Introduction
Li-Fraumeni syndrome (LFS) (OMIM #151623), a
heterogeneous inherited autosomal dominant disorder, manifested by
the occurrence of soft tissue sarcomas, early-onset breast cancer
and other neoplasms in children and young adults (1). There is an exceptionally high risk of
developing multiple primary cancers in members of families with LFS
(2). For example, among 200 LFS
family members diagnosed with cancer, 15% developed a second, 4% a
third and 2% developed a fourth cancer (2). The genetic defect responsible for LFS
lies in the germline mutations in the tumor suppressor gene
TP53 (3). Germline mutations
in CHEK2 may predispose to a few LFS-like families (4). A third LFS locus (LFS3) has
been mapped to chromosome 1q23 but no specific gene has yet been
identified (5).
A 45-year old female patient, who had a family
history of breast cancer, developed neuroblastoma,
rhabdomyosarcoma, breast, lung, thyroid, and gastric cancer,
osteosarcoma, as well as acute myeloid leukemia (AML) in a period
of 38 years. This is a rare case of LFS with nine metachronous
tumors. The family presented with features suggesting LFS (the
Chompret criteria). It is unknown how the patient developed nine
malignant tumors, but survived 45 years.
In the present study, we performed whole-genome
sequencing from blood specimens obtained a year prior to the
diagnosis of AML, as well as whole-exome sequencing from blood
specimens at the time of diagnosis of AML in the subject. We aimed
to obtain a better understanding of: i) how rare mutations in the
genome contribute to the development of the cancer phenotypes; ii)
how the cancer risk can be retrospectively estimated by common
variants; and iii) why the patient survival was longer than
expected in LFS patients.
Materials and methods
DNA samples
DNA samples from the patient's whole blood were
banked for next-generation sequencing under a protocol approved by
the institutional review board (IRB) at the Peking Union Medical
College Hospital (PUMCH). One sample was obtained a year prior to
the diagnosis of acute myeloid leukemia (AML)-M2
(pre-AML), and another sample was obtained at the time of the
diagnosis of AML-M2. The patient provided written
informed consent.
DNA extraction, library construction and
next-generation sequencing
Genomic DNA was isolated from two samples of whole
blood (pre-AML-DNA and AML-DNA). We performed whole-genome
sequencing (WGS) and whole-exome sequencing (WES) for pre-AML-DNA
and AML-DNA, respectively. All sequencing was carried out at the
Beijing Genomic Institute at Shenzhen (BGI-Shenzhen, Shenzhen,
China). A detailed description of library preparation and
sequencing is available upon request.
Sanger sequencing for TP53
We used the International Agency for Research on
Cancer (IARC) protocol
(p53.iarc.fr/Download/TP53_DirectSequencing_IARC.pdf) for detection
and validation of mutations in exons (2-11) of
TP53 by Sanger sequencing.
Identification of single nucleotide
variants (SNVs), structural variants (SVs) and somatic
mutations
A computational pipeline was used for variant
discovery. The pipeline integrates software tools widely used for
analyzing next-generation sequencing data. For detecting structural
variants, we used BreakDancer (6)
to predict a wide variety of structural variants including
large-scale indels, inversions and intra-/inter-chromosomal
translocations. We used Control-FREEC (7) for assessing copy number variants. The
program MuTect (8) was used to
identify somatic mutations leading to leukemic transformation by
comparing pre-AML DNA and AML DNA.
Analysis of rare mutations
In the case of only a single dominantly affected
individual available, we designed a pipeline to prioritize the
filtered mutations based on a candidate strategy (9). Analysis-ready variants were annotated
using SeattleSeq annotation (http://snp.gs.washington.edu/SeattleSeqAnnotation138/).
We focused on three areas: i) variants associated with genes for
hereditary cancer predisposition syndrome, i.e., syndromes of
inherited cancer predisposition in clinical oncology syndrome
(10); ii) variants in DNA repair
genes (11); and iii) mutations in
TP53 and genes in the p53 signaling pathway (KEGG,
www.genome.jp/kegg-bin/show_pathway?map04115). We
performed database queries, biophysical prediction and analyses of
non-coding regions to screen for known and novel mutations. For
rare novel mutations, we prioritized variants for stop, frame-shift
and splice site mutations. Polyphen (12) was used to predict the functional
effect of missense mutations.
Genetic risk prediction based on common
variants
To retrospectively estimate the cancer risk, we
performed genetic risk prediction using the VARiants Informing
MEDicine (VARIMED) database, a manually curated database of human
SNP-disease associations (13). The
current VARIMED database contained 466,890 unique SNPs associated
with 6,691 diseases and related traits extracted from 17,088
publications. We extracted all variants from VARIMED that were
associated with phenotypes containing terms of 'cancer',
'lymphoma', 'leukemia', 'melanoma', 'glioma', 'carcinoma',
'neuroblastoma', 'myeloma', 'sarcoma' and 'glioblastoma'. We only
used SNP-disease associations that were observed in at least two
genome-wide association studies at the significance level
P<10−6 with total number of at least 2,000
participants.
We estimated the pre-test probability of developing
different tumors in Chinese women using the incidence
(age-standardized incidence rate) reported in 2012 GLOBOCAN
(globocan.iarc.fr/Pages/fact_sheets_population.aspx). For all SNPs
associated with a given cancer, we emitted all genotypes (variant
calls and reference-reference calls) from the WES data. We
determined likelihood ratio (LR) for each SNP-disease association
based on VARIMED, correcting for the effects of linkage
disequilibrium among multiple loci (14). Finally, we combined the pre-test
probabilities and LRs to calculate the post-test probabilities
(14). We performed risk prediction
analysis for the patient, as well as for all Chinese individuals
from the 1000 Genomes Project phase 1 as a background (CHB and CHS
populations, n=197) (15).
We assigned a unique risk allele for each
cancer-related SNPs (if an SNP was associated with multiple cancers
with different risk alleles, we chose the risk allele linked to the
majority of the cancers). The total number of risk alleles was
calculated in the patient's exome and in each of the 197 genomes of
Chinese populations from the 1000 Genomes data.
