Introduction
A common feature of progressive cancers is
metastasis to other tissues. For migration out from their primary
site, cancer cells transform into cells that exhibit mesenchymal
phenotypes, and this transition is called epithelial-to-mesenchymal
transition (EMT). Physiologically, EMT has been associated with
development, wound healing, inflammation, fibrosis and cancer-cell
invasion and migration (1–3). In numerous cancers, EMT presents
several disadvantages to patients, such as radioresistance,
chemoresistance and maintenance of cancer stem cells (4–6). Thus,
EMT signaling is considered to be a favorable therapeutic target
for cancer treatment (7,8).
EMT is induced by diverse secreted molecules such as
fibroblast growth factor (FGF), hepatocyte growth factor, epidermal
growth factor (EGF), platelet-derived growth factor and
transforming growth factor-β (TGF-β), and among these, TGF-β is
well characterized as a key inducer of EMT in various types of
cancers (1,6). The TGF-β signaling pathway involves
the following steps: TGF-β binds to TGF-β receptor II (TβRII),
which then heterodimerizes with TGF-β receptor I (TβRI). Next, the
TGF-β receptors are phosphorylated and they activate downstream
signaling pathways, including the canonical Smad signaling pathway
and the non-Smad signaling pathway. Subsequently, various EMT
markers and associated molecules are transcriptionally regulated by
EMT-regulating transcription factors such as SNAI1 (snail family
zinc finger 1; SNAIL) and SNAI2 (snail family zinc finger 2; SLUG),
zinc finger E-box-binding homeobox 1 and 2 (ZEB1 and ZEB2) and
twist family bHLH transcription factor 1 (TWIST1) (1–3,9).
FGF receptors (FGFRs) constitute another family of
receptors associated with several cellular processes in cancer,
including metastasis, stemness, proliferation, anti-apoptosis, drug
resistance and angiogenesis (10,11);
thus, FGFRs have recently been considered as a potential
therapeutic target for cancer patients (10–12).
Four major FGFRs are expressed by cells, and among these, FGFR1 is
highly involved in metastasis and FGFR3 contributes to cancer-cell
metastasis. Moreover, the FGFR4 SNP variant Gly388Arg was shown to
be associated with metastasis, while FGFR2 acts as an antagonist
and suppresses the EMT process (13–15).
Previously, we demonstrated that
3-(2-chlorobenzyl)-1,7-dimethyl-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione
(IM-412) inhibits TGF-β-induced fibroblast differentiation through
both Smad-dependent and -independent pathways in human lung
fibroblasts (16). Since IM-412
exerted an anti-fibrotic effect by inhibiting the TGF-β signaling
pathway, in the present study, we investigated whether IM-412 also
affects EMT signaling and the invasiveness of cancer cells.
Materials and methods
Materials
We obtained primary antibodies against the following
proteins from commercial sources: p-Smad2 (Ser245/250/255), p-Smad2
(Ser465/467), Smad2, p-Smad3, Smad3, p-p38MAPK, p-Akt, Akt, p-JNK,
JNK, p-ERK, p-c-Jun and c-Jun (Cell Signaling Technology, Danvers,
MA, USA); e-cadherin, fibronectin, collagen type 1 A2, p38MAPK, ERK
and TβRI (Santa Cruz Biotechnology, Santa Cruz, CA, USA);
N-cadherin, SNAIL, SLUG and ZEB1 (BD biosciences, Palo Alto, CA,
USA); vimentin and α-SMA were obtained from Sigma (St. Louis, MO,
USA); p-FGFR3, FGFR3 and TβRII (Abcam, Cambridge, MA, USA); and
GAPDH (AbFrontier, Seoul, Korea). Horseradish peroxidase
(HRP)-conjugated secondary antibodies were purchased from Thermo
(Pittsburgh, PA, USA). For use in the in vitro invasion
assay, Transwells were purchased from Corning and Matrigel was
purchased from BD Biosciences. IM-412
(C16H14ClN5O2, MW: 344;
ID 9073517) was isolated from a chemical library of screening
compounds and was purchased from ChemBridge Corporation (San Diego,
CA, USA).
Cell culture
The human breast cancer cell line MDA-MB-231 was
purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA). The cells were maintained at 37°C in RPMI-1640
supplemented with 10% fetal bovine serum (Gibco-BRL, Grand Island,
NY, USA), 100 U/ml penicillin and 100 µg/ml streptomycin in
a humidified atmosphere containing 5% Co2.
MTT assay
Cell viability was assessed using the
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT)
assay (Sigma). MDA-MB-231 cells were plated at a density of
8×103 cells/well into 24-well plates (SPL, Gyeonggi,
Korea), and the seeded cells were stabilized for 24 h, and were
subsequently treated with each indicated dose of chemicals. MTT was
directly added to each well at a final concentration of 0.5 mg/ml,
and after 4 h, the medium was removed, the formazan crystals in
MDA-MB-231 cells were dissolved in dimethylsulfoxide and the
absorbance of the formazan solution was measured using a microplate
reader (Multiskan EX; Thermo LabSystems, waltham, MA, USA) equipped
with a 540-nm filter. each sample was assayed in triplicate, and
the experiment was repeated thrice.
In vitro wound migration assay
Cells were seeded into 6-well plates (SPL) at 90%
confluency (5×105 cells/well) and incubated overnight.
On the next day, the culture medium was aspirated, and the cells
were scratched using a 200-µl pipette tip. After wounding,
the cultures were washed with phosphate-buffered saline (PBS) and
treated with IM-412. The cells were allowed to migrate for 24 h and
were subsequently stained with crystal violet. Migration patterns
were examined under a microscope and photographed at a
magnification of ×40.
