Introduction
Osteosarcoma is a malignant tumor which seriously
affects the physical and mental health of young adults. Current
major treatment methods for osteosarcoma include surgical resection
and neoadjuvant chemotherapy. However, the side effects and drug
resistance to chemotherapy have not been eradicated yet, and this
has affected the development of chemotherapy to a certain
extent.
Drug resistance is a complicated process consisting
of multiple factors. One of the key factors is aberrant apoptosis.
But in recent years, autophagy alone or accompanied by apoptosis
affects cancer chemotherapy sensitivity. Autophagy is a highly
conserved (1,2) process in which the cytoplasm,
including excess or aberrant organelles, is sequestered into
double-membrane vesicles and delivered to degradative organelles,
lysosomes/vacuoles, for breakdown and eventual recycling of the
resulting macromolecules. Therefore, autophagy is essential for
maintaining cell homeostasis and seems to play a pro-survival role
as well. Excessive autophagy could lead to autophagic cell death,
also known as type II programmed cell death (3,4).
Under drug stimuli or stress conditions, generation
of reactive oxygen species (ROS) (5) and/or phosphorylation of the MAPK
signaling pathway (6) are vital for
the induction of autophagy. Conventional MAPKs are constituted by
ERK, JNK, and p38. Wang et al (7) reported that N-acetyl cysteine (NAC),
an ROS inhibitor, blocked rhArg-induced autophagy which plays a
cytoprotective role in triple-negative breast cancer cells in
vitro. Furthermore, Li et al (8) found that palmitate-induced autophagy
protected H9c2 cells against apoptosis through ROS-dependent JNK
and p38 MAPK. Consistent with these findings, Zhou et al
(9) also demonstrated that the
ROS-mediated JNK signaling pathway modulated cytoprotective
autophagy in Ciclopirox-treated rhabdomyosarcoma.
Cinobufagin is one of the active ingredients in the
anticancer Chinese medicine called 'Chan Su', which contains
bufalin, resibufogenin and cinobufagin. Chan Su and its derivatives
(10) have been reported to have
anticancer activities in various types of cancer such as leukemia
(11), hepatocellular cancer
(12), lung cancer (13), prostate cancer (14), colon cancer (15), and cervical cancer (16). Data showed that cinobufagin was able
to induce mitochondrial-dependent apoptosis (17) in hepatocellular cancer cells, exert
pro-apoptotic effects in multiple myeloma (MM) U266 cells (18) through modulation of the ROS-mediated
MAPK signaling pathway and was also able to induce osteosarcoma
cell apoptosis (19) via the
GSK-3β/NF-κB pathway.
Recently, it has been reported that bufalin induced
cytoprotective autophagy in glioma cells through endoplasmic
reticulum stress (20). Xie et
al (21) demonstrated that
ROS/JNK activation contributes to bufalin-induced autophagic cell
death in colon cells. However, the role played by autophagy depends
on the concentration and duration of stimuli as well as the type of
cancer cells. To date, there is no literature reporting that
cinobufagin induces autophagy in osteosarcoma cells. This study
aimed to clarify several points: i) whether cinobufagin could
induce autophagy in osteosarcoma cells; and ii) the interplay
between autophagy and apoptosis and its potential mechanism.
Materials and methods
Cinobufagin was obtained from the National Institute
for the Control of Pharmaceutical and Biological Products (Beijing,
China) and dissolved in Dulbecco's modified Eagle's medium (DMEM)
(Gibco, Grand Island, NY, USA) to each working dose. Fetal bovine
serum (FBS) was purchased from Hangzhou Sijiqing Biological
Engineering Material Co., Ltd. (Hangzhou, Zhejiang, China).
Chloroquine (CQ),
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
N-acetyl cysteine (NAC) and 3-methyladenine (3-MA) were purchased
from Sigma-Aldrich (St. Louis, MO, USA). The Annexin V-FITC
Apoptosis Detection Kit I was obtained from BD Biosciences (San
Diego, CA, USA). Hoechst 33258 was purchased from Promega Corp.
(Madison, Wi, USA). Lipofectamine 2000 transfection reagent was
purchased from Invitrogen Life Technologies (Carlsbad, CA, USA).
Antibodies against caspase-3, caspase-8, caspase-9, cleaved PARP,
phospho-JNK, total JNK, phospho-p38, total p38, LC3B, p62, GAPDH,
enhanced chemiluminescence (ECL) and western blotting kits were
purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA).
Cell culture and viability assay
The U2OS cells were purchased from Shanghai
Institute of Biochemistry and Cell Biology (Chinese Academy of
Sciences, Shanghai, China) and cultured in DMEM containing 10% FBS,
100 µg/ml penicillin and 100 µg/ml streptomycin at
37°C in a 5% CO2 incubator.
MTT assay
The cells were maintained in 96-well flat bottom
microtiter plates at 1×104 cells/well overnight, and
then treatment with cinobufagin at concentrations of 0, 15, 30 60
and 120 mg/l, respectively. MTT (20 µl) solution (5 g/l) was
added to the cells and incubation was carried out for 4 h at 37°C.
Automatic multiwell spectrophotometer was used to calculate the
absorbance value/well at 570 nm. All MTT assays were performed
three times. The inhibitory rate of U2OS cell proliferation was
calculated according to the formula: (1 − experimental absorbance
value/control absorbance value) × 100%. IC50 values (50%
inhibition concentration) were then calculated using the
Statistical Package for the Social Sciences (SPSS, Inc., Chicago,
IL, USA).
Detection of apoptosis
U2OS cells were washed with PBS, and then stained
following the manufacturer's instructions. The number of apoptotic
cells was measured by flow cytometry and analyzed using Cell Quest
™ software (Becton-Dickinson, San Jose, CA, USA). Each group was
calculated in triplicate.