Results
Patient
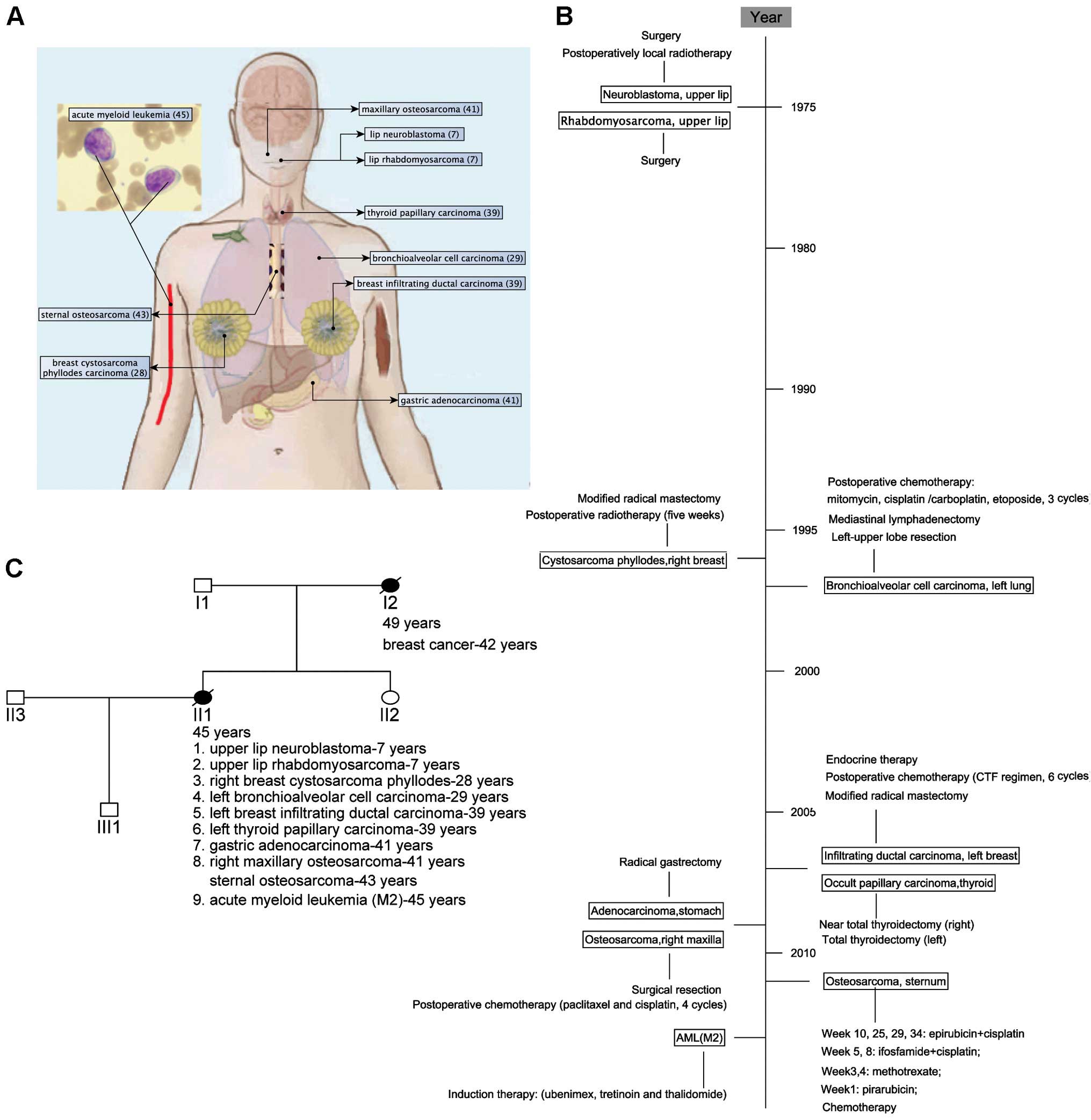
The patient was a 45-year old female Han Chinese,
who presented with nine primary malignant neoplasms (Fig. 1A and B) and a family history of
breast cancer (Fig. 1C). At the age
of 7, the patient was subsequently operated for upper lip
neuroblastoma (received postoperatively local radiotherapy) and
rhabdomyosarcoma. Twenty-one years later, she was treated for
cystosarcoma phyllodes of the right breast by radical mastectomy
and postoperative radiotherapy. At the age of 29, after a lung mass
was found, the left-upper lobe resection and mediastinal
lymphadenectomy revealed bronchioalveolar cell carcinoma with hilar
lymphatic metastases (T3N1M0, IIIA). She received postoperatively
adjuvant chemotherapy. Ten years later (at the age of 39), she
found a left breast mass by self-examination and a modified radical
mastectomy was performed. The pathology analysis showed a
middle-differentiated infiltrating ductal carcinoma (T2N0M0, IIA).
She was given adjuvant chemotherapy and endocrine therapy. Five
months later, after pharynx discomfort, she was treated for thyroid
nodule with total thyroidectomy for the left thyroid and near total
thyroidectomy for the right thyroid. Pathology reports indicated
nodular goiter and papillary carcinoma. At the age of 41, she
received gastric endoscopy due to black stool and found
preventriculus ulcer. She was treated by radical gastrectomy and
indicated gastric adenocarcinoma (T4N1M0, IIIA), and she received
four cycles of chemotherapy (paclitaxel and cisplatin). Two months
later, the bone scanning indicated 'abnormality in the maxilla' and
surgical resection of the mass in the right maxilla showed
osteosarcoma. Two years later (at the age of 43), the patient found
a lump on the sternum and the bone scanning showed an abnormal
radioactive accumulation in the right sternoclavicular
articulation. Sternal biopsy indicated spindle cell sarcoma
(T1N0M0G3, IIA). We could not confirm that the sternal spindle cell
sarcoma was a primary or a metastasis from the maxillary
osteosarcoma. Without a chance for operation, the patient was
administered chemotherapy and then local radiotherapy. At the age
of 45, analysis of her bone marrow revealed 30% myeloblasts and
13.5% promyelocytes and a complex karyotype involving monosomy 13,
16, and X, trisomy 8, 11, del(5q31), ? del(17p10), t(1:?)(q11:?),
ins(1p10), ?ins(17q10) and two marker chromosomes that could not be
resolved by standard cytogenetic analysis. The patient developed
respiratory infection and died five months later after the
diagnosis of AML-M2. All clinical information, including
photomicrographs in pathology, has been uploaded to the
Phenomecentral database at https://phenomecentral.org/P0001643.
Thus, a total of at least nine primary malignant
neoplasms developed within a 38-year period. This rare LFS patient
provided a model to identify susceptibility rare mutations and
estimate cancer risk based on common variants with the advent of
next-generation sequencing. We performed whole-genome sequencing
(WGS) from blood specimens obtained a year prior to the diagnosis
of AML to create a reference sequence (germline) for this patient
with a relatively lower coverage (~×30) and used this reference
sequence to investigate germline abnormity, including SNVs and
aberrations. In order to estimate cancer-risk of this patient, we
then performed whole-exome sequencing (WES) from blood specimens at
the time of diagnosis of AML with a relatively higher coverage
(105×). We can also detect somatic mutations underlying leukemic
transformation in the patient by comparing with the reference
sequence obtained a year prior to the diagnosis of AML.