In vitro Transwell invasion assay
The invasiveness of the cultured cells was
determined using a 24-well Transwell system (pore size,
8-µm) (Corning Incorporated, Corning, NY, USA). The upper
side of the Transwell membrane was coated with 1 mg/ml Matrigel (in
10 µl) and then cells were seeded into the upper chamber at
a density of 5×104 cells in 100 µl of media; the
complete medium was added to the lower chamber. Cells were
incubated for 24 h at 37°C in 5% Co2, and then the cells
that had not penetrated the filter were wiped away using cotton
swabs; the cells that had invaded the lower surface of the filter
were fixed with methanol, stained with crystal violet, examined
under a microscope, and photographed at a magnification of ×40. The
numbers of invaded cells were determined by counting the cells in 5
randomly selected regions.
Western blot analysis
Cells were lysed with RIPA buffer (50 mM Tris-Cl, pH
7.4, 1% NP-40, 150 mM NaCl, 1 mM EDTA) supplemented with protease
inhibitors (1 mM phenylmethylsulfonyl fluoride, 1 µg/ml
aprotinin and 1 µg/ml leupeptin) and phosphatase inhibitors
(1 mM Na3VO4 and 1 mM NaF). Proteins in the
whole-cell lysates were separated on 8–15% SDS-polyacrylamide gels
and transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA,
USA). The membranes were blocked (1 h) with 5% skim milk in
tris-buffered saline containing 0.1% Tween-20 and then probed with
primary antibodies (overnight, 4°C). After multiple washes, the
membranes were incubated with HRP-conjugated secondary antibodies,
and then the immunoreactive bands were detected using enhanced
chemiluminescence reagents according to the manufacturer's
recommendations (GE Healthcare; Little Chalfont, UK). The
experiments were repeated at least thrice.
Immunocytochemical staining
MDA-MB-231 cells (5×105 cells) were grown
on glass coverslips, stabilized for 24 h and then treated for 24 h
with the indicated doses of IM-412. Next, the cells were washed
with Hank's balanced salt solution (HBSS), fixed with 3.7%
formaldehyde in HBSS for 10 min at room temperature, and then
washed and permeabilized with 0.1% Triton X-100 in HBSS for 15 min
at room temperature. Mouse α-SMA antibody (1:500) was used as the
primary antibody and it was detected using an Alexa 546-conjugated
secondary antibody. Nuclei were counterstained with 300 ng/ml
4′,6-diamidino-2-phenylindole (DAPI; Invitrogen, Carlsbad, CA,
USA). Coverslips were mounted on glass slide with Gel/Mount™
(biomeda, Foster City, CA, USA), and images were obtained using a
laser-scanning confocal microscope (LSM710; Carl Zeiss, oberkochen,
Germany) at a magnification of ×400.
Three-dimensional (3D) cultures and
immunohistochemical staining
We prepared 3D cultures of cells as described with a
few modifications (17). To mimic
the in vivo environment, a type I collagen (Col I) gel
matrix was reconstituted according to the manufacturer's
specifications (Nitta Gelatin, Osaka, Japan), and then 250
µl of this mixture was plated into 12-mm polycarbonate
filter chambers (3.0 µm Millicell; Millipore, Temecula, CA,
USA). MDA-Mb-231 cells were seeded at a density of 2×105
cells/chamber and cultured in the submerged state in culture medium
for 7 days, and then in the air-liquid interface state for 24 h.
During this period of air-liquid interface culture, cells were
concurrently treated with or without IM-412. After the 1-day
air-liquid interface culture, the 3D cultures were fixed in
Carnoy's solution (ethanol:chloroform:acetic acid, 6:3:1) for 30
min at 4°C, and then the fixed samples were embedded in paraffin
and sectioned (3 µm). For staining, sections were
deparaffinized and the endogenous peroxidase activity was blocked.
Mouse α-SMA antibody (1:500) was used as the primary antibody and
the sections were developed using the Cap-Plus™ Detection kit
(Invitrogen); after the development step, samples were
counterstained with hematoxylin (Dako, Carpinteria, CA, USA). The
stained samples were examined under a microscope and photographed
at a magnification of ×200.
Modeling of IM-412-FGFR binding
We performed calculations using Discover
2.98/Insight II with CVFF force field as described by Hagler et
al (18). The crystal structure
of FGFR3 bound to an ATP mimetic molecule (phosphomethylphosphonic
acid adenylate ester) was used as the computational model
(4K33.pdb). IM-412 was built from the coordinates of an adenine
group, as found in the crystal structure using the Builder Module
in Insight II. The computational complex model was initially
minimized using 1,000 steps of steepest decent and 3,000 steps of
conjugated gradient with a 14.0 Å non-bonded cut-off distance.
Data analysis
Data are represented as means ± SD. Statistical
comparisons between groups were performed using the Student's
t-test. Data were considered statistically significant at
P<0.05, P<0.01 or P<0.001 as indicated in the figure
legends.
Results
IM-412 inhibits the invasion and
migration in MDA-MB-231 cells
TGF-β is widely recognized to exhibit pleiotropic
activity in biological processes ranging from development,
proliferation, differentiation, angiogenesis and immune regulation
to tumor biology. Moreover, TGF-β signaling plays a prominent role
in EMT (1,3,19).
