Introduction
Selenium compounds have received a great deal of
attention in cancer treatment and chemoprevention. The possible
selective inhibitory effect of selenium on tumor cells makes
selenium a promising candidate for controlling tumorigenesis
(1). Despite this potential, little
scientific evidence exists to describe the exact mechanism
underlying the anticancer effect of selenium (2). A variety of genetic studies have
revealed that cancer cell proliferation, invasion and metastasis
can be suppressed through at least three possible areas of
modulation, including the cell cycle and apoptosis, signaling
pathways and target genes (3–5). Human
clinical trials have revealed dose-limiting toxicity when selenium
compounds are administered at doses of up to 0.8 mg/day, and at the
maximum dose, plasma concentrations reached 601 µg selenium
(6).
One of the promising target molecules of selenium is
β-catenin. β-catenin is a transcription factor that plays a pivotal
role in cells, regulating a large set of genes involved in cell
development, differentiation, growth and metastasis (7). The canonical β-catenin pathway begins
with the stabilization of β-catenin. In the inactivated state of
the Wnt ligand, a low plasma level of β-catenin is maintained and
controlled by the activity of a multiprotein destruction complex
that targets β-catenin for ubiquitination and proteolytic
degradation (8). Upon binding to
Wnt ligands, β-catenin inhibits the formation of the multiprotein
complex, and phosphorylated β-catenin is translocated into the
nucleus to regulate expression of certain genes such as c-myc,
c-Jun and cyclin D1 (9–11). Inappropriate regulation of the
Wnt/β-catenin pathway is associated with hepatocellular carcinoma
tumorigenesis (12). Several
studies have shown that the tumor-suppressing inactivated form of
β-catenin occurs concomitantly with the activation of 5′ adenosine
monophosphate-activated protein kinase (AMPK) (13,14).
In the present study, we explored crosstalk between
β-catenin and AMPK to elucidate the molecular basis of
selenium-induced cancer cell control. AMPK was found to be a
crucial regulator initiating selenium-induced inhibition of
insulin-like growth factor 1 (IGF-1)-stimulated β-catenin
expression. We also discovered that selenium inhibits
phosphorylation of glycogen synthase kinase 3β (GSK3β) at Ser9 and
β-catenin at Ser552, and the selenium-induced activation of AMPK
led to the attenuated nuclear localization of β-catenin.
Materials and methods
Cells and reagents
The Hep3B hepatocellular carcinoma cell line was
purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA) and was cultured in Dulbecco's modified Eagle's
medium (DMEM) with 10% fetal bovine serum (Gibco, Gaithersburg, MD,
USA). EGCG, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) and Hoechst 33342 were obtained from Sigma (St.
Louis, MO, USA). Compound C was purchased from Calbiochem (San
Diego, CA, USA). Monoclonal antibodies specific for p-AMPKα1
(Thr172), AMPKα1, p-GSK3β (Ser9), GSK3β, p-β-catenin (Ser33/37),
p-β-catenin (Ser552) and β-catenin were purchased from Cell
Signaling Technology (Beverly, MA, USA). Antibody against lamin B1
was purchased from Santa Cruz (San Diego, CA, USA) and the β-actin
antibody was obtained from Sigma.
Cell proliferation measurements
Cells seeded into 96-well microplates at
4×103 cells/well, were incubated with test compounds at
the indicated concentrations for the indicated time periods.
Following incubation with the test compound, the medium was
removed, and the cells were then incubated with 10 µl MTT
solution (5 mg/ml MTT in PBS) for 1 h. The samples were solubilized
in DMSO. The purple formazan dye, converted from MTT by viable
cells was quantified by absorbance at 595 nm.
Apoptosis detection
Apoptosis was measured using a FITC-Annexin V
apoptosis detection kit (BD Pharmingen, San Diego, CA, USA) or
Hoechst 33342 chromatin staining dye. For Annexin V/PI staining
after treatment with selenium, cells were harvested by
trypsinization, washed with ice-cold phosphate-buffered saline
(PBS), and suspended in a binding buffer at a density of
1×106 cells/ml. Cells were stained with Annexin V-FITC
and propidium iodide (PI) and analyzed by flow cytometry
(Becton-Dickinson Biosciences, Franklin Lakes, NJ, USA). To examine
chromatin condensation, cells were stained with 10 µM
Hoechst 33342 for 30 min and fixed with 3.7% formaldehyde for 15
min. Changes in chromatin condensation were observed by
fluorescence microscopy (Olympus Optical Co., Tokyo, Japan).
Western blot analysis
Cells were seeded into six-well plates and treated
with test compounds. Total proteins were extracted using a RIPA
lysis buffer [50 mM Tris-HCl (pH 8.0), 1% NP-40, 0.5% sodium
deoxycholate, 150 mM NaCl, 1 mM PMSF] and subjected to western blot
analysis with specific antibodies. The proteins were then
visualized by enhanced chemiluminescence (Intron, Kyunggi, Korea)
and detected using a LAS4000 chemiluminescence detection system
(Fuji, Tokyo, Japan).
Cytoplasmic and nuclear
fractionation
Cells were seeded into six-well plates and treated
with test compounds. Cytoplasmic and nuclear proteins were
extracted using ProteoExtract® Subcellular Proteome
Extraction kit (Calbiochem) and subjected to western blot analysis
with specific antibodies. The proteins were then visualized by
enhanced chemiluminescence (Intron) and detected using a LAS4000
chemiluminescence detection system.