Whole-genome sequencing (WGS)
We performed four paired-end sequencing runs for
pre-AML-DNA and produced 1,079,697,548 clean sequence reads (219.7
GB). In total, 96.88% of the reads were mapped to the hg19,
resulting 99.14% total coverage of the reference genome. The
distribution of read depth indicated that the average depth was
29.44 and 98.8% of genomic positions had read depth ≥4.
We analyzed 2.84 Gbp of the reference genome for
SNVs and identified 3,565,277 filtered SNVs (autosomes and
chromosome X). The number was in the range of 3–4 million SNVs in
an average person (16). Of these,
4.8% were not represented in the dbSNP database (build 137) and
were considered to be novel. The numbers of homozygous and
heterozygous SNVs were 1,477,141 and 2,084,985, respectively. In
protein-coding regions, 10,223 missense and 99 nonsense mutations
were identified (Table I).
 | Table INumber of SNVs identified by whole
genome sequencing and whole exome sequencing. |
Table I
Number of SNVs identified by whole
genome sequencing and whole exome sequencing.
| Class | Number of SNVs
|
|---|
| Whole-genome
sequencing | Whole-exome
Sequencing |
|---|
| Missense | 10,223 | 9,826 |
| Nonsense | 99 | 87 |
| SNPs in splice
sites | 65 | 52 |
| Synonymous | 11,111 | 10,972 |
| SNPs in coding (not
mod 3) | 27 | 28 |
| SNPs in a UTR | 28,659 | 10,126 |
| SNPs near a
gene | 72,332 | 11,910 |
| Intronic | 1,256,089 | 200,160 |
| Intergenic
SNPs | 2,184,672 | 208,248 |
| SNPs in
microRNAs | 123 | 105 |
| SNPs in dbSNP | 3,480,654 | 446,277 |
| SNPs not in
dbSNP | 82,623 | 5,132 |
| Total | 3,565,277 | 451,409 |
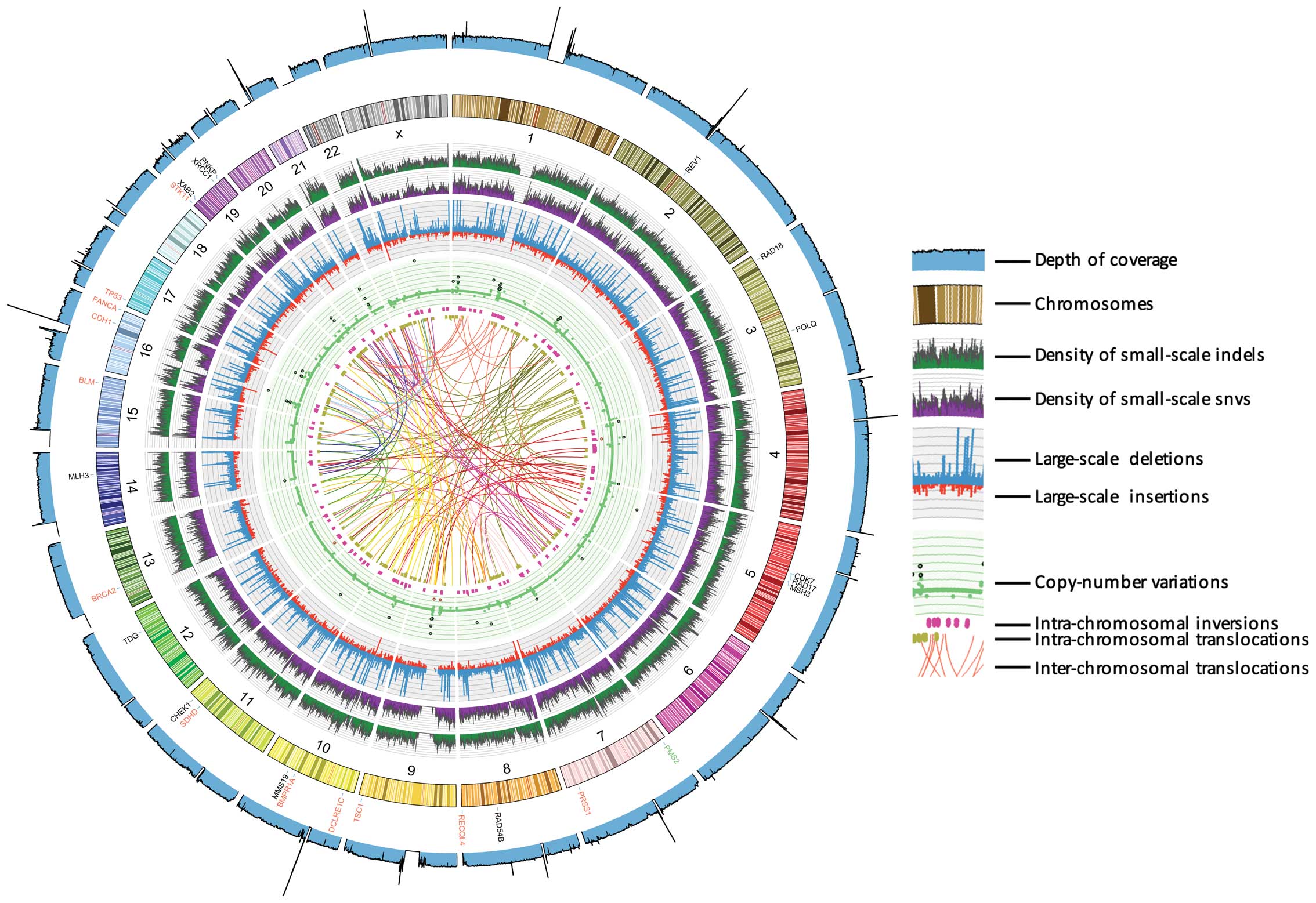
The overall patterns of genome alterations,
including density of SNVs, small indels, and structural variants of
whole chromosomes, were seen in this patient (Fig. 2). The mode of density plots of SNVs
and indels, in each interval of 0.25 MB, are similar. There were
171 inter-chromosomal and 263 intra-chromosomal translocations,
indicating that intra-chromosomal translocations outnumber
inter-chromosomal translocations. We identified 3,778 large-scale
insertions and 11,540 large-scale deletions, respectively. In
total, 6,663 CNVs were identified, which varied from −2 to 30 and
most of the CNVs were around 2. As expected, high value of CNVs
commonly involves high sequence depth. On the centromere and/or
chromosomes-end regions, the depth of coverage is low or
lacking.