Hoechst 33258 staining
After being fixed with 3.7% paraformaldelyde for 30
min, the cells were stained with 10 mg/l Hoechst 33258 at 37°C for
15 min. Apoptotic cells were detected using a fluorescence
microscope equipped with a UV filter. The presence of condensed or
fragmented nuclei stained bright blue were observed in the
cells.
Green fluorescent protein (GFP)-LC3 dot
assay
Cells were transfected with GFP-LC3 plasmids
following the manufacturer's manual. After transfection for 24 h,
the cells were treated with cinobufagin at various times. The
induction of autophagy was identified by counting the percentage of
cells emitting green fluorescence.
Western blot analysis
Cells were washed in phosphate buffered saline, and
resuspended at room temperature. After treatment on ice for 30 min,
the lysate was centrifuged at 14,0009 × g at 4°C. Protein
concentration was measured with the Bradford protein assay reagent
and bovine serum albumin as a standard. The membranes were
incubated overnight at 4°C with the designated primary antibodies
and secondary antibodies at room temperature for 2 h. GAPDH was
used as the control.
Transmission electron microscopy
Cells were fixed in 2% paraformaldehyde, and then
with 1% osmium tetroxide overnight at 4°C. The cells were then
dehydrated in an increased dose of alcohol and embedded in Agar 100
epoxy resin. Ultrathin sections were mounted on Cu grids and
stained first with uranyl acetate and post-stained with lead
citrate.
ROS detection
After cinobufagin treatment at various times, U2OS
cells were incubated with 5 mM DCFH-DA for 30 min in the dark, and
washed three times. The fluorescence was excited at the wavelength
of 485 nm and the corresponding emission wavelength was 520 nm. ROS
fluorescence intensity was examined under a Leica DM2500
fluorescence microscope using a suitable analysis software
system.
RNA interference
The cells were transfected with 60 nM of specific or
nontargeting siRNA using Lipofectamine 2000 (Invitrogen Life
Technologies) according to the manufacturer's instructions. Cells
were treated with 120 mg/l cinobufagin at various times and used in
other experiments. The siRNAs were obtained from GenePharma, Ltd.
(Shanghai, China). The si-beclin-1 targeting sequence was
5′-UUCAACACUCUUCAGCUCAUCAUCC-3′ and the scrambled siRNA sequence
was 5′-UUCUCCGAACGUGUCACGUTT-3′.
Statistical analysis
All data are presented as the mean ± SD. The
differences between groups were analyzed using the Student's t-test
and statistical analyses were performed using SPSS software version
16.0 (SPSS, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
Cinobufagin inhibits U2OS cell
proliferation and induces caspase-dependent apoptosis
As shown in Fig. 1,
the MTT assay results proved that cinobufagin inhibited the
proliferation of U2OS cells in a dose- and time-dependent manner
after treatment with 0, 15, 30, 60 and 120 mg/l for 12, 24 and 48
h. The IC50 value of cinobufagin treatment at 24 h was