Immunofluorescence staining
The cells were seeded into a 12-well plate with
cover glasses. Following treatment at the indicated time and dose,
the cells were fixed in 3.7% formaldehyde for 20 min at room
temperature (RT) and were permeabilized in 0.2% Triton X-100 for 20
min at RT. Then cells were blocked with 1% bovine serum albumin for
1 h. Next, the cells were incubated overnight with primary antibody
against either AMPKα1 or β-catenin. After washing, the cells were
incubated with Alexa546-conjugated anti-rabbit IgG and Alexa
488-conjugated anti-mouse IgG (both from Molecular Probes, Eugene,
OR, USA) for 1 h at RT. Next, cell nuclei were stained with 10
µM Hoechst 33342 for 10 min and then observed with a
confocal microscope (Carl Zeiss, Thornwood, NY, USA).
Transient transfection with small
interfering RNAs (siRNAs)
Specific siRNAs, targeting AMPKα1 (PRKAA1) and
GSK3β, and non-specific control siRNAs were purchased from
Dharmacon (Chicago, IL, USA). For transient transfection, cells
were seeded at a density of 5×104 cells/ml in
antibiotic-free medium, and siRNAs were transfected using the
DharmaFECT 4 transfection reagent (Dharmacon) according to the
manufacturer's instructions. After incubation for 72 h, the cells
were analyzed via immunofluorescence staining or western
blotting.
Tumor formation
Five-week-old male BALB/c nu/nu mice were obtained
from SLC (Tokyo, Japan) and were housed in sterile filter-topped
cages. Hep3B hepato-carcinoma cells (1×106 cells/150
µl) were subcutaneously injected into the left flank of the
mice. One week after the injection of Hep3B cells, selenium was
dissolved in PBS and administered intraperitoneally (30 mg/kg/day)
for 20 days. The control animals were injected with vehicle (PBS)
alone. Tumor size was measured using a caliper at 2-day intervals,
and the volume was calculated by the modified formula V = 1/2
(length x width2). After the 18 day treatment, tumors
were removed and frozen in liquid nitrogen for western blot
analysis or fixed with formalin for immunohistochemistry. All
animal experiments were approved by the Ethics Committee for Animal
Experimentation, Hannam University.