Analyses of rare mutations
Mutations associated with hereditary
cancer predisposition syndromes
We first searched for evidence of known/novel rare
missense mutations (<5% of the minor allele frequency in Asians
from HapMap) that would predispose the patient to hereditary cancer
predisposition syndromes (10),
including LFS cancers. We identified at least 20 known and 2 novel
rare missense mutations within 13 genes lead to hereditary cancer
predisposition syndromes, which may be associated with increased
risk of cancer in the patient (Table
II). High interest genes include: i) a mutation (rs28934576,
R273H) in TP53, which was previously identified in LFS
patients with soft-tissue sarcoma and gastric carcinoma (17,18);
ii) a mutation (rs144848, N372H) in BRCA2, associated with
an increased risk of breast cancer (19); and iii) a probably-damaging missense
mutation (rs2276331, L630V) in CDH1, where a heterozygous
insertion in CDH1 was found in the germline of a woman who
developed lobular breast cancer (20) and a mutation in CDH1
increased risk of developing gastric cancer and other cancers
associated with human diffuse gastric cancer (HDGC) (21). Additionally, several rare missense
mutations were identified in the RecQ helicase family, i.e.,
RECQL4 (RecQ protein-like 4), and BLM (Bloom
syndrome, RecQ helicase-like). DNA helicase deficiencies has been
found to be associated with chromosome instability and cancer
predisposition (22). Mutations in
RECQL4 and BLM have been shown to cause hereditary
disorders, i.e., Rothmund-Thomson Syndrome (23), and Bloom Syndrome (24), respectively. Recently, WES
identified rare deleterious mutations in BLM as potential
breast cancer susceptibility alleles (25). We also noted several rare missense
mutations in genes associated hereditary gastrointestinal
malignancies, e.g., Juvenile polyposis (BMPR1A), Turcot
syndrome (PMS2) and Peutz-Jeghers syndrome (STK11).
However, function effects of these missense mutations needed to be
further studied.
| Table IIIdentified rare missense mutations in
genes implicated in hereditary cancer predisposition syndromes and
mismatch repair genes. |
Table II
Identified rare missense mutations in
genes implicated in hereditary cancer predisposition syndromes and
mismatch repair genes.
| Chr_posa | Rs ID | AA changeb | PolyPhenc | Asian
HapMapFreqd | Gene |
|---|
| Hereditary cancer
predisposition syndromes |
| 7:6026942 | rs1805323 | T->K | 0 | NA | PMS2 |
| 7:6026988 | rs1805321 | P->S | 0.018 | 0 | PMS2 |
| 7:142460335 | rs201550522 | K->E | 0 | NA | PRSS1 |
| 7:142460369 | rs201719096 | S->N | 0 | NA | PRSS1 |
| 7:142460744 | rs150930992 | C->S | 0.185 | NA | PRSS1 |
| 7:142460752 | rs140793689 | Q->E | 0 | NA | PRSS1 |
| 7:142460764 | rs200902389 | V->I | 0 | NA | PRSS1 |
| 8:145737816 | rs4251691 | R->Q | 0.035 | NA | RECQL4 |
| 8:145741702 | rs4244612 | E->D | 0.002 | NA | RECQL4 |
| 8:145742514 | rs2721190 | S->P | Unknown | 0 | RECQL4 |
| 9:135772897 | – | I->T | 0.77 | NA | TSC1 |
| 10:88635779 | rs11528010 | P->T | 0 | NA | BMPR1A |
| 10:14976727 | rs35441642 | P->R | 1 | NA | DCLRE1C |
| 11:111963860 | rs592626 | Q->R | Unknown | NA | SDHD |
| 13:32929387 | rs169547 | V->A | Unknown | 0.7 | BRCA2 |
| 15:91354521 | rs7167216 | V->I | 0.01 | 3.3 | BLM |
| 16:68856080 | rs2276331 | L->V | 1 | 1.7 | CDH1 |
| 16:89813029 | – | C->S | 1 | NA | FANCA |
| 16:89836323 | rs7195066 | G->D | 0 | 1.2 | FANCA |
| 16:89866043 | rs7190823 | T->A | 0 | 1.9 | FANCA |
| 17:7577120 | rs28934576 | R->H | 1 | NA | TP53 |
| 19:1223125 | rs59912467 | F->L | 0.062 | NA | STK11 |
| Mismatch repair
genes |
| 2:100055158 | rs3087399 | N->S | 0.069 | 3.6 | REV1 |
| 3:8977648 | – | S->P | 0.937 | NA | RAD18 |
| 3:121263720 | rs702017 | R->I | 0 | 0 | POLQ |
| 5:68531253 | rs2972388 | T->I | Unknown | NA | CDK7 |
| 5:68695940 | rs1045051 | L->R | 1 | 0 | RAD17 |
| 5:80149981 | rs184967 | Q->R | 0 | 0 | MSH3 |
| 7:6026942 | rs1805323 | T->K | 0 | NA | PMS2 |
| 7:6026988 | rs1805321 | P->S | 0.018 | 0 | PMS2 |
| 8:95403868 | rs114216685 | N->S | 1 | NA | RAD54B |
| 10:99240758 | rs2275586 | A->G | 0 | 2.8 | MMS19 |
| 11:125525195 | rs506504 | I->V | 0 | 0 | CHEK1 |
| 12:104380734 | rs2888805 | V->M | 0.833 | NA | TDG |
| 14:75513883 | rs175081 | N->D | 0.001 | 0.4 | MLH3 |
| 19:7692266 | – | T->P | 0.961 | NA | XAB2 |
| 19:44047826 | rs2682557 | N->Y | 0 | NA | XRCC1 |
| 19:50370425 | rs201218221 | E->Q | 0.004 | NA | PNKP |
Mutations in DNA repair genes
Defects of DNA repair are a common cause of
inherited cancer susceptibility. DNA mismatch repair (MMR) is a
system for recognizing and repairing mismatch (e.g., erroneous
indels and mismatched bases) during DNA replication and
recombination, as well as repairing some forms of DNA damage
(26). We investigated whether the
patient had mutations in DNA repair genes (Table II). We identified two rare missense
mutations in PMS2, as well as one each in MSH3 and
MLH3. The rs1805321 (P470S) in PMS2 was previously
found in three out of seven patients with colorectal cancer without
family history (27). We
additionally found a common variant in the MLH1 promoter
(rs1800734, A/G), which was previously found to be heterozygous in
a patient with metachronous carcinomas that had microsatellite
instability and lacked MLH1 expression (28).