120 mg/l. To further confirm cinobufagin-triggered apoptosis in
U2OS cells, flow cytometry, Hoechst 33258 and western blotting were
performed in turn.
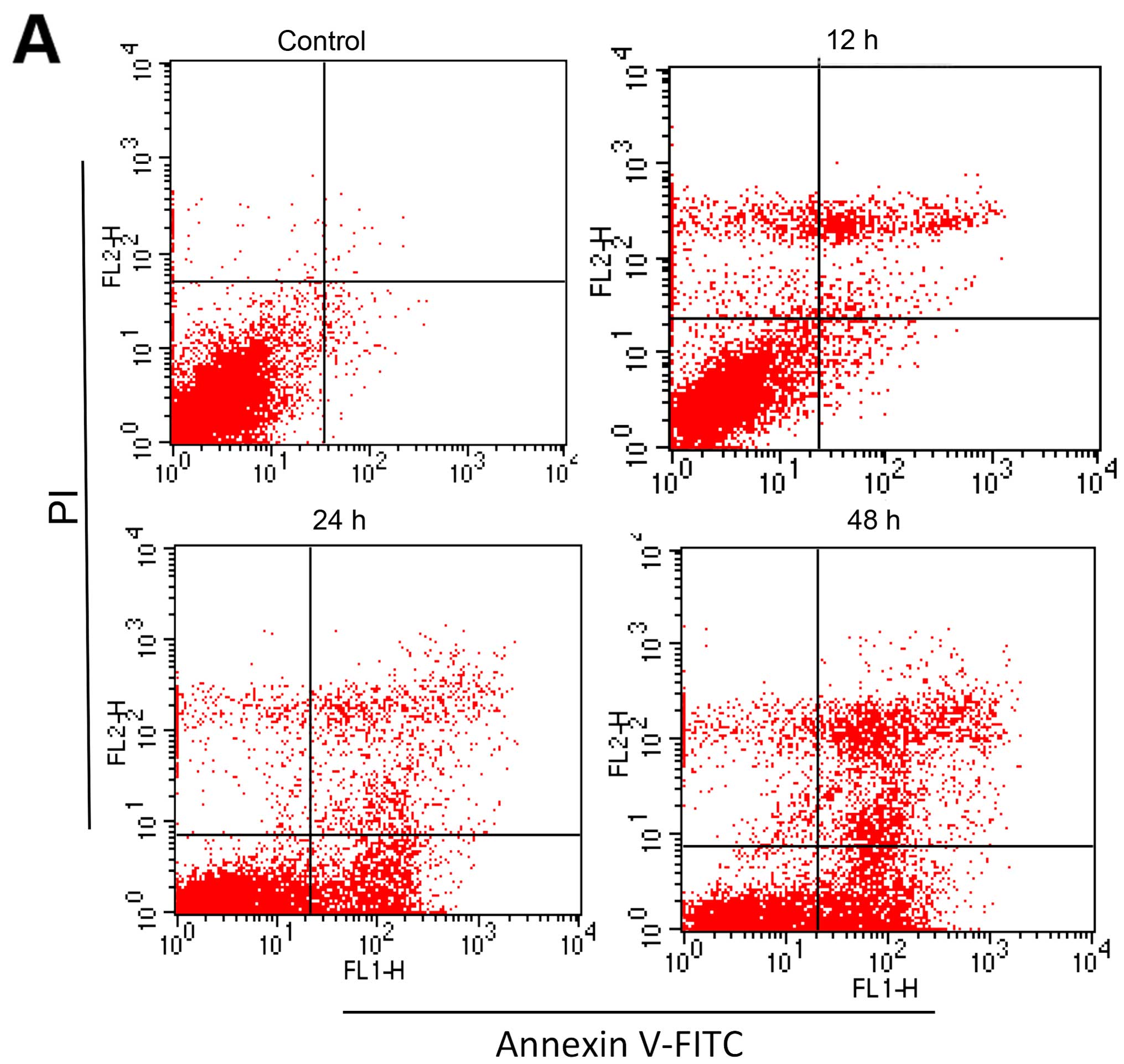
As shown in Fig. 2A and
B, the percentage of apoptotic cells in the control group was
2.71±0.24%, and the apoptotic ratio gradually increased with
prolonged treatment time. Then morphological changes in the cell
nuclei were observed under a fluorescence microscope by Hoechst
33258 staining. Shrunken, condensed nuclei with bright blue
fluorescence were tagged as apoptotic cells. As shown in Fig. 2C and D, more apoptotic cells with
brighter fluorescence were observed as the treatment of cinobufagin
progressed. To further characterize apoptosis, western blotting was
performed. As shown in Fig. 2E and
F, caspase-3, -8, -9 and cleaved PARP were upregulated
following treatment. In the presence of the specific caspase
inhibitors Z-IETD-fmk, Z-LEHD-fmk and Z-VAD-fmk, respectively, cell
viability was upregulated (Fig.
2G). The aforementioned results indicated that cinobufagin is
cytotoxic against U2OS cells and induces caspase-dependent
apoptosis.
Cinobufagin triggers U2OS cell autophagy
flux
We clearly observed a multitude of autophagic
vacuoles engulfing damaged organelles and macromolecular substances
triggered by 120 mg/l cinobufagin under TEM (Fig. 3A; black arrows). In order to further
elucidate cinobufagin-induced autophagy, GFP-LC3 transient
transfection and western blot analysis were employed.
At a low level of autophagy, autophagy marker
protein GFP-LC3 plasmids are dispersed. GFP-LC3 fluorescent
particles during activation of autophagy are increased in both
brightness and density with increased treatment time (Fig. 3B and C). During autophagy,
microtubule-associated protein 1 light chain 3 (LC3)/Atg8 converts
18-kDa LC3-I to 16-kDa LC3-II. The ratio of LC3-II/LC3-I correlates
with activation of autophagic activity. p62 (22) accumulates the ubiquitin protein
selectively to increase autophagy, and eventually to complete the
protein degradation by fusion with the lysosomes. Consistent with
the GFP-LC3 plasmid results, western blot analysis indicated an
increase in the levels of LC3-II/LC3-I, and a reduction in p62
expression in a time-dependent manner (Fig. 3D and E).
Furthermore autophagy is a dynamic, multi-step
process. And when blocked at the final stage, LC3-II/LC3-I is also
accumulated. To further confirm the induction of autophagy flux by
cinobufagin, we verified the accumulation of LC3-II/LC3-I with or
without autophagic inhibitor CQ. As shown in Fig. 3F and G, when compared to the control
group, the CQ group alone exhibited no statistical significant
difference. The levels of LC3-II/LC3-I were increased in the
cinobufagin+CQ group following pretreatment with CQ for 1 h
compared to the cinobufagin group. All the above results indicated
that cinobufagin induced autophagy flux in the U2OS cells.