Since our previous results demonstrated that the novel compound
IM-412 effectively inhibited TGF-β signaling in human lung
fibroblasts (16), we aimed to
investigate whether IM-412 prevents the migration and invasion of
cancer cells. First, we examined the cytotoxic effect of IM-412 on
MDA-MB-231 cells, which are highly metastatic human breast
carcinoma cells. At concentrations above 2 µM, IM-412
markedly reduced the viability of the cells, but at concentrations
below 2 µM, the viability levels were the same as those in
untreated control cells (Fig. 1A).
Next, the changes in cell motility induced by IM-412 were
investigated using the wound migration (Fig. 1B) and the Transwell invasion assays
(Fig. 1C) under comparatively less
cytotoxic conditions. IM-412 drastically reduced the abilities of
migration and invasion of MDA-MB-231 cells in a dose-dependent
manner.
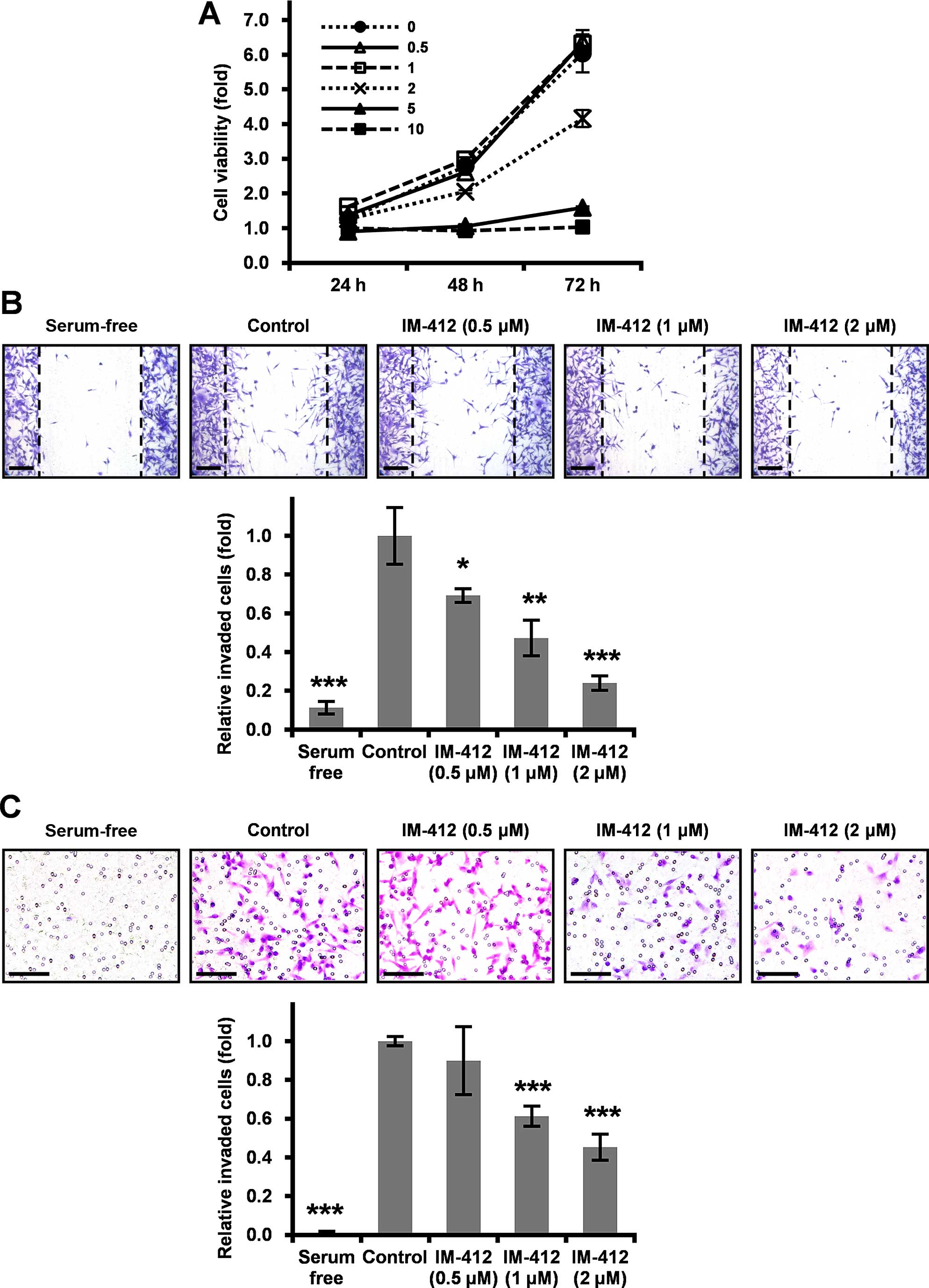 | Figure 1IM-412 inhibits the migration and
invasion of MDA-MB-231 cells. (A) MTT assays were performed to
examine cell viability. MDA-MB-231 cells were seeded at
8×103 cells/well into 24-well plates and treated with
the indicated concentrations of IM-412 for 24, 48 or 72 h. Each
sample was assayed in triplicate, and the experiment was repeated
thrice. (B) To measure the effect of IM-412 on cell migration,
MDA-MB-231 cells were seeded at 5×105 cells/well into
6-well plates. The cells were scratched using a 200-µl
pipette tip and treated with the indicated concentrations of IM-412
for 24 h, after which the cells were stained with crystal violet
and photographed under a microscope at a magnification of ×40.