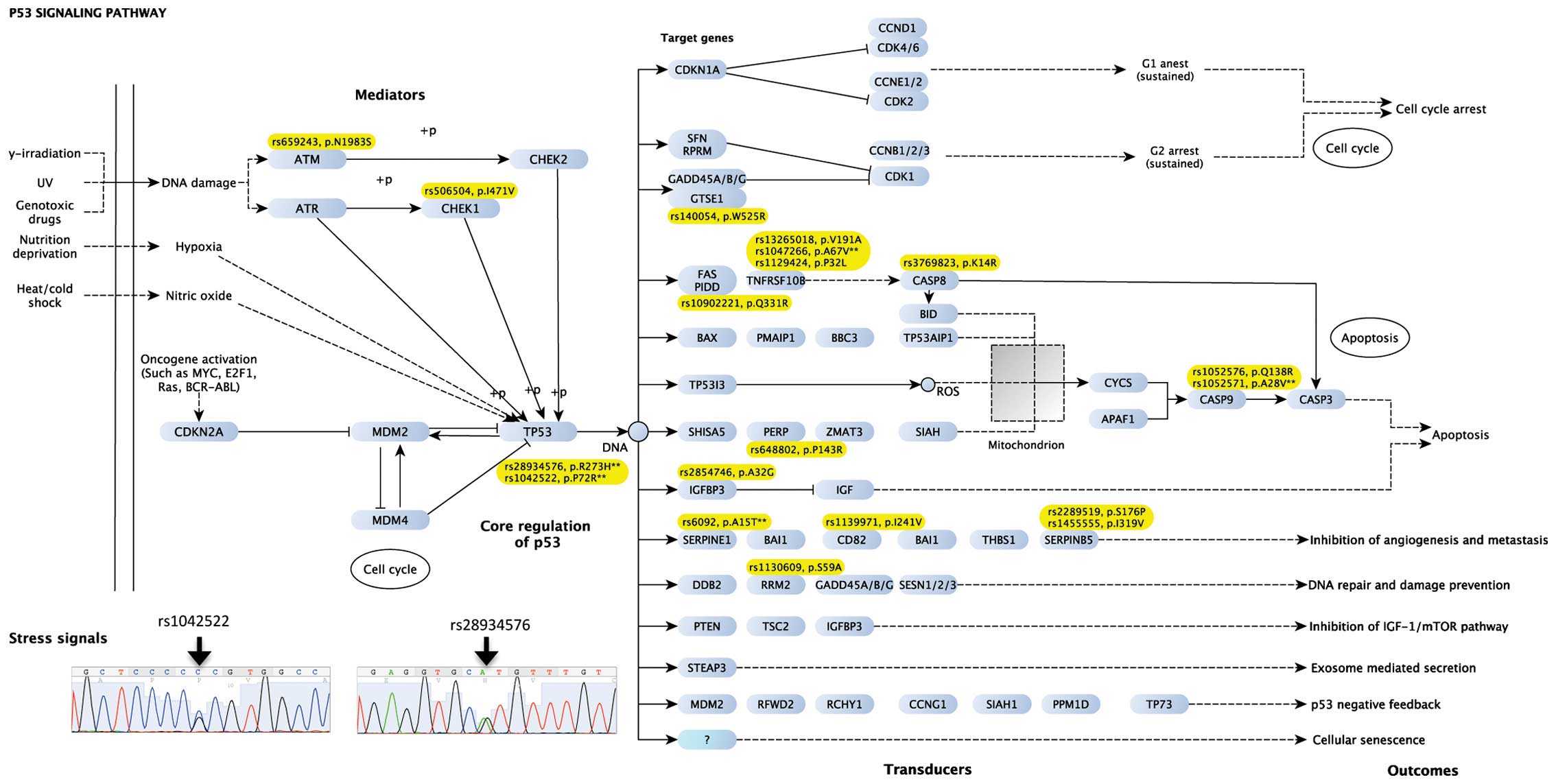
Variants in TP53 and genes in the
p53-signaling pathway
As the signaling cascades are genetically
dysregulated tumor dependencies, we characterized variants in the
p53-signaling pathway. Of 68 genes in the p53-signaling pathway, 19
missense mutations in 14 genes were identified, which was
significantly higher than expected (Fisher's exact test, P=0.02)
(Fig 3). The mutated genes were
clustered in: i) the mediators of p53 signaling pathway (e.g.,
ATM and CHEK1); ii) TP53; and iii) transducers
leading to the outcomes of 'apoptosis' and 'inhibition of
angiogenesis and metastasis'. Of note, two missense mutations
(rs1042522 and rs28934576) in TP53 were identified and
confirmed by Sanger sequencing. The rs28934576 (codon 272,
His293Arg) was identified previously to be present in a case of LFS
with gastric cancer (18). We did
not note other missense mutations in TP53.
Whole-exome sequencing (WES)
WES was performed for AML-DNA with a relatively
higher depth of coverage (105×); resulting in an output of 30 GB of
raw sequence and 99.4% exome regions were covered at least four
times. A summary of the identified variants was shown in Table I, including 451,409 and 19,995
filtered SNVs and small indels, respectively.
Concordance of SNVs identified by WGS
and WES
The genotype concordance between WGS and WES was
high. Of 438,261 SNVs called in both sequencing data, genotypes in
395,824 SNVs (90.32%) were consistent. Nearly all of inconsistent
genotypes (42,327 out of 42,437, 99.7%) resulted from a low
sequence depth in either WGS or WES data, leading to
alternative-allele homozygous calls. The remaining sites of
inconsistent genotypes may be introduced by sequencing errors.
Therefore, we estimated the sequence error rate to be 0.024%
(110/42437). Of 36 SNVs listed in Table II, genotypes of 31 SNVs were
consistently identified in both sequencing, and the remaining 5
were only identified in WGS. We also estimated the false discovery
rate of novel SNV identification by randomly selecting 53 novel
SNVs identified in both WGS and WES for validation by Sanger
sequencing. We obtained 47 and 49 PCR products for pre-AML and
AML-DNA, respectively. Of 41 and 39 SNVs with successful
sequencing, only one SNV for each were found to be false positive
(false discovery rate, 2.4 and 2.6%, respectively).