Immunohistochemistry
Tumor specimens from mice were fixed in 10%
formaldehyde, embedded in paraffin and sectioned into 5-µm
thick slices. Sections were deparaffinized with xylene and
dehydrated with 98% ethanol. Serial sections were stained using
standard immunoperoxidase techniques with primary antibodies
against β-catenin (1:50) and p-AMPKα1 (1:50). For epitope
retrieval, specimens were microwave-treated for 25 min before
incubation with primary antibodies. Pre-immune serum was used as a
negative control for immunostaining, and positive-staining was
visualized with diaminobenzidine, followed by a light
counter-staining with hematoxylin. All findings were evaluated by a
pathologist blinded to the treatment conditions, and samples were
evaluated on the basis of stain intensity and percentage of
reactive cells. Images of representative results were recorded.
Statistical analysis
Cell viability and tumor volume data were
statistically analyzed using an unpaired t-test (SPSS, Inc.,
Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant result.
Results
Selenium-induced inhibition of
IGF-1-stimulated β-catenin is associated with activation of
AMPK
A large body of evidence suggests that β-catenin is
often aberrantly overexpressed in hepatocellular carcinoma. IGF-1
has recently been identified as being capable of increasing
β-catenin expression and its transcriptional activity via
phosphorylation of GSK3β (10). We
examined this effect of IGF-1 on hepatocellular carcinoma growth
through regulation of the GSK3β/β-catenin survival pathway. Our
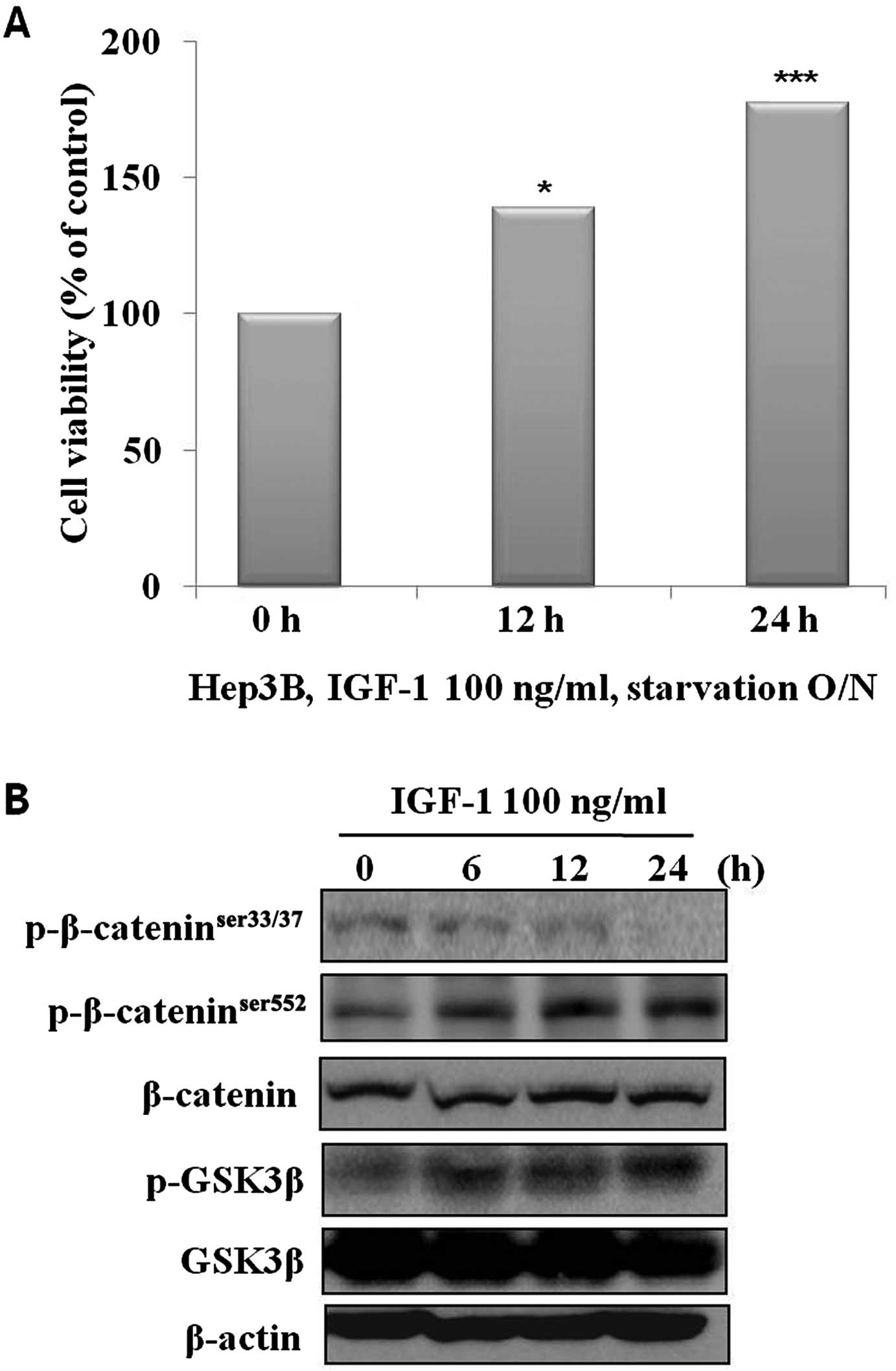
results showed that IGF-1 effectively increased cell growth in a
time-dependent manner (Fig. 1A). We
analyzed the molecular changes in the control and selenium-treated
cells. The phosphorylation of β-catenin on the Ser552 residue and
GSK3β were significantly increased, and phosphorylation of
β-catenin on the Ser33/37 residue was significantly decreased in a
time-dependent manner (Fig. 1B). To
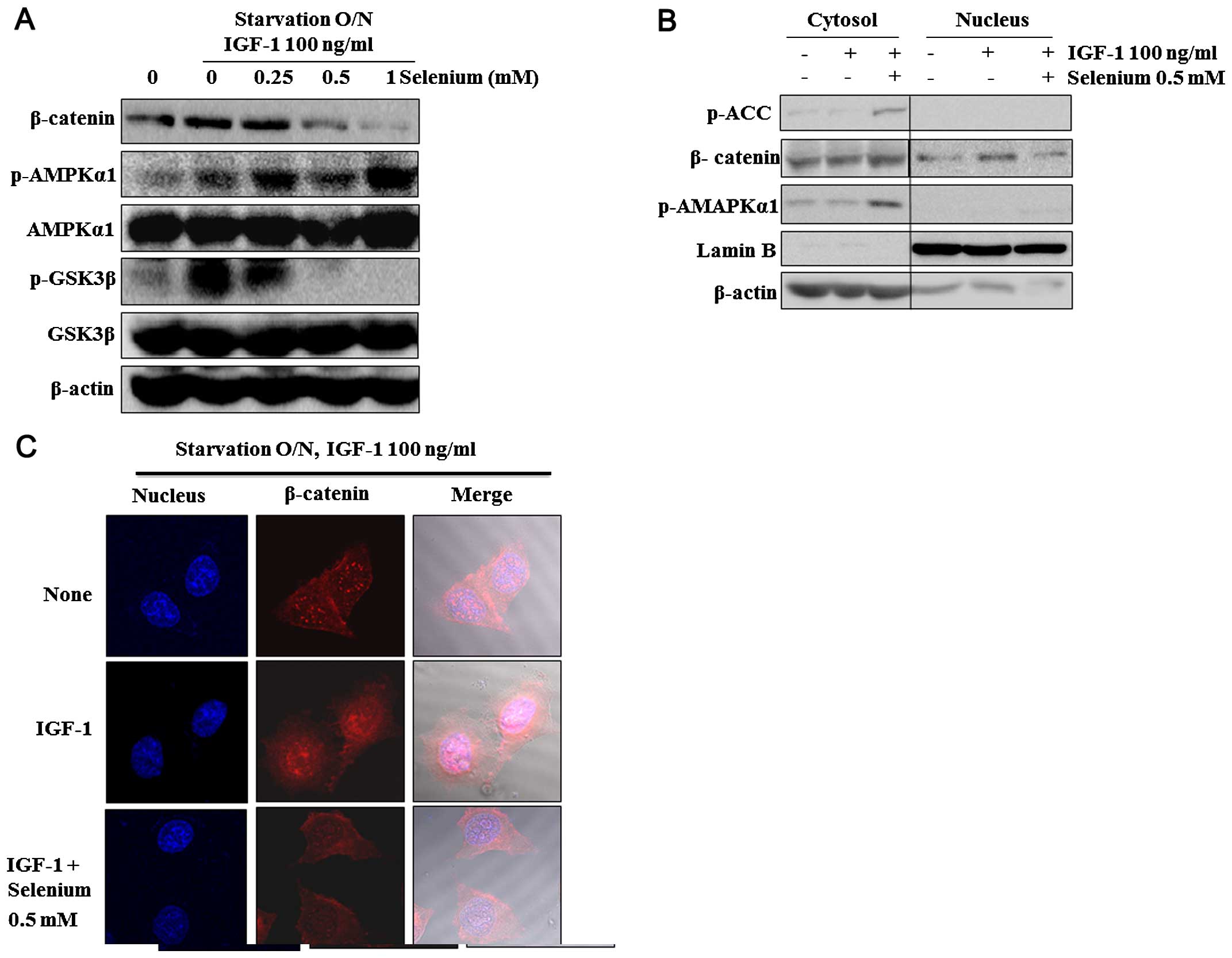
examine whether selenium exerts inhibitory effects on β-catenin and
GSK3β, we analyzed changes in their phosphorylation and expression.