Scale bar, 100 µm. Results were quantified by counting the
migrated cells in each experiment. Data shown are means ± SD of
three independent experiments performed in triplicate;
*P<0.05 vs. control, **P<0.01 vs.
control, ***P<0.001 vs. control. (C) To examine the
IM-412-induced invasiveness of cells, the Transwell invasion assay
was performed. MDA-MB-231 cells were seeded at 5×104
cells/well in the Matrigel-coated upper chamber. For visualization,
the cells were stained with crystal violet and photographed under a
microscope at ×40 magnification. Scale bar, 100 µm. Results
were quantified by counting the invaded cells in 5 randomly
selected regions in each experiment. Data shown are means ± SD of
three independent experiments performed in triplicate;
***P<0.001 vs. control. |
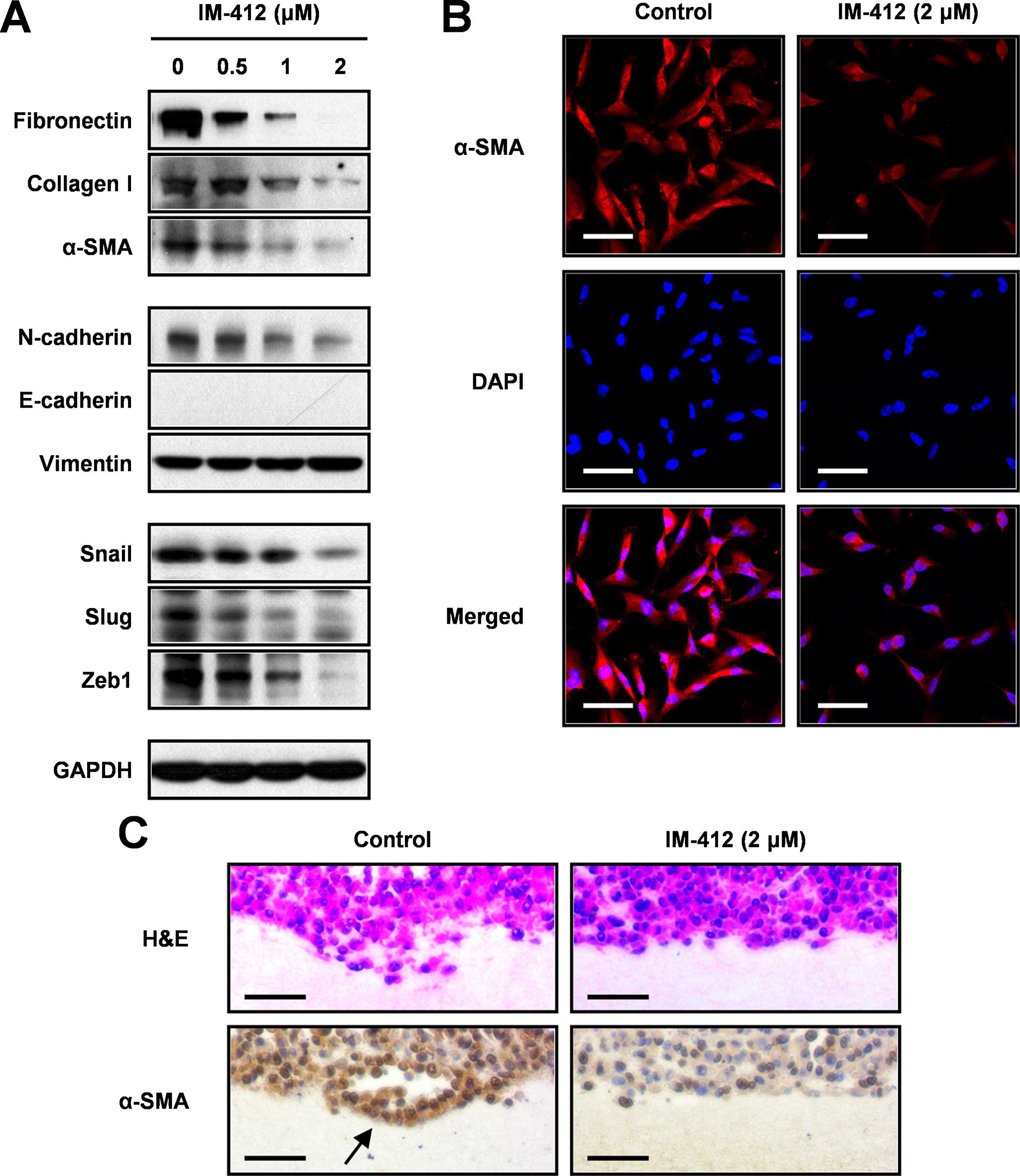
IM-412 suppresses the expression of
EMT-associated markers in MDA-MB-231 cells
To evaluate the capacity of IM-412 to affect
EMT-related markers during MDA-MB-231 cell invasion, we determined
the expression levels of EMT-associated molecules by performing
western blot analysis (Fig. 2A).
IM-412 markedly reduced the expression of mesenchymal markers such
as fibronectin, collagen type 1 A2 and α-SMA (ACTA2; α-smooth
muscle actin). The expression level of N-cadherin was also
decreased following IM-412 treatment, but increased expression of
E-cadherin, an epithelial marker, was not detected in the
MDA-MB-231 cells, which were previously reported to be
E-cadherin-negative cells (20).
The expression of vimentin, another well-known EMT marker, also
remained unchanged in response to IM-412. In close agreement with
these results, the expression of EMT-activating transcription
factors such as SNAIL, SLUG and ZEB1 was drastically suppressed by
IM-412 in a dose-dependent manner.
We also verified the effect of IM-412 on the
downregulation of the representative molecule α-SMA by performing
immunocytochemical and immunohistochemical staining. At 2
µM, IM-412 significantly downregulated the cellular
expression of α-SMA (Fig. 2B).