WES-based cancer risk evaluation:
Risk-O-Gram analysis
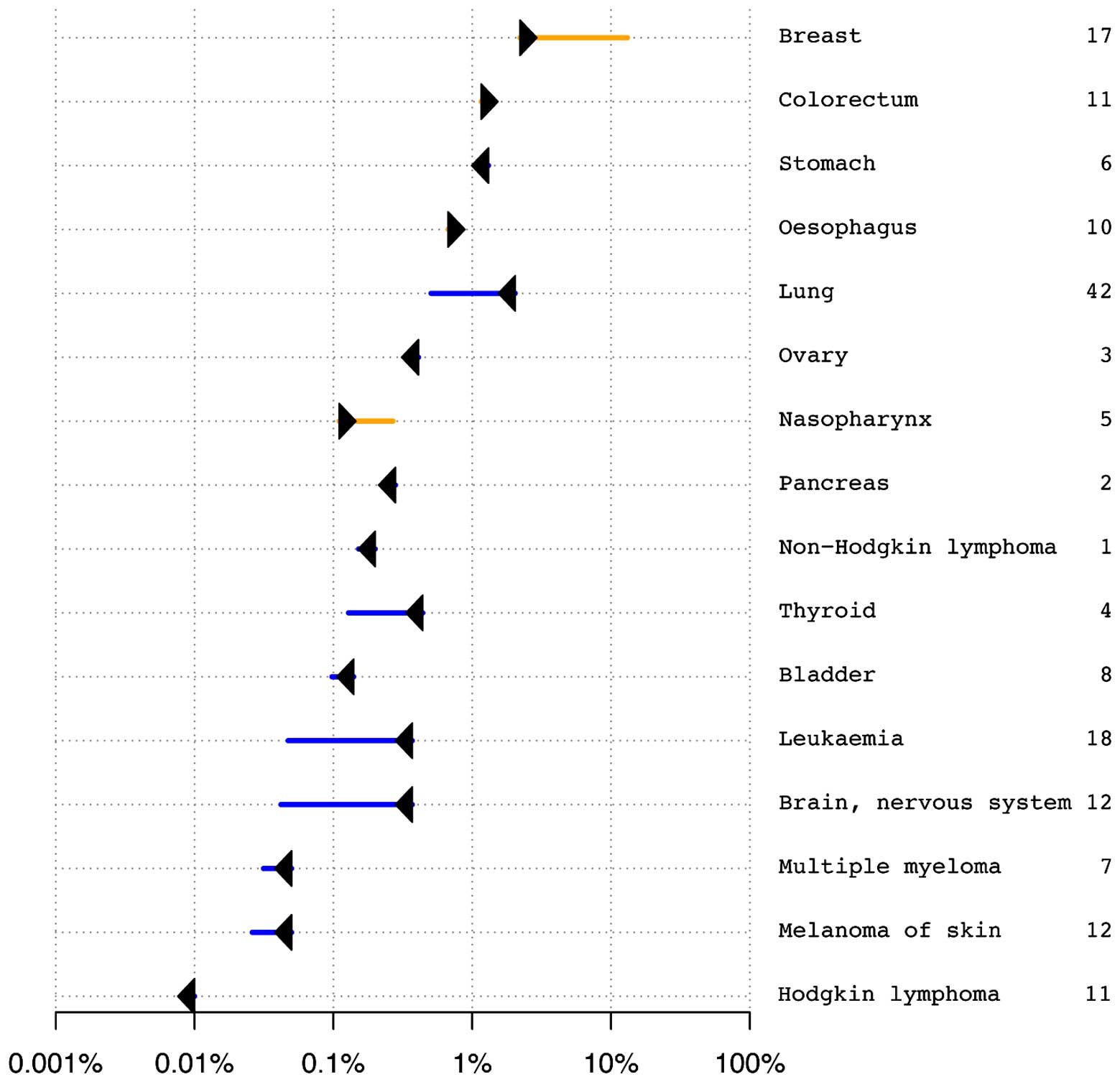
It is unknown whether the phenotypes of multiple
primary malignant neoplasms in the studied patient were caused by
an unusual accumulation of common cancer risk variants.
VARIMED-based analysis of the genetic risk of cancer did not reveal
increased risk for multiple cancers (Fig. 4). Risk-O-Gram plots for four cancers
(breast, gastric, neuroblastoma and thyroid) diagnosed in the
patient were available upon request. Our results suggested that the
patient yielded an average overall risk of cancer, with an
increased risk of some cancers and decreased risk of others. For
example, a significantly increased risk of breast cancer was
estimated to be over six times greater than expected. Only 2% of
control samples have higher predicted risk of breast cancer. The
patient developed right breast cystosarcoma phyllodes at 28 years
and left breast infiltrating ductal carcinoma at 39 years, which
confirmed our predictions. An increased risk of developing
nasopharyngeal cancer was predicted, which, however, was not
diagnosed in the patient. Regarding other diagnosed and undiagnosed
cancers, the patient showed either average risk, not deviating from
the median of the control samples (e.g. neuroblastoma and lung
cancer), or significantly decreased risk (e.g. thyroid cancer and
melanoma) (data not shown). The remaining cancers diagnosed in the
patient did not have enough significant associations in
VARIMED.
In addition, in order to assess the patient's risk
with respect to the control population, we performed the risk
calculation for all Chinese samples of the 1000 Genomes data
(15). The fractions of control
samples (CHB and CHS populations from the 1000 Genomes Project)
have lower, equal or higher predicted genetic risk than the cancer
risk of the studied patient (data not shown).
Somatic mutations implicated in
leukemic transformation
We compared the sequencing data of AML-DNA with
pre-AML-DNA, and identified at least 60 mis-/non-sense somatic
mutations. Of note, 12 missense mutations in 11 genes have been
reported in COSMIC database (29),
including MLL3, that may contribute to development of
AML-M2. It has been suggested that MLL3 is a
haploinsufficient 7q tumor suppressor in AML (30), and a germline mutation in
MLL3 was identified in a pedigree of colorectal cancer and
AML (31).
Discussion
We presented a rare case of Li-Fraumeni syndrome
(LFS), who had developed at least nine primary malignant neoplasms
over a period of 38 years. The characteristics of this patient,
i.e., presenting with cancers at an early age (7 years), with
multiple primary cancers, and with a family history, suggested an
increased inherited cancer susceptibility risk. The identification
of the genetic basis for cancer susceptibility has important
clinical implications for the early diagnosis and prevention (e.g.,
change of lifestyle) of associated neoplasms. A recent study has
shown that regular screening in LFS families led to a survival
benefit (32). The advent of
next-generation sequencing, a comprehensive, unbiased approach,
made it possible to identify mutations in cancer susceptibility
genes. In the present study, we identified rare mutations leading
to multiple malignant neoplasms, retrospectively estimated cancer
risk, and explored the underlying mechanism of her relatively
longer life expectancy.
We found mutations in cancer susceptibility genes of
hereditary breast cancer syndromes (e.g., LFS), including
TP53 and BRCA2. The mutations in TP53 may
impair or reduce the function of p53 due to dominant negative
activity (33). Mutations in
TP53 correlated with 'complex' karyotypes in AML (34) as shown in the bone marrow of the
studied patient. The number of mutated genes in the p53 signaling
pathway was significantly greater than expected, which suggested
that, the p53 stress response pathway harbors functional inherited
variants acting together to affect p53 signaling in cells (35). The patient contained rare mutations
in five genes (CDH1, BMPR1A, STK11,
PRSS1 and PMS2), which were implicated in hereditary
gastrointestinal malignancies. A mutation in CDH1 was found
to be associated with an increased risk developing gastric cancer,
e.g., hereditary diffuse gastric cancer (36).