The influence of autophagy inhibition on
cinobufagin-induced apoptosis and cell viability in U2OS cells
Autophagy is a multi-step process and can be
inhibited at different stages. To determine the role of autophagy
induced by cinobufagin, autophagy inhibitors (CQ and 3-MA) were
used in the following experiments. 3-MA blocks class III
phosphatidylinositol 3-kinases (PI-3Ks) leading to inhibition of
autophagosome (23) formation at an
early stage. CQ is generally used to block autophagosome
combination (24) with lysosomes
and finally degradation obstruction in the late stage. As shown in
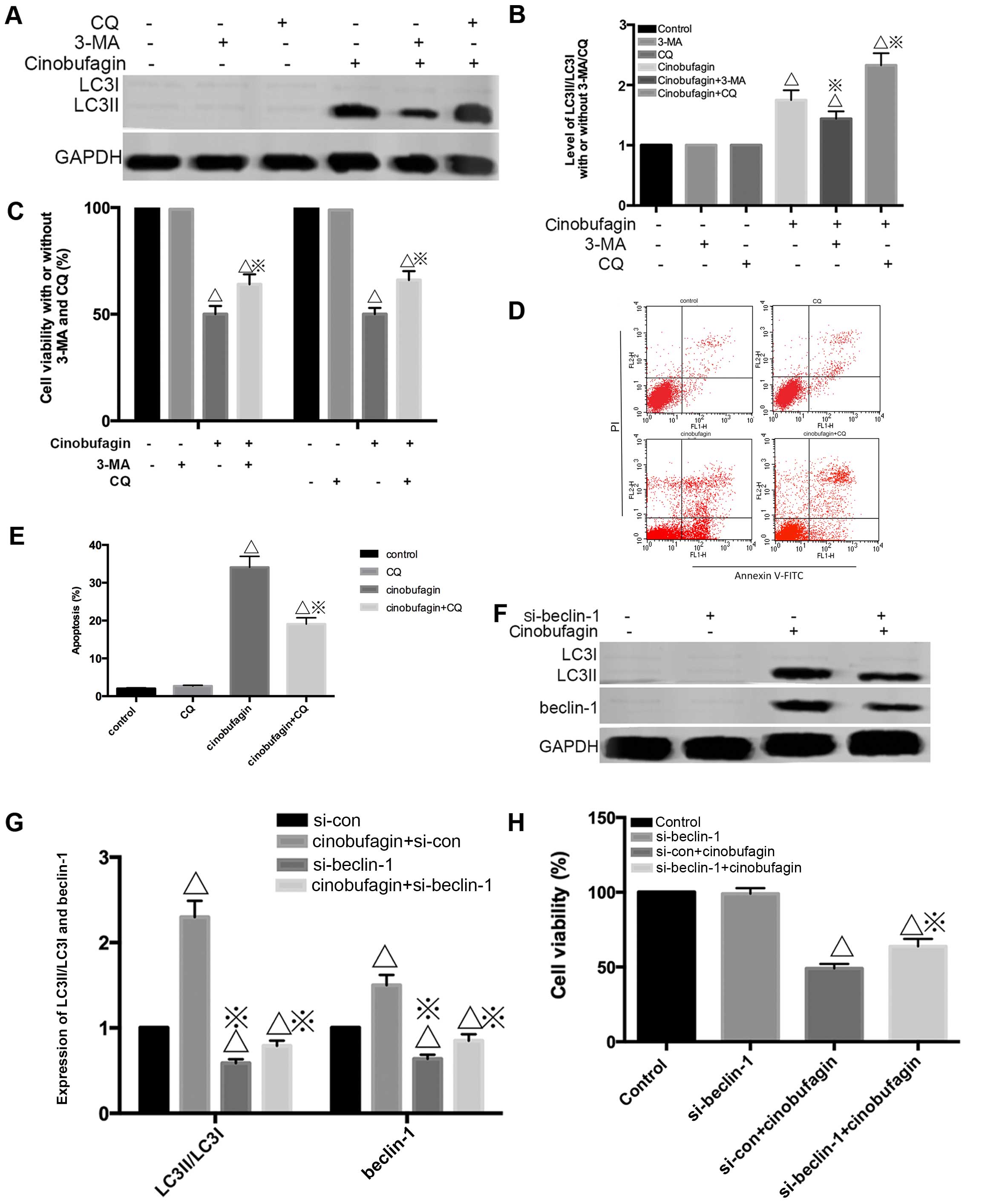
Fig. 4A and B, preincubation with
3-MA resulted in the decrease of LC3-II/LC3-I. Meanwhile,
inhibition of autophagy using CQ enhanced LC3-II/LC3-I compared
with cinobufagin alone. However, a marked reduction in
cinobufagin-induced cell growth inhibition was also observed when
autophagy was inhibited by CQ and 3-MA (Fig. 4C). Consistently, we also found that
pretreatment with CQ decreased the proportion of apoptotic cells
(56.11%) compared with the cinobufagin group alone (28.19%)
(Fig. 4D and E).
We further inhibited autophagy by silencing the
beclin 1 gene, as beclin-1 is an important autophagy-related gene.
siRNA was used. Transfection with si-beclin-1 resulted in a marked
reduction in beclin-1 and LC3-II/LC3-I protein expression, while
the scrambled siRNA-control group showed no change (Fig. 4F and G). As shown in Fig. 4H, beclin-1 knockdown markedly
reduced the viability of the cells treated with cinobufagin. Thus,
beclin-1 has a positive effect on the growth of U2OS cells.