Immunohistochemical analysis of 3D collagen-gel-cultured MDA-MB-231
cells revealed numerous invaded cells in the collagen matrix.
However, the cells treated with IM-412 did not exhibit invasive
activity and expressed α-SMA at a low level (Fig. 2C). These results indicated that
IM-412 effectively suppresses cell motility by inhibiting the
expression of EMT-associated transcription factors and mesenchymal
markers.
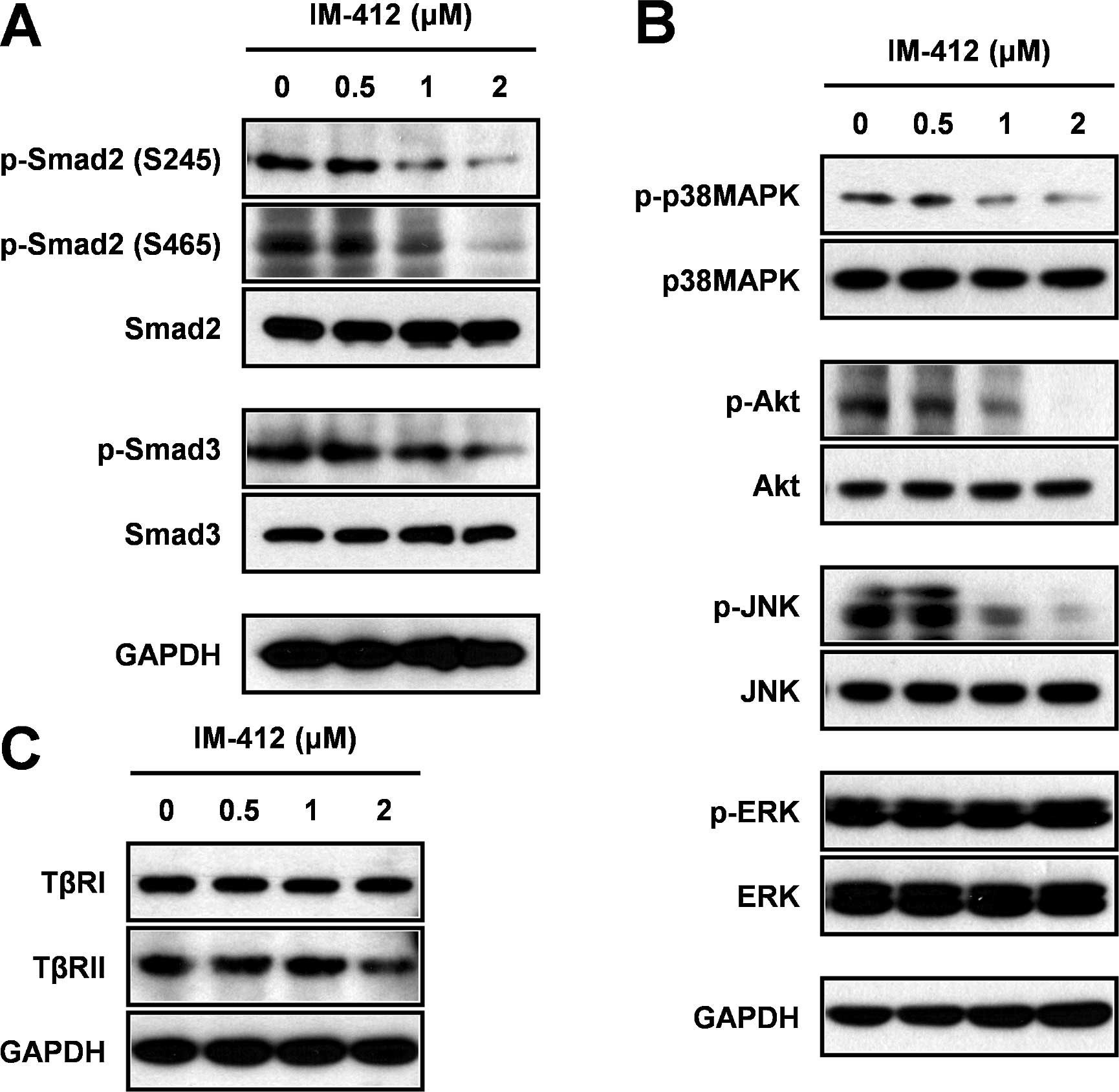
IM-412 inhibits Smad and non-Smad
signaling pathways
To investigate the molecular mechanism underlying
IM-412 inhibition of cancer-cell invasion, we first examined the
major TGF-β downstream signaling pathway, the Smad-dependent
pathway. As expected, IM-412 significantly suppressed the
phosphorylation of both Smad2 and Smad3 in a dose-dependent manner
(Fig. 3A). Since the non-canonical
signaling pathway of TGF-β has also been clearly shown to fully
activate the EMT process (1–3), we
examined how non-Smad signaling was affected by IM-412; the
phosphorylation levels of p38MAPK, Akt and JNK were significantly
decreased by IM-412 in a concentration-dependent manner, but the
level of ERK phosphorylation was not affected (Fig. 3B). To determine whether IM-412
influences the upstream molecules of the TGF-β signaling pathways,
we examined the expression of TβRII and TβRI. In contrast to our
previous findings in lung fibroblasts, IM-412 did not alter the
expression of TβRII and TβRI (Fig.
3C).
