication and then by recruiting DNMT1 to ensure
faithful maintenance of DNA-methylation patterns in daughter cells
(47,48). DNA hypomethylation contributes to
oncogenesis through multiple mechanisms, including chromosomal
instability, derepression of imprinted genes, retrotransposon
activation and aberrant gene expression, including induction of
oncogenes (20). The precise
mechanism by which DNA hypomethylation causes cancer remains
elusive. Both apoptosis and senescence serve to limit the
propagation of cells with aberrant DNA methylation. DNMT1 and UHRF1
are significantly altered across cancer types. Change in the
expression of UHRF1 is sufficient to alter the cancer cell
methylome and drive carcinogenesis. However, there is no evidence
suggesting the relationship between mTORC1 signaling and DNA
methylation. To our knowledge we are the first to report that
mTORC1 signaling can regulate the expression of UHRF1. Our data
indicated that the downregulation of UHRF1 by Tοrin-2 was at least
partially due to the reduced UHRF1 mRNA levels. Since mTORC1
classically modulates metabolism and protein synthesis, it is
plausible to speculate that mTORC1 regulates the translation of
unknown factors which subsequently influences the transcription of
UHRF1. However, we cannot exclude the possibility that Torin-2
suppresses UHRF1 expression independently of mTORC1. The mechanisms
modulating UHRF1 expression are still very poorly elucidated.
Further studies will be performed to dissect the signaling pathways
causing this change. We confirmed that with decreased UHRF1
expression, expression of DNMT1 was upregulated. This may be due to
the fact that destabilization of DNMT1 by UHRF1 is compromised as
UHRF1 itself is downregulated.
Collectively, the present study demonstrated the
effect of Torin-2 on the inhibition of HCC cell proliferation and
survival. The inhibitory efficacy can be ascribed to blockage of
mTORC1 signaling, subsequently enhancing autophagy and leading to
autophagic cell death. Meanwhile, blockage of mTORC1 also
suppressed the cell entry into the S phase, thus causing
G0/G1 arrest. Torin-2 also downregulated the
expression of UHRF1, which consequently impacted the oncogenesis of
HCC. Our research provides new clues in the search for therapeutic
strategies for HCC.
References
|
1
|
Olsen SK, Brown RS and Siegel AB:
Hepatocellular carcinoma: Review of current treatment with a focus
on targeted molecular therapies. Therap Adv Gastroenterol. 3:55–66.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Breuhahn K, Longerich T and Schirmacher P:
Dysregulation of growth factor signaling in human hepatocellular
carcinoma. Oncogene. 25:3787–3800. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Moeini A, Cornellà H and Villanueva A:
Emerging signaling pathways in hepatocellular carcinoma. Liver
Cancer. 1:83–93. 2012. View Article : Google Scholar
|
|
4
|
Sanz-Cameno P, Trapero-Marugán M, Chaparro
M, Jones EA and Moreno-Otero R: Angiogenesis: From chronic liver
inflammation to hepatocellular carcinoma. J Oncol. 2010:2721702010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Matter MS, Decaens T, Andersen JB and
Thorgeirsson SS: Targeting the mTOR pathway in hepatocellular
carcinoma: Current state and future trends. J Hepatol. 60:855–865.
2014. View Article : Google Scholar :
|
|
6
|
Zhou Q, Lui VW and Yeo W: Targeting the
PI3K/Akt/mTOR pathway in hepatocellular carcinoma. Future Oncol.
7:1149–1167. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Newell P, Toffanin S, Villanueva A, Chiang
DY, Minguez B, Cabellos L, Savic R, Hoshida Y, Lim KH,
Melgar-Lesmes P, et al: Ras pathway activation in hepatocellular
carcinoma and anti-tumoral effect of combined sorafenib and
rapamycin in vivo. J Hepatol. 51:725–733. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guertin DA and Sabatini DM: Defining the
role of mTOR in cancer. Cancer Cell. 12:9–22. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wörns MA and Galle PR: HCC therapies -
lessons learned. Nat Rev Gastroenterol Hepatol. 11:447–452. 2014.
View Article : Google Scholar
|
|
10
|
Martelli AM, Chiarini F, Evangelisti C,
Cappellini A, Buontempo F, Bressanin D, Fini M and McCubrey JA: Two
hits are better than one: Targeting both phosphatidylinositol
3-kinase and mammalian target of rapamycin as a therapeutic
strategy for acute leukemia treatment. Oncotarget. 3:371–394.
2012.PubMed/NCBI
|
|
11
|
Zhu AX, Kudo M, Assenat E, Cattan S, Kang
YK, Lim HY, Poon RT, Blanc JF, Vogel A, Chen CL, et al: Effect of
everolimus on survival in advanced hepatocellular carcinoma after
failure of sorafenib: The EVOLVE-1 randomized clinical trial. JAMA.