Selenium at concentrations of >500 µM effectively
inhibited IGF-1-stimulated β-catenin and phosphorylation of GSK3β
and enhanced AMPK phosphorylation (Fig.
2A). We investigated the effects of selenium on the β-catenin
trans-location from the cytosol to the nucleus. We discovered that
selenium decreased IGF-1-increased β-catenin translocation in the
nucleus at 6 h, whereas no marked difference occurred in the
expression of β-catenin in the cytosol (Fig. 2C). To analyze the localization
pattern of β-catenin in this system further, we immunostained
β-catenin and detected it using fluorescence microscopy. Consistent
with western blot results, β-catenin decreased in the nucleus after
selenium treatment for 6 h (Fig.
2B). Taken together, we inferred that selenium may redistribute
IGF-1-increased β-catenin protein in the cytoplasm and nuclei.
Selenium-activated AMPK directly
suppresses β-catenin
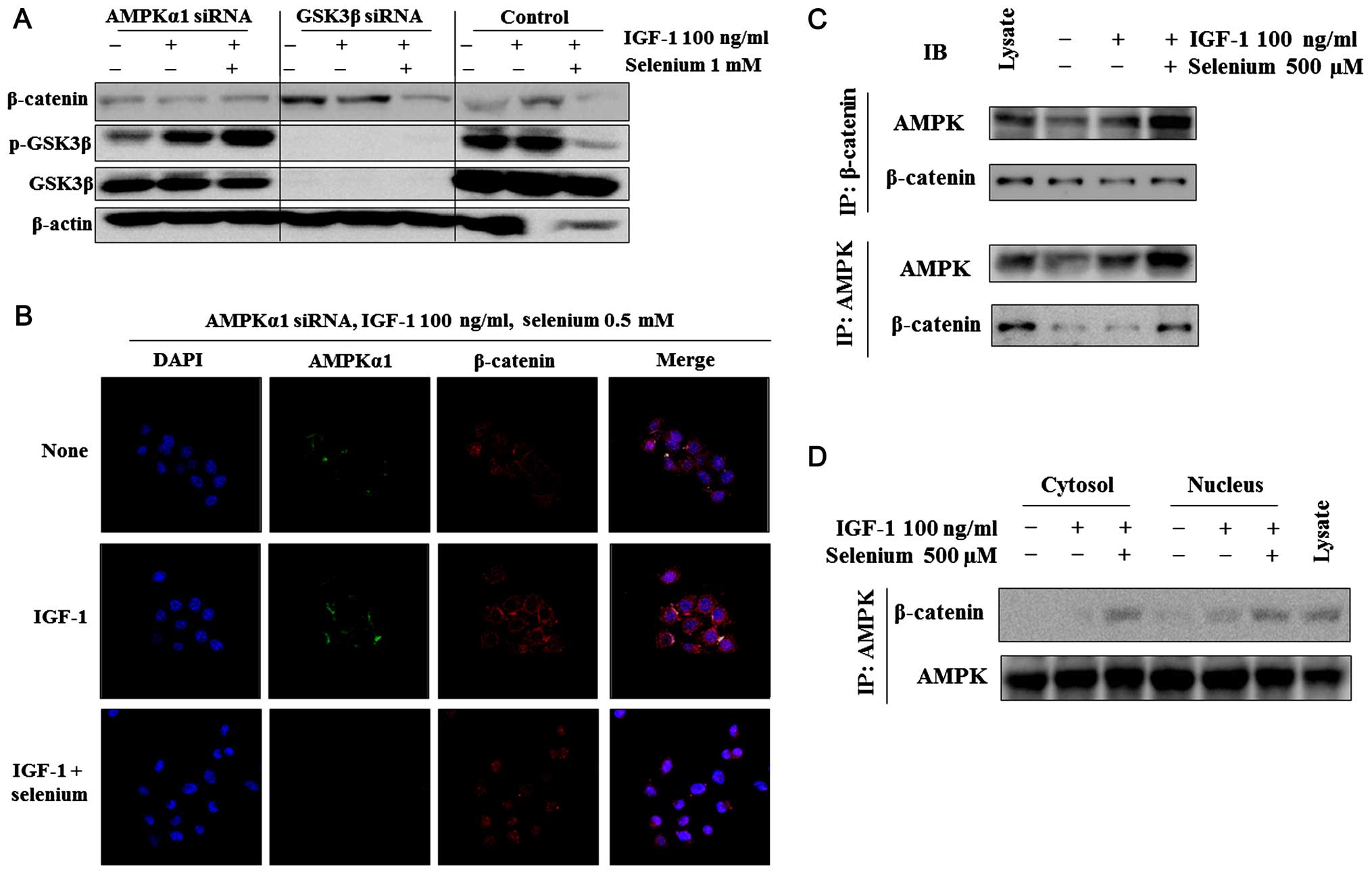
We investigated whether selenium-reduced β-catenin
expression in Hep3B cells is associated with the activation of
AMPK. We examined the effects of selenium on the activity of
β-catenin in AMPK or GSK3β siRNA-transfected Hep3B cells. To
examine whether selenium-reduced β-catenin levels are AMPK
dependent, we determined the effects of selenium on β-catenin after
knockdown of AMPK using siRNA in Hep3B cells. Selenium did not
regulate β-catenin and GSK3β in the absence of AMPK. However,
selenium regulated β-catenin in the absence of GSK3β (Fig. 3A). β-catenin was regulated not only
by the GSK3β signaling pathway but also by the PI3K/Akt pathway.