ROS participates in cinobufagin treatment
in U2OS cells
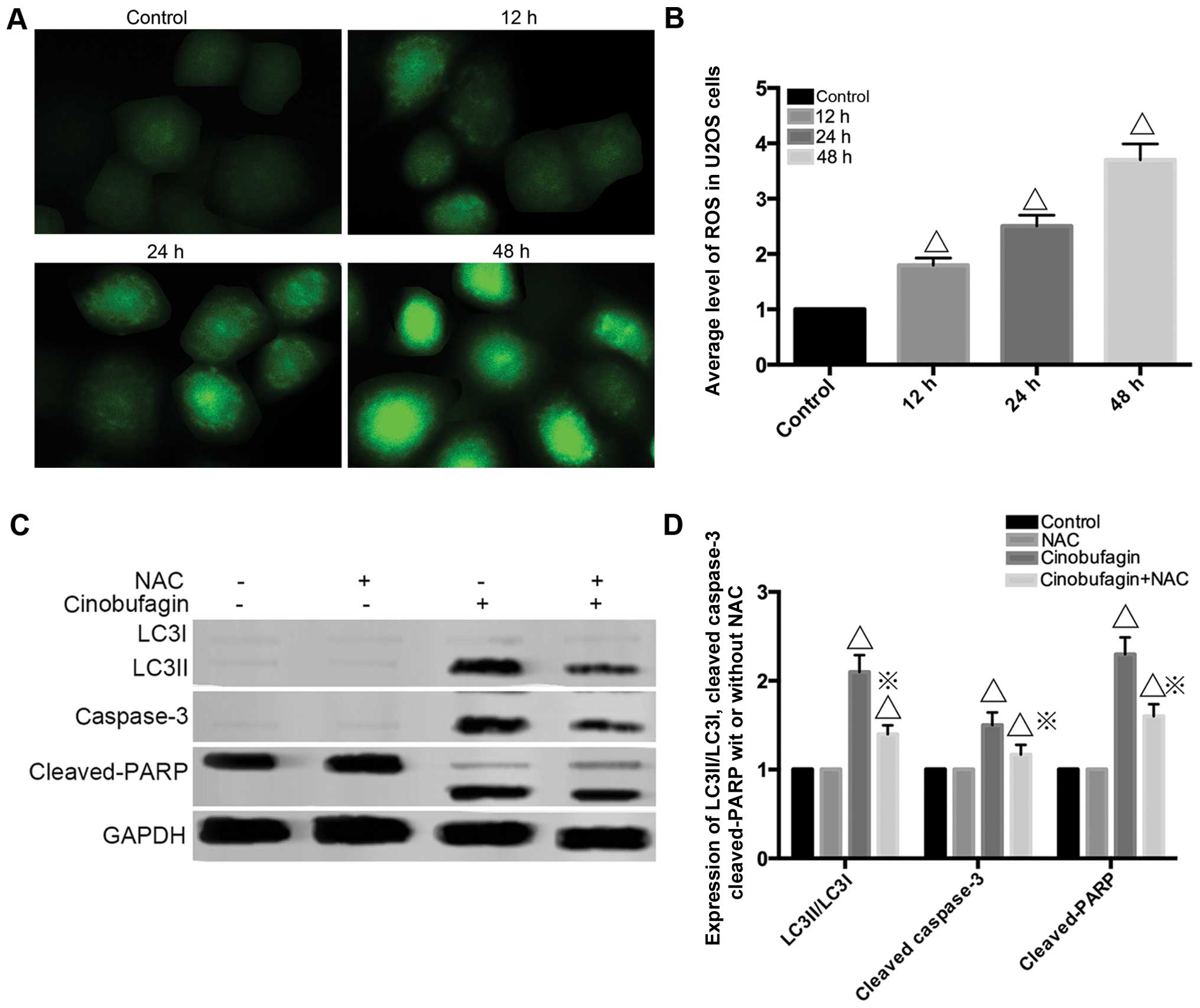
As shown in Fig. 5A and
B, the intensity of green fluorescence representing the degree
of ROS became more intense as treatment time progressed, which
indicated that ROS were involved in cinobufagin treatment. NAC, an
ROS exclusive inhibitor, was preincubated for 1 h to clarify the
effects of ROS on cinobufagin-induced cytotoxicity in U2OS cells.
The western blot analysis assay demonstrated that the levels of
LC3-II/LC3-I, cleaved caspase-3 and cleaved PARP were reduced in
the cinobufagin+NAC group compared to the cinobufagin group
(Fig. 5C and D) indicating that NAC
antagonized cinobufagin treatment. All these results revealed that
ROS are the crucial factor of cinobufagin-induced apoptosis and
autophagy.
The activation of the JNK signaling
pathway following cinobufagin treatment in U2OS cells
As shown in Fig. 6A and
B, western blot analysis was employed to assess the expression
of JNK, p-JNK, p-p38 and p38. Cinobufagin cleaved the
phosphorylation of the JNK and p-38 signaling pathway. In addition,
pretreatment with the JNK and p-38 inhibitors SP600125 and SB203580
significantly attenuated LC3-II/LC3-I, cleaved caspase-3 as well as
cleaved PARP levels acccompanied by downregulation of p-JNK/p-p38
compared to the cinobufagin group (Fig.
6C and D). These data indicated that the JNK/p38 signaling
pathway was upstream in cinobufagin cytotoxicity. To study the
relationship between ROS and JNK/p38 signaling pathway, the ROS
inhibitor NAC was used. As expected, NAC suppressed the level of
p-JNK and p-p38 (Fig. 6E and F).
Thus, the ROS/JNK/p38 signaling pathway plays a central role in
cinobufagin-triggered apoptosis and autophagy.