312:57–67. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou Q, Wong CH, Lau CP, Hui CW, Lui VW,
Chan SL and Yeo W: Enhanced antitumor activity with combining
effect of mTOR inhibition and microtubule stabilization in
hepatocellular carcinoma. Int J Hepatol. 2013:1038302013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu Q, Wang J, Kang SA, Thoreen CC, Hur W,
Ahmed T, Sabatini DM and Gray NS: Discovery of
9-(6-aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl)benzo[h][1,6]naphthyridin-2(1H)
- one (Torin2) as a potent, selective, and orally available
mammalian target of rapamycin (mTOR) inhibitor for treatment of
cancer. J Med Chem. 54:1473–1480. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu Q, Xu C, Kirubakaran S, Zhang X, Hur
W, Liu Y, Kwiatkowski NP, Wang J, Westover KD, Gao P, et al:
Characterization of Torin2, an ATP-competitive inhibitor of mTOR,
ATM, and ATR. Cancer Res. 73:2574–2586. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ahmed M, Hussain AR, Bavi P, Ahmed SO, Al
Sobhi SS, Al-Dayel F, Uddin S and Al-Kuraya KS: High prevalence of
mTOR complex activity can be targeted using Torin2 in papillary
thyroid carcinoma. Carcinogenesis. 35:1564–1572. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bonder MJ, Kasela S, Kals M, Tamm R, Lokk
K, Barragan I, Buurman WA, Deelen P, Greve JW, Ivanov M, et al:
Genetic and epigenetic regulation of gene expression in fetal and
adult human livers. BMC Genomics. 15:8602014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Robertson KD: DNA methylation,
methyltransferases, and cancer. Oncogene. 20:3139–3155. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bianchi C and Zangi R: UHRF1 discriminates
against binding to fully-methylated CpG-Sites by steric repulsion.
Biophys Chem. 171:38–45. 2013. View Article : Google Scholar
|
|
19
|
Bashtrykov P, Jankevicius G, Jurkowska RZ,
Ragozin S and Jeltsch A: The UHRF1 protein stimulates the activity
and specificity of the maintenance DNA methyltransferase DNMT1 by
an allosteric mechanism. J Biol Chem. 289:4106–4115. 2014.
View Article : Google Scholar :
|
|
20
|
Mudbhary R, Hoshida Y, Chernyavskaya Y,
Jacob V, Villanueva A, Fiel MI, Chen X, Kojima K, Thung S, Bronson
RT, et al: UHRF1 overexpression drives DNA hypomethylation and
hepatocellular carcinoma. Cancer Cell. 25:196–209. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pi JT, Lin Y, Quan Q, Chen LL, Jiang LZ,
Chi W and Chen HY: Overexpression of UHRF1 is significantly
associated with poor prognosis in laryngeal squamous cell
carcinoma. Med Oncol. 30:6132013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Choi YJ, Li X, Hydbring P, Sanda T,
Stefano J, Christie AL, Signoretti S, Look AT, Kung AL, von Boehmer
H, et al: The requirement for cyclin D function in tumor
maintenance. Cancer Cell. 22:438–451. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Averous J, Fonseca BD and Proud CG:
Regulation of cyclin D1 expression by mTORC1 signaling requires
eukaryotic initiation factor 4E-binding protein 1. Oncogene.
27:1106–1113. 2008. View Article : Google Scholar
|
|
24
|
Simioni C, Cani A, Martelli AM, Zauli G,
Tabellini G, McCubrey J, Capitani S and Neri LM: Activity of the
novel mTOR inhibitor Torin-2 in B-precursor acute lymphoblastic
leukemia and its therapeutic potential to prevent Akt reactivation.
Oncotarget. 5:10034–10047. 2014.PubMed/NCBI
|
|
25
|
Kimura T, Takabatake Y, Takahashi A and
Isaka Y: Chloroquine in cancer therapy: A double-edged sword of
autophagy. Cancer Res. 73:3–7. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Polivka J Jr and Janku F: Molecular
targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol
Ther. 142:164–175. 2014. View Article : Google Scholar
|
|
27
|
Villanueva A, Chiang DY, Newell P, Peix J,
Thung S, Alsinet C, Tovar V, Roayaie S, Minguez B, Sole M, et al:
Pivotal role of mTOR signaling in hepatocellular carcinoma.
Gastroenterology. 135:1972–1983. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ashworth RE and Wu J: Mammalian target of
rapamycin inhibition in hepatocellular carcinoma. World J Hepatol.
6:776–782. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Knox JJ, Qin R, Strosberg JR, Tan B,
Kaubisch A, El-Khoueiry AB, Bekaii-Saab TS, Rousey SR, Chen HX and
Erlichman C: A phase II trial of bevacizumab plus temsirolimus in
patients with advanced hepatocellular carcinoma. Invest New Drugs.
33:241–246. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kelley RK, Nimeiri HS, Munster PN, Vergo
MT, Huang Y, Li CM, Hwang J, Mulcahy MF, Yeh BM, Kuhn P, et al:
Temsirolimus combined with sorafenib in hepatocellular carcinoma: A
phase I dose-finding trial with pharmacokinetic and biomarker
correlates. Ann Oncol. 24:1900–1907. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhou Q, Lui VW, Lau CP, Cheng SH, Ng MH,
Cai Y, Chan SL and Yeo W: Sustained antitumor activity by
co-targeting mTOR and the microtubule with temsirolimus/vinblastine
combination in hepatocellular carcinoma. Biochem Pharmacol.