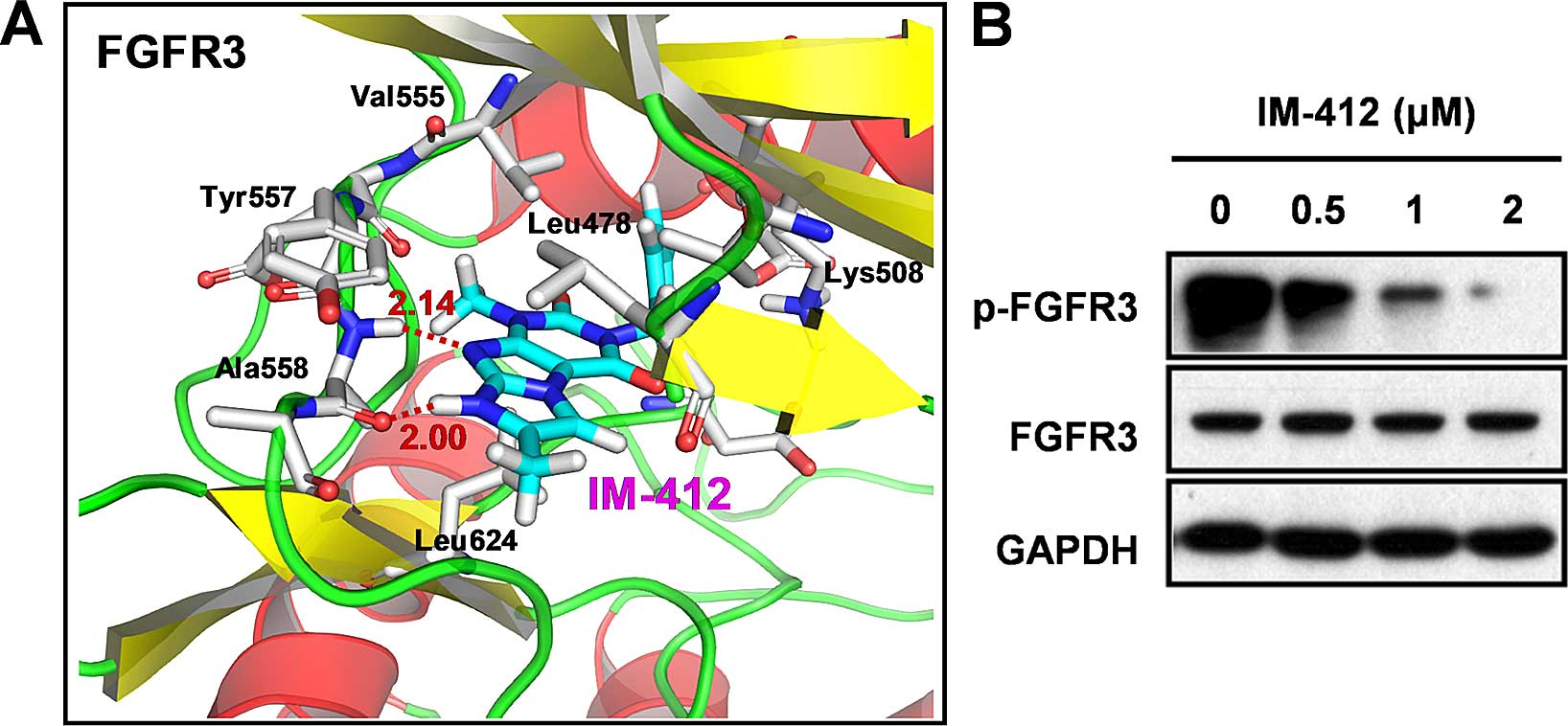
IM-412 inhibits FGFR3
phosphorylation
In addition to TGF-β-family proteins, several other
growth factors can induce partial or full EMT by activating
receptor tyrosine kinases and triggering downstream signaling
cascades (2,6); among the receptor tyrosine kinases,
FGFRs, in particular, are involved in diverse cellular processes,
including metastasis and EMT (10).
In our unpublished data, we demonstrating that another derivative
compound of IM-412 [one fluoryl and one methyl group was added to
IM-412,
3-(2-chloro-6-fluorobenzyl)-1,6,7-tri-methyl-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione]
exhibited the highest inhibitory activity on FGFR3 among 65 kinases
and 2-fold higher inhibitory activity against FGFR3 compared with
FGFR1 in an in vitro kinase assay. Therefore, we focused on
FGFR3 as it appeared to be the most probable target molecule of
IM-412. We employed computational prediction to identify the
binding site of FGFR3 for IM-412 (Fig.
4A). The molecular-modeling results obtained for the
FGFR3/IM-412 complex showed the two strong hydrogen bonds between
the N-H imidazo group of IM-412 and the C=O backbone of Ala558 (2.0
Å), the nitrogen of the purine dione group, and the N-H backbone of
Ala558 (2.14 Å) at the hinge backbone of FGFR3. In the structure,
the 2-chlorobenzyl is directed toward the gatekeeper residue
Val555. Hydrophobic interactions were observed between the benzyl
ring and the gatekeeper residue Val555 in addition to the several
hydrophobic interactions observed with the 2-chloro substitutions
(Lys508 and Ala634). Thus, we considered the imidazopurine
structure of IM-412 to be highly active in blocking the
ATP-binding-pocket region of FGFR3.
To confirm the potential ability of IM-412 to block
the activation of FGFR3, we examined the levels of FGFR3 and
phosphorylated FGFR3. The phosphorylation of FGFR3 was
significantly inhibited by IM-412, while the expression of FGFR3
protein was not changed (Fig.
4B).
Discussion
From a cell-based screening of 8,000 small compounds
performed using a p3TP-Lux reporter-assay system, IM-412 was
isolated as a new inhibitor of TGF-β responses (16). Since the TGF-β signaling pathway is
strongly associated with EMT, we sought to investigate the effect
of IM-412 on the EMT pathway in aggressive human breast cancer
cells.