Thus, selenium regulates β-catenin via a GSK3β-independent pathway.
Furthermore, the immunostaining results showed that selenium had no
effect on β-catenin localization in AMPK-transfected Hep3B cells
(Fig. 3B).
Next, we examined the direct relationship of
AMPK/β-catenin using immunoprecipitates.
Co-immunoprecipitation/western blot experiments performed with
Hep3B cells showed identical results (Fig. 3C). To further characterize the
specificity of the β-catenin-AMPK interaction, we performed
additional experiments with antibodies specific for β-catenin and
AMPK isoforms in the lysate of untreated or selenium-treated cells.
Treatment with selenium led to the appearance of β-catenin or AMPK
in the Hep3B cell lysate (Fig. 3C).
These results suggest that selenium directly regulates the
interaction between β-catenin and AMPK.
Selenium suppresses cell proliferation
and induces apoptosis through AMPK activation in vitro
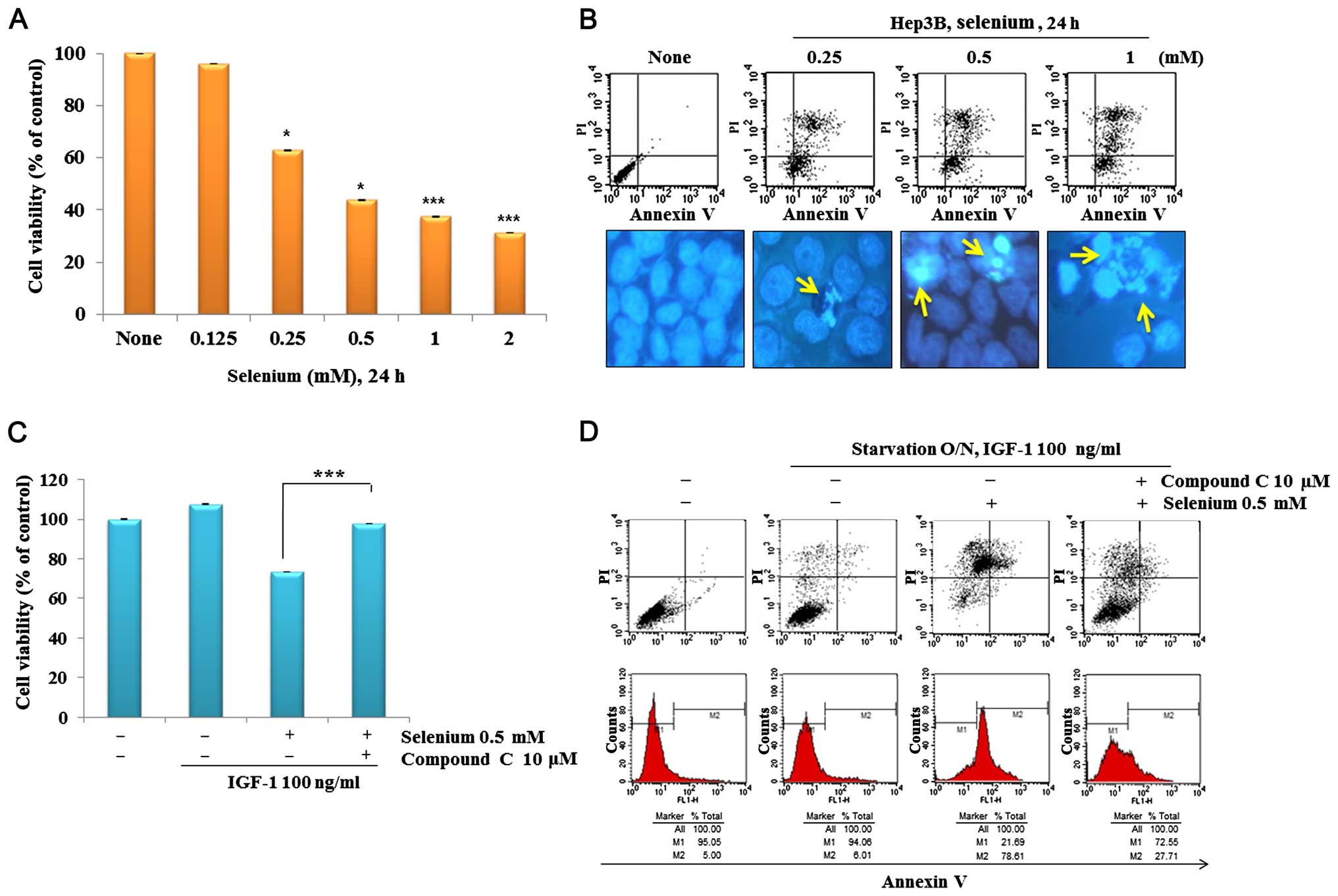
To examine whether selenium exerts anticancer
activity in Hep3B cells, we examined the effect of selenium on cell
proliferation and apoptosis. Selenium effectively inhibited cell
growth in a dose-dependent manner (Fig.