It is possible that an accumulation of common
germline variants associated with cancer may determine inherited
predisposition to cancer. To test the hypothesis, we estimated
cancer risk by integrating information from multiple alleles
associated with cancer risk. We found that the phenotype of the
studied patient, characterized by the development of multiple
cancers, was unlikely to be caused by the unusual accumulation of
known cancer-associated common variants (Fig. 4), although the patient did have an
increased risk of breast cancer when compared with the control
populations in the 1000 Genomes Project. However, the
post-probabilities did not take into account the exactly matching
background population; for example, the predicted genetic risk of
lung cancer was decreased not only in the studied patient but also
in the studied background population. It is unknown whether rare
mutations with high penetrance and common low penetrant variants
have synergistic effects in the development of multiple cancers in
the patient.
Life expectancy is substantially reduced in LFS,
which is likely to be below 40 years of age on average (37). Given the development of nine
malignant tumors in the patient, the survival (45 years) was better
than expected. One reason was that the patient received excellent
health care at the PUMCH. Second, we could consider an alternative
hypothesis that the patient's genome is depleted of cancer risk
alleles and enriched for cancer protective alleles. Our
disease-based analysis of known variants seemed to partially
support this hypothesis in that the total number of risk alleles in
the patient was less than the average in control samples. However,
VARIMED does not really provide evidence that the patient was
protected from dying from cancer (SNPs in VARIMED are the risk SNPs
for developing a cancer but not for survival).
Other possible explanations for the occurrence of
nine primary metachronous malignant neoplasms in the patient
included: radiation-induced cancers have been suggested to be more
common in patients with LFS (38,39);
and therapy-related AML is a rare but well-described complication
of chemotherapy (40). In the
setting of DNA repair defects due to germline TP53 mutation,
the effect of therapeutic ionizing radiation is a major concern. We
could not exclude the possibility that AML in this patient is
secondary after etoposide therapy for bronchioalveolar cell
carcinoma (41).
It has been suggested that whether a TP53
germline mutation carrier will develop cancer and where the
cancer(s) will develop depends upon where the second mutation
occurs in which cell type. It is unclear whether there was a second
TP53 mutation in the patient. Typically, a tumor contains
two to eight of 'driver gene' mutations (42), thus, it is unknown whether there
were driver somatic mutations or massive chromosome rearrangements
underlying the development of different tumors. Discovering
mutation driver genes for different tumors in the studied patient
remains challenging. Rausch et al (43) uncovered massive, complex chromosome
rearrangements (termed 'chromothripsis') in a Sonic-Hedgehog
medulloblastoma brain tumor from an LFS patient. The association
between TP53 mutation and chromothripsis in Sonic-Hedgehog
medulloblastoma was revealed in additional patients (43). More work is needed to characterize
structural variants, especially for chromothripsis, in different
tumors.
In conclusion, we identified multiple rare mutations
in genes implicated in hereditary cancer predisposition syndromes,
which may lead to multiple malignant neoplasms in the studied
patient. Of note, we found that the number of mutated genes in p53
signaling pathway was significantly greater than expected. However,
the phenotype of multiple primary neoplasms in this patient was
unlikely to be caused by an unusual accumulation of common cancer
risk variants. Further studies are needed to characterize mutation
patterns, including somatic mutations and structural variants in
different tumors developed in this patient.
Acknowledgments
We thank Di Ma, Xiao Chen, Wenxun Huang, Xia Tang,
Wenwei Yin and Shiwen Tong for technical support. The study is
supported by the Recruitment Program of Global Youth Experts in
China (KD) and a start-up fund from the Second Affiliated Hospital
of Chongqing Medical University (KD). Conflict of interest: A.
Patwardhan and R. Chen are employees of Personalis, Inc., the
company licenced to develop the VARIMED database.
References
|
1
|
Li FP and Fraumeni JF Jr: Soft-tissue
sarcomas, breast cancer, and other neoplasms. A familial syndrome?
Ann Intern Med. 71:747–752. 1969. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hisada M, Garber JE, Fung CY, Fraumeni JF
Jr and Li FP: Multiple primary cancers in families with Li-Fraumeni
syndrome. J Natl Cancer Inst. 90:606–611. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Olivier M, Hollstein M and Hainaut P: TP53
mutations in human cancers: Origins, consequences, and clinical
use. Cold Spring Harb Perspect Biol. 2:a0010082010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bell DW, Varley JM, Szydlo TE, Kang DH,
Wahrer DC, Shannon KE, Lubratovich M, Verselis SJ, Isselbacher KJ,
Fraumeni JF, et al: Heterozygous germ line hCHK2 mutations in
Li-Fraumeni syndrome. Science. 286:2528–2531. 1999. View Article : Google Scholar
|
|
5
|
Bachinski LL, Olufemi S-E, Zhou X, Wu CC,
Yip L, Shete S, Lozano G, Amos CI, Strong LC and Krahe R: Genetic
mapping of a third Li-Fraumeni syndrome predisposition locus to
human chromosome 1q23. Cancer Res. 65:427–431. 2005.PubMed/NCBI
|
|
6
|
Chen K, Wallis JW, McLellan MD, Larson DE,
Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, et
al: BreakDancer: An algorithm for high-resolution mapping of
genomic structural variation. Nat Methods. 6:677–681. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Boeva V, Popova T, Bleakley K, Chiche P,
Cappo J, Schleiermacher G, Janoueix-Lerosey I, Delattre O and
Barillot E: Control-FREEC: A tool for assessing copy number and
allelic content using next-generation sequencing data.
Bioinformatics. 28:423–425. 2012. View Article : Google Scholar :
|
|
8
|
Cibulskis K, Lawrence MS, Carter SL,
Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES
and Getz G: Sensitive detection of somatic point mutations in
impure and heterogeneous cancer samples. Nat Biotechnol.
31:213–219. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gilissen C, Hoischen A, Brunner HG and
Veltman JA: Disease gene identification strategies for exome
sequencing. Eur J Hum Genet. 20:490–497. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Garber JE and Offit K: Hereditary cancer
predisposition syndromes. J Clin Oncol. 23:276–292. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wood RD, Mitchell M, Sgouros J and Lindahl
T: Human DNA repair genes. Science. 291:1284–1289. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ramensky V, Bork P and Sunyaev S: Human
non-synonymous SNPs: Server and survey. Nucleic Acids Res.
30:3894–3900. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen R, Davydov EV, Sirota M and Butte AJ:
Non-synonymous and synonymous coding SNPs show similar likelihood
and effect size of human disease association. PLoS One.
5:e135742010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ashley EA, Butte AJ, Wheeler MT, Chen R,
Klein TE, Dewey FE, Dudley JT, Ormond KE, Pavlovic A, Morgan AA, et
al: Clinical assessment incorporating a personal genome. Lancet.