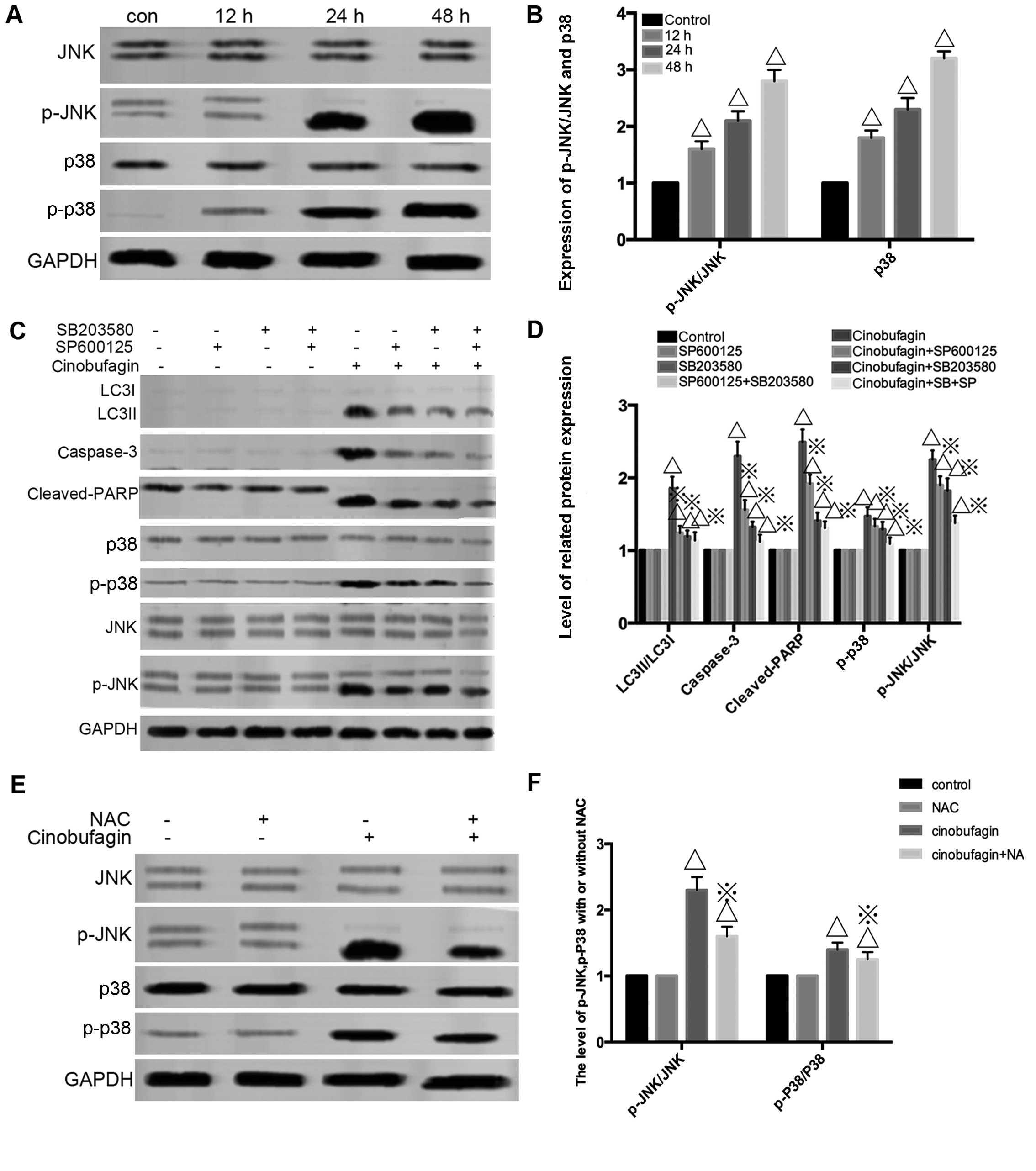 | Figure 6Reactive oxygen species
(ROS)-activated MAPK and JNK/P38 pathways contribute to
cinobufagin-induced autophagy and apoptosis. (A and B) U2OS cells
were incubated with 120 mg/l cinobufagin at various times and
measurement of p-JNK, JNK, p-38 and p-p38 by western blot analysis
followed. ΔP<0.05 vs. the control group. (C and D)
U2OS cells were pretreated with SP600125 (10 µM) or SB203580
(0.5 µM) for 1 h, and then incubated with 120 mg/l
cinobufagin for 24 h to determine the level of autophagy-related
protein LC3II/LC3I, apoptotic-related protein cleaved caspase-3,
cleaved PARP as well as p-JNK, t-JNK, p-p38, t-P38.
ΔP<0.05 vs. the control group, ※P<0.05
vs. the cinobufagin group. (E and F) NAC attenuated
cinobufagin-induced JNK and p-38 activation. U2OS cells were
pretreated with NAC (5 mM) for 1 h, and then incubated with 120
mg/l of cinobufagin for 24 h to detect the effects of ROS on
JNK/p38 signaling. GAPDH was used as loading control. Data are
represented as the mean ± SEM of three independent experiments.
ΔP<0.05 vs. the control group, ※P<0.05
vs. the cinobufagin group. |
Discussion
In our research, the data showed that cinobufagin
suppressed the proliferation of U2OS cells in a concentration- and
time-dependent manner. Research showed that cinobufagin/bufalin
induced osteosarcoma apoptosis by triggering the mitochondrial
pathway (25) and downregulating
Hsp27 (26). Yin et al
(19) demonstrated that
cinobufagin-induced apoptosis was attributed to the activation of
the GSK-3β/NF-κB pathway in osteosarcoma cells. Our experimental
results revealed that the percentage of apoptotic cellswas
increased at the indicated treatment times by flow cytometry and
Hoechst 33258 staining (Fig. 2A–D).
The process of apoptosis is carried out by two pathways: i)
extrinsic stimuli through cell surface death receptor (caspase-8)
pathway or ii) intrinsic stimuli through the mitochondrial
signaling pathway (caspase-9). However, for either pathway,
caspase-3/cleaved-PARP are both activated and are related to cell
structure, cell cycle regulation, DNA repair and other related
proteins, ultimately causing irreversible cell death (27). Cao et al found that
cinobufagin (28) induced
osteosarcoma cell mitochondrial-dependent apoptosis accompanied by
caspase-7 activation. However, Xie et al (21) did not find any increase in caspase-3
and PARP cleavage following bufalin treatment in colon cells. In
our experiments, cleaved caspase-3, -8, -9 as well as cleaved PARP
were simultaneously upregulated. Following pretreatment with
caspase inhibitors, cell viability increased to different degrees.
In short, cinobufagin triggered caspase-dependent apoptosis in our
experiment.
Autophagy, also known as type II cell death, is a
non-caspase-dependent cell death form distinct from apoptosis.