MDA-MD-231 is a representative cell line of triple
negative breast cancer (TNBC), which lacks the expression of
estrogen receptor (ER), progesterone receptor (PR) and human EGF
receptor 2 (HER2) (and is therefore commonly referred to as
ER-/PR-/HER2-) (21). TNBC is
widely recognized for its aggressive phenotype and poor prognosis,
and thus researchers are attempting to identify novel targets and
develop inhibitors for them as new treatment options for TNBC
patients (22). Recently,
inhibitors of androgen, EGF and VEGF receptors, FGFR, PARP and
tyrosine kinases have been actively studied for use in targeted
cancer therapy, and some of these inhibitors are currently being
used (22,23). However, the occurrence of resistance
to targeted drugs has emerged as a major challenge in the clinic
and has highlighted the necessity for developing a novel class of
anticancer drugs (24–27).
In the present study, IM-412 potently inhibited the
migration and invasion of MDA-MB-231 cells. Although IM-412 did not
alter the expression of vimentin and E-cadherin (which are typical
EMT markers) relative to their expression in untreated control
cells, the compound effectively suppressed the expression of
EMT-activating transcription factors (SNAIL, SLUG and ZEB1) and
mesenchymal makers (N-cadherin, fibronectin, collagen type 1 A2 and
α-SMA). Furthermore, IM-412 treatment led to a reduction in Smad2/3
phosphorylation and also p38MAPK, Akt and JNK phosphorylation,
which indicates that IM-412 can block both canonical and
non-canonical TGF-β signaling pathways. However, the expression of
TβRII and TβRI was not suppressed by IM-412. We also observed that
the phosphorylation of TβRII was not changed with or without IM-412
(data not shown). Similarly, our previous study showed that the
levels of TβRI and TβRII were not changed by IM-412 in human lung
fibroblast, but IM-412 inhibited TGF-mediated TβRI and TβRII
upregulation (16). These findings
suggest that the EMT-related signaling molecules that are in common
may function distinctly depending on the cell type and on the
stimuli and the existing conditions. Although TGF-β receptors
signal directly upstream of Smad2/3 in the EMT signaling pathway,
other kinases such as p38MAPK, Akt, ERK and JNK can also activate
Smad signaling through the phosphorylation of the Smad2 linker
region (Ser245/250/255), which is induced by various growth-factor
receptors, including FGFR (28,29).
Moreover, stimulation of these kinases can lead to the induction of
EMT-activating transcription factors through the Smad-independent
signaling cascade (2,8).
On the basis of the results of computational
prediction, we verified that IM-412 effectively inhibited the
phosphorylation of FGFR3, but did not affect the receptor's protein
level per se. In addition, IM-412 treatment also slightly
reduced the phosphorylation of FGFR1 which share a conserved
structure and a high level of homology with FGFR3 (data not shown).
Given the results of above-mentioned in vitro kinase assay
that most of kinases, such as AKT, p38MAPK, ERK, JNK, Src, RAF,
RAS, JAK and so on, were not influenced by IM-412 derivative, we
carefully considered that IM-412 may not be a general kinase
inhibitor.
FGFRs are well-characterized EMT-inducing
growth-factor receptors (13) that
are frequently overexpressed in cancer cells and are associated
with its aggressive phenotype (14,30,31).
The FGFR-family proteins contain a common extracellular
immunoglobulin-like domain and a cytoplasmic tyrosine kinase
domain. Phosphorylated FGFR activates phospholipase C-γ and FGFR
substrate 2, and this results in the activation of downstream
signaling pathways such as the PKC, STAT and MAPK signaling
pathways (14,15,31).
Thus, the use of targeted therapies against FGFRs, such as those
involving an FGFR1-IIIc-specific antibody (IMC-A1) and
FGFR3-specific antibodies (PRO-001 and R3Mab), is considered a new
approach for treating cancer patients (11). Moreover, the FGFR1 inhibitor
PD173074 has been shown to prevent EMT in head and neck
squamous-cell carcinoma (32). In
the present study, one limitation was that we did not obtain direct
evidence of crosstalk between FGFR/MAPK signaling and Smad
cascades, and thus further investigation must be conducted to
clarify this potential connection between the signaling
pathways.
Collectively, our results demonstrated that the
newly identified small molecule IM-412 inhibited the migration and
invasion of MDA-MB-231 cells by suppressing FGFR3 signaling. We
suggest that further preclinical evaluation of this compound may
facilitate the development of a novel therapeutic agent for
treating malignant cancers.
Acknowledgments
The present study was supported by the Nuclear
Research and Development Program (2012M2A2A7010422), and in part by
the Basic Science Research Program (2013R1A1A2012886) through the
National Research Foundation of Korea (NRF) grant funded by the
Korean government (MSIP).
References
|
1
|
Derynck R, Muthusamy BP and Saeteurn KY:
Signaling pathway cooperation in TGF-β-induced
epithelial-mesenchymal transition. Curr Opin Cell Biol. 31:56–66.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fan YL, Zheng M, Tang YL and Liang XH: A
new perspective of vasculogenic mimicry: EMT and cancer stem cells
(Review). Oncol Lett. 6:1174–1180. 2013.PubMed/NCBI
|
|
5
|
Kurrey NK, Jalgaonkar SP, Joglekar AV,
Ghanate AD, Chaskar PD, Doiphode RY and Bapat SA: Snail and slug
mediate radioresistance and chemoresistance by antagonizing
p53-mediated apoptosis and acquiring a stem-like phenotype in
ovarian cancer cells. Stem Cells. 27:2059–2068. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee JK, Joo KM, Lee J, Yoon Y and Nam DH:
Targeting the epithelial to mesenchymal transition in glioblastoma:
The emerging role of MET signaling. Onco Targets Ther. 7:1933–1944.