4A) and induced apoptosis, as measured by Annexin V/PI and
Hoechst 33342 staining (Fig.
4B).
Numerous studies have identified AMPK as a central
factor inducing apoptosis in various cancer cells. Our prior study
together with that of others has also implicated AMPK as a key
regulator of selenium-induced apoptosis in cancer cells (15). To further validate whether AMPK
inhibition by compound C is associated with cell proliferation and
apoptosis of hepatocel-lular carcinoma cells. As shown in Fig. 4C, inhibition of AMPK by compound C
treatment abolished the growth-stimulatory effects of IGF-1.
Furthermore, fluorescence-activated cell sorter results revealed
that the apoptotic rate in compound C-treated cells declined from
73.61 to 22.71% after treatment compared with that in
selenium-treated cells in the control group (Fig. 4D). All of these data argue for the
critical involvement of AMPK in selenium-induced apoptosis in Hep3B
cells.
Selenium inhibits the GSK3β/β-catenin
survival pathway in hepatocellular carcinoma xenograft tumors
We built an in vivo hepato-carcinoma
xenograft tumor model by inoculating Hep3B cells into male nude
mice subcutaneously. We primarily discovered that treatment with
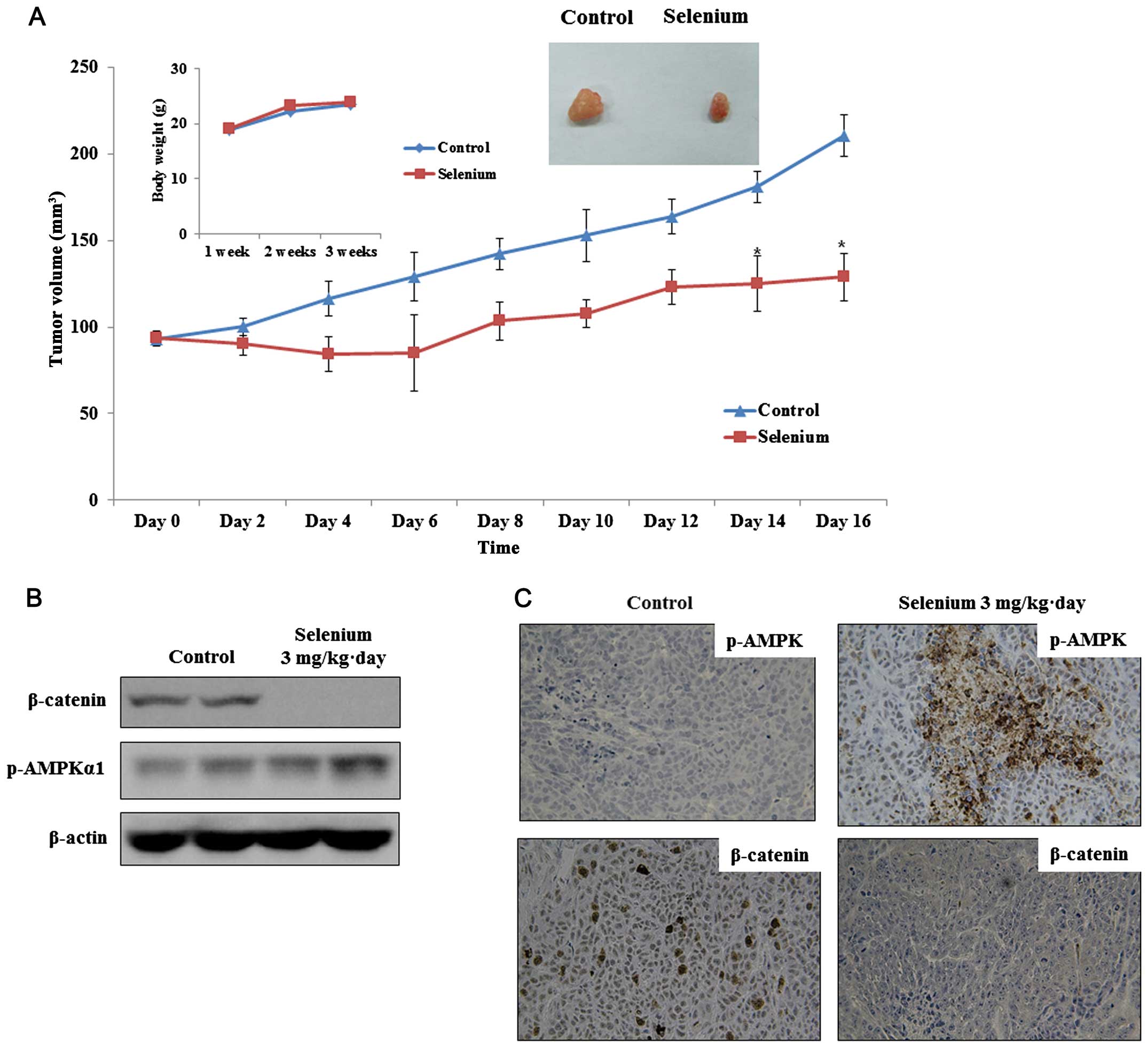
selenium (3 mg/kg·day) markedly attenuated tumor growth without
adverse effects on body weight and activity compared with that in
the control group (Fig. 5A). We
analyzed the molecular changes in the control and selenium-treated
cells. The phosphorylation of GSK3β and expression of β-catenin
were significantly suppressed, but the phosphorylation of AMPK was
significantly increased in the selenium treatment group relative to
that in the control group (Fig.