83:1146–1158. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang Z, Jin W, Jin H and Wang X: mTOR in
viral hepatitis and hepatocellular carcinoma: Function and
treatment. Biomed Res Int. 2014:7356722014.PubMed/NCBI
|
|
33
|
Bockstaele L, Kooken H, Libert F, Paternot
S, Dumont JE, de Launoit Y, Roger PP and Coulonval K: Regulated
activating Thr172 phosphorylation of cyclin-dependent kinase
4(CDK4): Its relationship with cyclins and CDK 'inhibitors̓. Mol
Cell Biol. 26:5070–5085. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Choi YJ and Anders L: Signaling through
cyclin D-dependent kinases. Oncogene. 33:1890–1903. 2014.
View Article : Google Scholar
|
|
35
|
Takuwa N, Fukui Y and Takuwa Y: Cyclin D1
expression mediated by phosphatidylinositol 3-kinase through
mTOR-p70(S6K)-independent signaling in growth factor-stimulated NIH
3T3 fibroblasts. Mol Cell Biol. 19:1346–1358. 1999.PubMed/NCBI
|
|
36
|
Yang ZJ, Chee CE, Huang S and Sinicrope
FA: The role of autophagy in cancer: Therapeutic implications. Mol
Cancer Ther. 10:1533–1541. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sui X, Chen R, Wang Z, Huang Z, Kong N,
Zhang M, Han W, Lou F, Yang J, Zhang Q, et al: Autophagy and
chemotherapy resistance: A promising therapeutic target for cancer
treatment. Cell Death Dis. 4:e8382013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li P, Du Q, Cao Z, Guo Z, Evankovich J,
Yan W, Chang Y, Shao L, Stolz DB, Tsung A, et al: Interferon-γ
induces autophagy with growth inhibition and cell death in human
hepatocellular carcinoma (HCC) cells through interferon-regulatory
factor-1 (IRF-1). Cancer Lett. 314:213–222. 2012. View Article : Google Scholar :
|
|
39
|
Tai WT, Shiau CW, Chen HL, Liu CY, Lin CS,
Cheng AL, Chen PJ and Chen KF: Mcl-1-dependent activation of Beclin
1 mediates autophagic cell death induced by sorafenib and SC-59 in
hepatocellular carcinoma cells. Cell Death Dis. 4:e4852013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Allen KD, Mata BA, Gabr MA, Huebner JL,
Adams SB Jr, Kraus VB, Schmitt DO and Setton LA: Kinematic and
dynamic gait compensations resulting from knee instability in a rat
model of osteoarthritis. Arthritis Res Ther. 14:R782012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou Y, Sun K, Ma Y, Yang H, Zhang Y, Kong
X and Wei L: Autophagy inhibits chemotherapy-induced apoptosis
through downregulating Bad and Bim in hepatocellular carcinoma
cells. Sci Rep. 4:53822014.PubMed/NCBI
|
|
42
|
Guo XL, Li D, Sun K, Wang J, Liu Y, Song
JR, Zhao QD, Zhang SS, Deng WJ, Zhao X, et al: Inhibition of
autophagy enhances anticancer effects of bevacizumab in
hepatocarcinoma. J Mol Med Berl. 91:473–483. 2013. View Article : Google Scholar :
|
|
43
|
Yang PM, Liu YL, Lin YC, Shun CT, Wu MS
and Chen CC: Inhibition of autophagy enhances anticancer effects of
atorvastatin in digestive malignancies. Cancer Res. 70:7699–7709.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo
Y, Song Z, Zheng Q and Xiong J: Autophagy promotes hepatocellular
carcinoma cell invasion through activation of
epithelial-mesenchymal transition. Carcinogenesis. 34:1343–1351.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li X, Wu D, Shen J, Zhou M and Lu Y:
Rapamycin induces autophagy in the melanoma cell line M14 via
regulation of the expression levels of Bcl-2 and Bax. Oncol Lett.
5:167–172. 2013.
|
|
46
|
Ravikumar B, Berger Z, Vacher C, O'Kane CJ
and Rubinsztein DC: Rapamycin pre-treatment protects against
apoptosis. Hum Mol Genet. 15:1209–1216. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Unoki M: Current and potential anticancer
drugs targeting members of the UHRF1 complex including epigenetic
modifiers. Recent Patents Anticancer Drug Discov. 6:116–130. 2011.
View Article : Google Scholar
|
|
48
|
Bronner C, Fuhrmann G, Chédin FL, Macaluso
M and Dhe-Paganon S: UHRF1 links the histone code and DNA
methylation to ensure faithful epigenetic memory inheritance. Genet
Epigenet. 2009:29–36. 2010.PubMed/NCBI
|