2014.PubMed/NCBI
|
|
7
|
Kotiyal S and Bhattacharya S: Breast
cancer stem cells, EMT and therapeutic targets. Biochem Biophys Res
Commun. 453:112–116. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tania M, Khan MA and Fu J: Epithelial to
mesenchymal transition inducing transcription factors and
metastatic cancer. Tumour Biol. 35:7335–7342. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Massagué J: TGFβ signalling in context.
Nat Rev Mol Cell Biol. 13:616–630. 2012. View Article : Google Scholar
|
|
10
|
Katoh M and Nakagama H: FGF receptors:
Cancer biology and therapeutics. Med Res Rev. 34:280–300. 2014.
View Article : Google Scholar
|
|
11
|
Turner N and Grose R: Fibroblast growth
factor signalling: From development to cancer. Nat Rev Cancer.
10:116–129. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brooks AN, Kilgour E and Smith PD:
Molecular pathways: fibroblast growth factor signaling: a new
therapeutic opportunity in cancer. Clin Cancer Res. 18:1855–1862.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Acevedo VD, Gangula RD, Freeman KW, Li R,
Zhang Y, Wang F, Ayala GE, Peterson LE, Ittmann M and Spencer DM:
Inducible FGFR-1 activation leads to irreversible prostate
adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer
Cell. 12:559–571. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Feng S, Zhou L, Nice EC and Huang C:
Fibroblast growth factor receptors: Multifactorial-contributors to
tumor initiation and progression. Histol Histopathol. 30:13–31.
2015.
|
|
15
|
Haugsten EM, Wiedlocha A, Olsnes S and
Wesche J: Roles of fibroblast growth factor receptors in
carcinogenesis. Mol Cancer Res. 8:1439–1452. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Park S, Ahn JY, Lim MJ, Kim MH, Lee SL,
Yun YS, Jeong G and Song JY: IM-412 inhibits transforming growth
factor beta-induced fibroblast differentiation in human lung
fibroblast cells. Biochem Biophys Res Commun. 399:268–273. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yi JY, Hur KC, Lee E, Jin YJ, Arteaga CL
and Son YS: TGFbeta1-mediated epithelial to mesenchymal transition
is accompanied by invasion in the SiHa cell line. Eur J Cell Biol.
81:457–468. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hagler AT, Lifson S and Dauber P:
Consistent force field studies of intermolecular forces in
hydrogen-bonded crystals. 2. A benchmark for the objective
comparison of alternative force fields. J Am Chem Soc.
101:5122–5130. 1979. View Article : Google Scholar
|
|
19
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cailleau R, Young R, Olivé M and Reeves WJ
Jr: Breast tumor cell lines from pleural effusions. J Natl Cancer
Inst. 53:661–674. 1974.PubMed/NCBI
|
|
21
|
Kao J, Salari K, Bocanegra M, Choi YL,
Girard L, Gandhi J, Kwei KA, Hernandez-Boussard T, Wang P, Gazdar
AF, et al: Molecular profiling of breast cancer cell lines defines
relevant tumor models and provides a resource for cancer gene
discovery. PLoS One. 4:e61462009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schmadeka R, Harmon BE and Singh M:
Triple-negative breast carcinoma: Current and emerging concepts. Am
J Clin Pathol. 141:462–477. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yadav BS, Sharma SC, Chanana P and Jhamb
S: Systemic treatment strategies for triple-negative breast cancer.
World J Clin Oncol. 5:125–133. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ciardiello F and Tortora G: EGFR
antagonists in cancer treatment. N Engl J Med. 358:1160–1174. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ellis LM and Hicklin DJ: VEGF-targeted
therapy: Mechanisms of anti-tumour activity. Nat Rev Cancer.
8:579–591. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Grose R and Dickson C: Fibroblast growth
factor signaling in tumorigenesis. Cytokine Growth Factor Rev.
16:179–186. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Takeuchi K and Ito F: Receptor tyrosine
kinases and targeted cancer therapeutics. Biol Pharm Bull.
34:1774–1780. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hough C, Radu M and Doré JJ: Tgf-beta
induced Erk phosphorylation of smad linker region regulates smad
signaling. PLoS One. 7:e425132012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kamato D, Burch ML, Piva TJ, Rezaei HB,
Rostam MA, Xu S, Zheng W, Little PJ and Osman N: Transforming
growth factor-β signalling: Role and consequences of Smad linker
region phos-phorylation. Cell Signal. 25:2017–2024. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wendt MK, Taylor MA, Schiemann BJ,
Sossey-Alaoui K and Schiemann WP: Fibroblast growth factor receptor
splice variants are stable markers of oncogenic transforming growth
factor β1 signaling in metastatic breast cancers. Breast Cancer
Res. 16:R242014. View
Article : Google Scholar
|
|
31
|
Tiong KH, Mah LY and Leong C-o: Functional
roles of fibroblast growth factor receptors (FGFRs) signaling in
human cancers. Apoptosis. 18:1447–1468. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nguyen PT, Tsunematsu T, Yanagisawa S,
Kudo Y, Miyauchi M, Kamata N and Takata T: The FGFR1 inhibitor
PD173074 induces mesenchymal-epithelial transition through the
transcription factor AP-1. Br J Cancer. 109:2248–2258. 2013.
View Article : Google Scholar : PubMed/NCBI
|