5B). Immunohistochemical analysis confirmed that the control
cells had high levels of β-catenin, and the selenium-treated cells
had reduced β-catenin levels. Furthermore, tumors in the selenium
treatment group showed strongly increased p-AMPK levels (Fig. 5C).
Discussion
Selenium affects numerous intracellular targets,
making selenium compounds desirable chemotherapeutic and
chemopreventive agents. Studies in both in vitro and in
vivo models have suggested that selenium suppresses components
of IGF-1-induced β-catenin (15).
The objective of the present study was to investigate the
inhibitory effects of selenium on β-catenin or GSK3β through the
activation of AMPK in hepato-carcinoma cells and xenograft tumors.
Prior in vitro and in vivo models have suggested that
selenium suppresses IGF-induced β-catenin through activation of
AMPK, which in turn may suppress cell proliferation and induce
apoptosis. Selenium is a nonmetallic trace element that is
essential for human health; selenium supplementation also appears
to work as an anti-carcinogenic agent (16).
The anticancer activity of selenium has been
attributed to various mechanisms, such as mitogen-activated protein
kinase suppression or modulation of Akt, mammalian target of
rapamycin or β-catenin (17–19).
Further, selenium has been shown to inhibit β-catenin accumulation
in the nucleus. In colon cancer models, selenium treatment resulted
in JNK suppression and subsequent inhibition of β-catenin (20). Our previous studies have revealed
that the apoptotic effect of selenium is dependent on the
AMPK-regulated extracellular signal-regulated
kinase/cyclooxygenase-2 pathway as well as the AMPK/Akt mammalian
target of rapamycin pathway (15,21).
Since abnormal β-catenin activation in many human
malignancies is well documented and overexpressed β-catenin may
have oncogenic effects in hepatocellular carcinoma (22), the interaction between β-catenin and
AMPK may represent an important mechanism for the regulation of
β-catenin signaling pathways with selenium in cancer. Knockdown of
AMPK using AMPK siRNA increased β-catenin in contrast with results
after selenium treatment. Importantly, the inhibition of AMPK
allows the increases in β-catenin as well as in GSK3β and Akt that
can be encountered in advanced hepatocellular carcinoma cells. The
present study did not reveal the exact mode of the regulation of
β-catenin activity by AMPK, although immunoprecipitation studies
have shown that the binding between AMPK and β-catenin occurs in
cytoplasm, and fluorescent microscopic examinations have pointed
out that AMPK is responsible for the inhibition of β-catenin into
the nucleus. Our data show that selenium increases AMPK via
phosphorylation of AMPK Thr172, which in turn leads to the
inhibition of β-catenin, indicating that selenium is capable of
suppressing β-catenin function via an AMPK-dependent pathway. In
addition to β-catenin suppression by selenium-induced AMPK
activation, we observed that activated AMPK regulates GSK3β. We
used inhibitors of AMPK and GSK3β to determine whether the
capability of AMPK to regulate β-catenin was required for selenium
to exert its antiproliferative functions. When AMPK was inhibited
by AMPK siRNA, selenium treatment failed to decrease β-catenin,
indicating that AMPK is required for selenium regulation of
β-catenin. By contrast, GSK3β siRNA-mediated inhibition of GSK3β
did not affect the capability of selenium to inhibit β-catenin,
indicating that direct inhibition of β-catenin by selenium may
occur without the involvement of GSK3β.
GSK3β/β-catenin is known to promote tumor growth
through its function in the tumor microenvironment, but its exact
method for converting normal cells into cancerous cells remains
undefined. β-catenin is a transcription factor that plays a pivotal
role in cells, regulating a large set of genes involved in cell
development, differentiation, growth and metastasis. The β-catenin
pathway seems to play a critical role against hepatocellular
caricinoma, possibly through alteration at multiple levels,
including mutation of β-catenin or its upstream or downstream
regulators/effectors such as GSK3b, Akt and T-cell factor/lymphoid
enhancer binding factor (23,24).
In the present study, we elucidated that selenium
down-regulates the β-catenin survival pathway through activation of
AMPK in hepatocellular carcinoma cells and xenograft tumor models.
We primarily discovered that selenium could inhibit β-catenin at
Ser552 and GSK3β at Ser9 in Hep3B cells; however, the attenuation
of nuclear localization of β-catenin occurred only under the
activation of AMPK. Taken together, these findings help illuminate
the molecular mechanisms underlying the anticancer effects of
selenium and highlight the regulation of β-catenin through an
AMPK-dependent pathway.
References
|
1
|
Wu M, Kang MM, Schoene NW and Cheng WH:
Selenium compounds activate early barriers of tumorigenesis. J Biol
Chem. 285:12055–12062. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Combs GF Jr: Current evidence and research
needs to support a health claim for selenium and cancer prevention.
J Nutr. 135:343–347. 2005.PubMed/NCBI
|
|
3
|
Song H, Hur I, Park HJ, Nam J, Park GB,
Kong KH, Hwang YM, Kim YS, Cho DH, Lee WJ, et al: Selenium inhibits
metastasis of murine melanoma cells through the induction of cell
cycle arrest and cell death. Immune Netw. 9:236–242. 2009.