According to the stage of the tumor as well as the tissue or the
cells targeted (29), autophagy can
exert a positive or negative effect on the growth of tumors. In our
experiments, we clearly observed activation of autophagy flux by
cinobufagin in ostersarcoma U2OS cells for the first time as
confirmed by the following results. i) An abundance of
autophagosomes were observed under TEM (Fig. 3A). ii) GFP-LC3 green fluorescent
particles aggregated with the progression of treatment time
(Fig 3B and C). iii) The expression
of LC3-II/LC3-I accumulated following pretreatment with CQ
(Fig. 3D and E). However, what role
autophagy plays has been largely debated. According to the
autophagy inhibitors 3-MA and CQ, respectively the inhibition
mechanism and the level of LC3-II/LC3-I changed compared to the
cinobufagin group alone (Fig. 4A and
B). Consistently, cell proliferation of the cinobufagin+3-MA/CQ
group was synchronously attenuated compared to the cinobufagin
group (Fig. 4C). Flow cytometry
results also revealed that the number of apoptotic cells was
diminished in the cinobufagin+CQ group more when compared with that
in the cinobufagin group alone revealing that cinobufagin triggered
autophagic cell death (Fig. 4D and
E).
To further clarify the viewpoint above, the siRNA
approach was used. Beclin-1 (a key autophagy regulator) was
silenced, and LC3-II/LC3-I were downregulated in the
cinobufagin+si-beclin-1 group compared with the scrambled control
(SCR) siRNA transfection group (Fig. 4F
and G). We also observed that silencing of beclin-1 decreased
cinobufagin-induced suppression of cell viability when compared
with the siSCR group (Fig. 4H).
Induction of autophagy, at least in part, was responsible for
cinobufagin cytotoxicity in osteosarcoma which was in line with the
findings of Xie et al (21).
ROS include a variety of oxygen-containing, reactive
and short-lived molecules (30)
which are thought to operate as signaling molecules in signal
transduction pathways regulating cell growth, differentiation,
survival, inflammation and immune response. The MAPK pathway can be
activated by extracellular stimuli, such as Gi protein-coupled
receptors (GPCR), ultraviolet irradiation (31,32),
genotoxic agents and oxidative stress. In our study, generation of
ROS was activated in a time-dependent manner. Consequently, we
confirmed that JNK/p-38 phosphorylation decreased by pretreatment
with ROS inhibitor NAC. Following preincubation with the ROS
inhibitor NAC, JNK inhibitor SP600125 and p-38 inhibitor SB230580
respectively, LC3-II/LC3-I, caspase-3, cleaved PARP as well as
p-JNK and p-p38 were decreased to different degrees compared with
the cinobufagin group, indicating that ROS activated the JNK/p38
signaling pathway and that cinobufagin induced autophagy and
caspase-dependent apoptosis. In agreement with our results, Li
et al (33) reported that
suppression of celastrol-induced autophagy diminished
caspase-dependent apoptosis. However, Xie et al demonstrated
that bufalin induced autophagic cell death without cleaved
caspase-3 and cleaved PARP generation. Shen et al (20) found that bufalin-induced autophagy
played a cytoprotective role in bufalin-induced ER stress and cell
death. Different types of cells caused stimulation to different
levels of severity.
However, there is no doubt that the underlying
mechanisms of cinobufagin in osteosarcoma therapy still requires
further investigation. For example, in this study we did not
address expression of the Bax/Bcl-2 and PI3kt/mTOR/Akt signaling
pathways, and moreover whether endoplasmic reticulum stress occurs
in cinobufagin treatment of osteosarcoma. Thus, further exploration
is needed to reveal the underlying mechanisms in the crosstalk
among apoptosis, autophagy and ER stress.
In conclusion, our study elucidated a detailed
mechanism involved in cinobufagin-induced apoptosis and autophagy
in osteosarcoma which will pave the way for further development of
the clinical application of this compound for treating
osteosarcoma.
References
|
1
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yorimitsu T and Klionsky DJ: Autophagy:
molecular machinery for self-eating. Cell Death Differ. 12(Suppl
2): 1542–1552. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Apel A, Zentgraf H, Büchler MW and Herr I:
Autophagy - A double-edged sword in oncology. Int J Cancer.
125:991–995. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rami A and Kögel D: Apoptosis meets
autophagy-like cell death in the ischemic penumbra: two sides of
the same coin? Autophagy. 4:422–426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dewaele M, Maes H and Agostinis P:
ROS-mediated mechanisms of autophagy stimulation and their
relevance in cancer therapy. Autophagy. 6:838–854. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kamiyama M, Naguro I and Ichijo H: In vivo
gene manipulation reveals the impact of stress responsive MAPK
pathways on tumor progression. Cancer Sci. 106:785–796. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang Z, Shi X, Li Y, Fan J, Zeng X, Xian
Z, Wang Z, Sun Y, Wang S, Song P, et al: Blocking autophagy
enhanced cytotoxicity induced by recombinant human arginase in
triple-negative breast cancer cells. Cell Death Dis. 5:e15632014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu J, Chang F, Li F, Fu H, Wang J, Zhang
S, Zhao J and Yin D: Palmitate promotes autophagy and apoptosis
through ROS-dependent JNK and p38 MAPK. Biochem Biophys Res Commun.
463:262–267. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou H, Shen T, Shang C, Luo Y, Liu L, Yan
J, Li Y and Huang S: Ciclopirox induces autophagy through reactive
oxygen species-mediated activation of JNK signaling pathway.