375:1525–1535. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
1000 Genomes Project Consortium; Abecasis
GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang
HM, Marth GT and McVean GA: An integrated map of genetic variation
from 1,092 human genomes. Nature. 491:56–65. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mwenifumbo JC and Marra MA: Cancer
genome-sequencing study design. Nat Rev Genet. 14:321–332. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Malkin D, Jolly KW, Barbier N, Look AT,
Friend SH, Gebhardt MC, Andersen TI, Børresen AL, Li FP, Garber J,
et al: Germline mutations of the p53 tumor-suppressor gene in
children and young adults with second malignant neoplasms. N Engl J
Med. 326:1309–1315. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sugano K, Taniguchi T, Saeki M, Tsunematsu
Y, Tomaru U and Shimoda T: Germline p53 mutation in a case of
Li-Fraumeni syndrome presenting gastric cancer. Jpn J Clin Oncol.
29:513–516. 1999. View Article : Google Scholar
|
|
19
|
Healey CS, Dunning AM, Teare MD, Chase D,
Parker L, Burn J, Chang-Claude J, Mannermaa A, Kataja V, Huntsman
DG, et al: A common variant in BRCA2 is associated with both breast
cancer risk and prenatal viability. Nat Genet. 26:362–364. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Masciari S, Larsson N, Senz J, Boyd N,
Kaurah P, Kandel MJ, Harris LN, Pinheiro HC, Troussard A, Miron P,
et al: Germline E-cadherin mutations in familial lobular breast
cancer. J Med Genet. 44:726–731. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kaurah P, MacMillan A, Boyd N, Senz J, De
Luca A, Chun N, Suriano G, Zaor S, Van Manen L, Gilpin C, et al:
Founder and recurrent CDH1 mutations in families with hereditary
diffuse gastric cancer. JAMA. 297:2360–2372. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mohaghegh P and Hickson ID: DNA helicase
deficiencies associated with cancer predisposition and premature
ageing disorders. Hum Mol Genet. 10:741–746. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kitao S, Shimamoto A, Goto M, Miller RW,
Smithson WA, Lindor NM and Furuichi Y: Mutations in RECQL4 cause a
subset of cases of Rothmund-Thomson syndrome. Nat Genet. 22:82–84.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ellis NA, Groden J, Ye TZ, Straughen J,
Lennon DJ, Ciocci S, Proytcheva M and German J: The Bloom's
syndrome gene product is homologous to RecQ helicases. Cell.
83:655–666. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Thompson ER, Doyle MA, Ryland GL, Rowley
SM, Choong DY, Tothill RW, Thorne H, Barnes DR, Li J, Ellul J, et
al: kConFab: Exome sequencing identifies rare deleterious mutations
in DNA repair genes FANCC and BLM as potential breast cancer
susceptibility alleles. PLoS Genet. 8:e10028942012. View Article : Google Scholar
|
|
26
|
Kunkel TA and Erie DA: DNA mismatch
repair. Annu Rev Biochem. 74:681–710. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakagawa H, Lockman JC, Frankel WL, Hampel
H, Steenblock K, Burgart LJ, Thibodeau SN and de la Chapelle A:
Mismatch repair gene PMS2: Disease-causing germline mutations are
frequent in patients whose tumors stain negative for PMS2 protein,
but paralogous genes obscure mutation detection and interpretation.
Cancer Res. 64:4721–4727. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hitchins MP, Wong JJL, Suthers G, Suter
CM, Martin DIK, Hawkins NJ and Ward RL: Inheritance of a
cancer-associated MLH1 germ-line epimutation. N Engl J Med.
356:697–705. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Forbes SA, Bindal N, Bamford S, Cole C,
Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al:
COSMIC: Mining complete cancer genomes in the Catalogue of Somatic
Mutations in Cancer. Nucleic Acids Res. 39(Database): D945–D950.
2011. View Article : Google Scholar :
|
|
30
|
Chen C, Liu Y, Rappaport AR, Kitzing T,
Schultz N, Zhao Z, Shroff AS, Dickins RA, Vakoc CR, Bradner JE, et
al: MLL3 is a haploinsufficient 7q tumor suppressor in acute
myeloid leukemia. Cancer Cell. 25:652–665. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li WD, Li QR, Xu SN, Wei FJ, Ye ZJ, Cheng
JK and Chen JP: Exome sequencing identifies an MLL3 gene germ line
mutation in a pedigree of colorectal cancer and acute myeloid
leukemia. Blood. 121:1478–1479. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Villani A, Tabori U, Schiffman J, Shlien
A, Beyene J, Druker H, Novokmet A, Finlay J and Malkin D:
Biochemical and imaging surveillance in germline TP53 mutation
carriers with Li-Fraumeni syndrome: A prospective observational
study. Lancet Oncol. 12:559–567. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Haferlach C, Dicker F, Herholz H,
Schnittger S, Kern W and Haferlach T: Mutations of the TP53 gene in
acute myeloid leukemia are strongly associated with a complex
aberrant karyotype. Leukemia. 22:1539–1541. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Grochola LF, Zeron-Medina J, Mériaux S and
Bond GL: Single-nucleotide polymorphisms in the p53 signaling
pathway. Cold Spring Harb Perspect Biol. 2:a0010322010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fitzgerald RC, Hardwick R, Huntsman D,
Carneiro F, Guilford P, Blair V, Chung DC, Norton J, Ragunath K,
Van Krieken JH, et al International Gastric Cancer Linkage
Consortium: Hereditary diffuse gastric cancer: Updated consensus
guidelines for clinical management and directions for future
research. J Med Genet. 47:436–444. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Evans DGR and Ingham SL: Reduced life
expectancy seen in hereditary diseases which predispose to
early-onset tumors. Appl Clin Genet. 6:53–61. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Limacher JM, Frebourg T, Natarajan-Ame S
and Bergerat JP: Two metachronous tumors in the radiotherapy fields
of a patient with Li-Fraumeni syndrome. Int J Cancer. 96:238–242.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Heymann S, Delaloge S, Rahal A, Caron O,
Frebourg T, Barreau L, Pachet C, Mathieu MC, Marsiglia H and
Bourgier C: Radio-induced malignancies after breast cancer
postoperative radiotherapy in patients with Li-Fraumeni syndrome.
Radiat Oncol. 5:1042010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Godley LA and Larson RA: Therapy-related
myeloid leukemia. Semin Oncol. 35:418–429. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Smith MA, Rubinstein L, Anderson JR,
Arthur D, Catalano PJ, Freidlin B, Heyn R, Khayat A, Krailo M, Land
VJ, et al: Secondary leukemia or myelodysplastic syndrome after
treatment with epipodophyllotoxins. J Clin Oncol. 17:569–577.
1999.PubMed/NCBI
|
|
42
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rausch T, Jones DTW, Zapatka M, Stütz AM,
Zichner T, Weischenfeldt J, Jäger N, Remke M, Shih D, Northcott PA,
et al: Genome sequencing of pediatric medulloblastoma links
catastrophic DNA rearrangements with TP53 mutations. Cell.
148:59–71. 2012. View Article : Google Scholar : PubMed/NCBI
|