View Article : Google Scholar
|
|
4
|
Venkateswaran V, Klotz LH and Fleshner NE:
Selenium modulation of cell proliferation and cell cycle biomarkers
in human prostate carcinoma cell lines. Cancer Res. 62:2540–2545.
2002.PubMed/NCBI
|
|
5
|
Zeng H and Combs GF Jr: Selenium as an
anticancer nutrient: Roles in cell proliferation and tumor cell
invasion. J Nutr Biochem. 19:1–7. 2008. View Article : Google Scholar
|
|
6
|
Burk RF, Norsworthy BK, Hill KE, Motley AK
and Byrne DW: Effects of chemical form of selenium on plasma
biomarkers in a high-dose human supplementation trial. Cancer
Epidemiol Biomarkers Prev. 15:804–810. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stein U, Arlt F, Walther W, Smith J,
Waldman T, Harris ED, Mertins SD, Heizmann CW, Allard D, Birchmeier
W, et al: The metastasis-associated gene S100A4 is a novel target
of beta-catenin/T-cell factor signaling in colon cancer.
Gastroenterology. 131:1486–1500. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li VS, Ng SS, Boersema PJ, Low TY,
Karthaus WR, Gerlach JP, Mohammed S, Heck AJ, Maurice MM, Mahmoudi
T, et al: Wnt signaling through inhibition of β-catenin degradation
in an intact Axin1 complex. Cell. 149:1245–1256. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cadigan KM and Nusse R: Wnt signaling: A
common theme in animal development. Genes Dev. 11:3286–3305. 1997.
View Article : Google Scholar
|
|
10
|
MacDonald BT, Tamai K and He X:
Wnt/beta-catenin signaling: Components, mechanisms, and diseases.
Dev Cell. 17:9–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
van de Wetering M, Sancho E, Verweij C, de
Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D,
Haramis AP, et al: The beta-catenin/TCF-4 complex imposes a crypt
progenitor phenotype on colorectal cancer cells. Cell. 111:241–250.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ihara A, Koizumi H, Hashizume R and
Uchikoshi T: Expression of epithelial cadherin and alpha- and
beta-catenins in nontumoral livers and hepatocellular carcinomas.
Hepatology. 23:1441–1447. 1996.PubMed/NCBI
|
|
13
|
Takatani T, Minagawa M, Takatani R,
Kinoshita K and Kohno Y: AMP-activated protein kinase attenuates
Wnt/β-catenin signaling in human osteoblastic Saos-2 cells. Mol
Cell Endocrinol. 339:114–119. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Subramaniam N, Sherman MH, Rao R, Wilson
C, Coulter S, Atkins AR, Evans RM, Liddle C and Downes M:
Metformin-mediated Bambi expression in hepatic stellate cells
induces prosurvival Wnt/β-catenin signaling. Cancer Prev Res.
5:553–561. 2012. View Article : Google Scholar
|
|
15
|
Lee YK, Park SY, Kim YM, Kim DC, Lee WS,
Surh YJ and Park OJ: Suppression of mTOR via Akt-dependent and
-independent mechanisms in selenium-treated colon cancer cells:
Involvement of AMPKalpha1. Carcinogenesis. 31:1092–1099. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ip C, Dong Y and Ganther HE: New concepts
in selenium chemoprevention. Cancer Metastasis Rev. 21:281–289.
2002. View Article : Google Scholar
|
|
17
|
Saifo MS, Rempinski DR Jr, Rustum YM and
Azrak RG: Targeting the oncogenic protein beta-catenin to enhance
chemotherapy outcome against solid human cancers. Mol Cancer.
9:3102010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee YC, Tang YC, Chen YH, Wong CM and Tsou
AP: Selenite-induced survival of HuH7 hepatoma cells involves
activation of focal adhesion kinase-phosphatidylinositol
3-kinase-Akt pathway and Rac1. J Biol Chem. 278:39615–39624. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jiang C, Kim KH, Wang Z and Lü J: Methyl
selenium-induced vascular endothelial apoptosis is executed by
caspases and principally mediated by p38 MAPK pathway. Nutr Cancer.
49:174–183. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fang W, Han A, Bi X, Xiong B and Yang W:
Tumor inhibition by sodium selenite is associated with activation
of c-Jun NH2-terminal kinase 1 and suppression of beta-catenin
signaling. Int J Cancer. 127:32–42. 2010. View Article : Google Scholar :
|
|
21
|
Hwang JT, Kim YM, Surh YJ, Baik HW, Lee
SK, Ha J and Park OJ: Selenium regulates cyclooxygenase-2 and
extracellular signal-regulated kinase signaling pathways by
activating AMP-activated protein kinase in colon cancer cells.
Cancer Res. 66:10057–10063. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen Ban K, Singh H, Krishnan R and Fong
Seow H: Comparison of the expression of β-catenin in hepatocellular
carcinoma in areas with high and low levels of exposure to
aflatoxin B1. J Surg Oncol. 86:157–163. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stauffer JK, Scarzello AJ, Andersen JB, De
Kluyver RL, Back TC, Weiss JM, Thorgeirsson SS and Wiltrout RH:
Coactivation of AKT and β-catenin in mice rapidly induces formation
of lipogenic liver tumors. Cancer Res. 71:2718–2727. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ban KC, Singh H, Krishnan R and Seow HF:
GSK-3beta phosphorylation and alteration of beta-catenin in
hepatocellular carcinoma. Cancer Lett. 199:201–208. 2003.
View Article : Google Scholar : PubMed/NCBI
|