Oncotarget. 5:10140–10150. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qi F, Li A, Inagaki Y, Kokudo N, Tamura S,
Nakata M and Tang W: Antitumor activity of extracts and compounds
from the skin of the toad Bufo bufo gargarizans Cantor. Int
Immunopharmacol. 11:342–349. 2011. View Article : Google Scholar
|
|
11
|
Zhu Z, Li E and Liu Y, Gao Y, Sun H, Wang
Y, Wang Z, Liu X, Wang Q and Liu Y: Bufalin induces the apoptosis
of acute promyelocytic leukemia cells via the downregulation of
survivin expression. Acta Haematol. 128:144–150. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xie RF, Li ZC, Chen PP and Zhou X:
Bufothionine induced the mitochondria-mediated apoptosis in H22
liver tumor and acute liver injury. Chin Med. 10:52015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang J, Jin Y, Xu Z, Zheng Z and Wan S:
Involvement of caspase-3 activity and surviving downregulation in
cinobufocini-induced apoptosis in A 549 cells. Exp Biol Med
(Maywood). 234:566–572. 2009. View Article : Google Scholar
|
|
14
|
Yu CH, Kan SF, Pu HF, Jea Chien E and Wang
PS: Apoptotic signaling in bufalin- and cinobufagin-treated
androgen-dependent and -independent human prostate cancer cells.
Cancer Sci. 99:2467–2476. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li C, Hashimi SM, Cao S, Mellick AS, Duan
W, Good D and Wei MQ: The mechanisms of chansu in inducing
efficient apoptosis in colon cancer cells. Evid Based Complement
Alternat Med. 2013:8490542013.PubMed/NCBI
|
|
16
|
Cao-Hong, Shibayama-Imazu T, Masuda Y,
Shinki T, Nakajo S and Nakaya K: Involvement of Tiam1 in apoptosis
induced by bufalin in HeLa cells. Anticancer Res. 27:245–249.
2007.PubMed/NCBI
|
|
17
|
Qi F, Inagaki Y, Gao B, Cui X, Xu H,
Kokudo N, Li A and Tang W: Bufalin and cinobufagin induce apoptosis
of human hepato-cellular carcinoma cells via Fas- and
mitochondria-mediated pathways. Cancer Sci. 102:951–958. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Baek SH, Kim C, Lee JH, Nam D, Lee J, Lee
SG, Chung WS, Jang HJ, Kim SH and Ahn KS: Cinobufagin exerts
anti-proliferative and pro-apoptotic effects through the modulation
ROS-mediated MAPKs signaling pathway. Immunopharmacol
Immunotoxicol. 37:265–273. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yin JQ, Wen L, Wu LC, Gao ZH, Huang G,
Wang J, Zou CY, Tan PX, Yong BC, Jia Q, et al: The glycogen
synthase kinase-3β/nuclear factor-kappa B pathway is involved in
cinobufagin-induced apoptosis in cultured osteosarcoma cells.
Toxicol. 218:129–136. 2013.
|
|
20
|
Shen S, Zhang Y, Wang Z, Liu R and Gong X:
Bufalin induces the interplay between apoptosis and autophagy in
glioma cells through endoplasmic reticulum stress. Int J Biol Sci.
10:212–224. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xie CM, Chan WY, Yu S, Zhao J and Cheng
CH: Bufalin induces autophagy-mediated cell death in human colon
cancer cells through reactive oxygen species generation and JNK
activation. Free Radic Biol Med. 51:1365–1375. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bjørkøy G, Lamark T, Brech A, Outzen H,
Perander M, Overvatn A, Stenmark H and Johansen T: p62/SQSTM1 forms
protein aggregates degraded by autophagy and has a protective
effect on huntingtin-induced cell death. J Cell Biol. 171:603–614.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nilsson JR: Does chloroquine, an
antimalarial drug, affect autophagy in Tetrahymena pyriformis? J
Protozool. 39:9–16. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang D and Bi Z: Bufalin inhibited the
growth of human osteosarcoma MG-63 cells via down-regulation of
Bcl-2/Bax and triggering of the mitochondrial pathway. Tumour Biol.
35:4885–4890. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xie XB, Yin JQ, Wen LL, Gao ZH, Zou CY,
Wang J, Huang G, Tang QL, Colombo C, He WL, et al: Critical role of
heat shock protein 27 in bufalin-induced apoptosis in human
osteosarcomas: a proteomic-based research. PLoS One. 7:e473752012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nicholson DW: Caspase structure,
proteolytic substrates, and function during apoptotic cell death.
Cell Death Differ. 6:1028–1042. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cao F, Kang X and Wang L: Effects of
cinobufagin on apoptosis in U-2OS osteosarcomas cells. Zhongguo Xiu
Fu Chong Jian Wai Ke Za Zhi. 28:349–353. 2014.In Chinese.
PubMed/NCBI
|
|
29
|
Jia LT, Chen SY and Yang AG: Cancer gene
therapy targeting cellular apoptosis machinery. Cancer Treat Rev.
38:868–876. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gupta RK, Patel AK, Shah N, Chaudhary AK,
Jha UK, Yadav UC, Gupta PK and Pakuwal U: Oxidative stress and
antioxidants in disease and cancer: a review. Asian Pac J Cancer
Prev. 15:4405–4409. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhou YY, Li Y, Jiang WQ and Zhou LF:
MAPK/JNK signalling: a potential autophagy regulation pathway.
Biosci Rep. 35:e001992015.PubMed/NCBI
|
|
32
|
Jalmi SK and Sinha AK: ROS mediated MAPK
signaling in abiotic and biotic stress- striking similarities and
differences. Front Plant Sci. 6:7692015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li HY, Zhang J, Sun LL, Li BH, Gao HL, Xie
T, Zhang N and Ye ZM: Celastrol induces apoptosis and autophagy via
the ROS/JNK signaling pathway in human osteosarcoma cells: an in
vitro and in vivo study. Cell Death Dis. 6:e16042015. View Article : Google Scholar : PubMed/NCBI
|