Introduction
Colorectal cancer (CRC) is one of the most common
types of cancers and is a leading cause of morbidity and mortality
worldwide (1). It accounts for more
than 9% of the total incidence of malignancies (2,3) and
accounted for approximately 8% of all new cases of malignancies in
2014 (4). Advances in the available
methods for CRC treatment have been made. Yet, due to limitations
in early cancer detection and accurate prognostic prediction, the
survival rates of CRC patients remain low. Thus, identification and
knowledge of biomarkers and effective molecular mechanisms
regarding CRC are crucial.
p53 is one of the most significative
tumor-suppressor genes. Mutations of p53 result in the loss of
wild-type p53 activity (5). These
mutant p53 proteins may also serve as negative inhibitors to play
an opposite role of wild-type p53. One study showed that expression
of mutant p53 dampened the restoration effect of wild-type p53
(6). These mutant p53 proteins can
display new functions in promoting tumorigenesis. Many studies have
also shown that p53 mutations act on various human tumors such as
colon, breast, liver, and lung cancers (7).
Epithelial-mesenchymal transition (EMT) is the
process involved in the regeneration of wound healing, fibrosis and
cancer progression (8–11). A large number of studies have shown
that the adhesion ability of tumor cells decreases the migration
capacity of tumor cells. Thus, EMT is associated with the invasion
and metastasis of tumors. In the process of EMT, the expression
level of cell adhesion molecules (such as E-cadherin) is reduced
while mesenchymal markers such as vimentin protein are increased
(12). The EMT process is involved
in the invasion, metastasis and drug resistance of tumors.
In the present study, we demonstrated that miR-300
and p53 were significantly increased in CRC tissues. miR-300 and
p53 promoted CRC cell proliferation, migration, and invasion in
vitro. In addition, miR-300 was found to be a direct positive
regulator of p53 through binding to the binding site in the 3'UTR
of the p53 gene in human CRC cells. miR-300 and p53 also induced
CRC cell EMT. Taken together, we demonstrated that miR-300 promoted
proliferation and EMT-mediated CRC migration and invasion by
targeting p53. Our findings aid in the understanding of the
underlying mechanisms of CRC metastasis and may elucidate effective
therapeutic targets.
Materials and methods
Cell lines
Human embryonic kidney 293T (HEK293T) cells and
human CRC cell lines (NCM460, HT29, SW480, SW620 HCT116 and DLD-1)
were purchased from the Type Culture Collection of the Chinese
Academy of Sciences (Shanghai, China). NCM460, SW480, HCT116 and
DLD-1 cells were cultured in RPMI-1640 medium, and SW620, HT29 and
HEK293T cells were cultured in Dulbecco's modified Eagle's medium
(DMEM) (all from Invitrogen, Carlsbad, CA, USA). Each medium
contained 10% fetal bovine serum (FBS) and 100 U/ml penicillin and
streptomycin (all from Invitrogen). The incubation condition was 5%
CO2 at 37°C.
Patients and clinical specimens
In the present study, we collected 93 pairs of CRC
tissues and matched adjacent noncancerous tissue samples from
patients who had undergone surgical treatment for CRC between 2013
and 2016 at Southwest Hospital, Third Military Medical University
(Chongqing, China). Written informed consent was provided by each
patient. CRC histological diagnosis was assessed according to the
World Health Organization (WHO). All tissue samples were washed
with sterile phosphate-buffered saline (PBS) and immediately
conserved at −80°C until use.
Lentiviral vector construction for p53
expression
For the construction of the lentiviral vector
expressing p53, we cloned the human p53 gene from HEK293T cell
genomic DNA by PCR with specific designed primers (the forward
primer contained a Xhol site:
5′-CCGCTCGATGGAGGAGCCGCAGTCGA-3′, and the reverse primer contained
a BamHI site: 5′-CGGGATCCTCAGTCTGAGTCAGGCCCTT-3′). A
lentiviral vector expressing EGFP was used as a control. Then, the
PCR products were inserted into a lentiviral vector. Packaging
vectors (pMD2.G, pMDL-G/P-RRE and pRSV-REV) and the transfer vector
(p53) were cotransfected into 293T cells for 48 h, and the
pseudotyped lentiviruses were produced. We performed virus harvest
and purification using ultracentrifugation. Thereafter, the SW480
and HT29 cells at a density of 10,000/well were seeded in 24-well
plates and transduced with the lentivirus (MOI of 5) and 8 µg/ml
polybrene (Sigma-Aldrich, St. Louis, MO, USA).
Transfection of microRNA (miRNA)
mimics
A total of 2×105 cells were seeded in
6-well plates with antibiotic-free complete medium. The cells were
grown overnight, and then transfected with 200 µl mature miRNA
mimic (100 nM) and Lipofectamine 3000 (Invitrogen) according to the
manufacturer's instructions for 72 h. The sequences for miR-300
were 5′-UAUACAAGGGCAGACUCUCUCU-3′ (sense) and
5′-AGAGAGAGUCUGCCCUUGUAUA-3′ (antisense).
RNA preparation and reverse
transcription
According to the manufacturer's instructions, total
RNA was extracted from the cells or the CRC tissue samples with
TRIzol reagent (Invitrogen). Each 1.0 µg of RNA was used as a
template to synthesize corresponding cDNA with random primers using
a RevertAid First Strand cDNA synthesis kit (Thermo Fisher
Scientific, Inc., Waltham, MA, USA).
Quantitative real-time reverse
transcription PCR (qRT-PCR) assay
As previously described (13), we performed all PCR reactions using
SYBR-Green PCR Master Mix kit and an ABI 7500 Real-Time PCR system
(Applied Biosystems, Warrington, UK). An endogenous housekeeping
gene GAPDH was selected as internal loading control. The primer
sequences for miR-300 were: miR-300 forward,
5′-TATACAAGGGCAGACTCTCTCT-3′ and U6 reverse,
5′-CGCAAGGATGACACGCAAATTCGT-3′; the primer sequences for p53 were:
p53 forward, 5′-GAGGTTGGCTCTGACTGTACC-3′ and reverse,
5′-TCCGTCCCAGTAGATTACCAC-3′; the primer sequences for GAPDH were:
GAPDH forward, 5′-CATGAGAAGTATGACAACAGCCT-3′ and reverse,
5′-AGTCCTTCCACGATACCAAAGT-3′. All results are showed as the mean ±
SD of three independent experiments.
Cell proliferation
Cell proliferation was detected using the
methylthiazol tetrazolium (MTT) assay. Firstly, cells were seeded
into 96-well plates at a density of 2×103 cells/well
with 100 µl complete medium and cultured for 24, 48, 72, 96 and 120
h. At the specified time of incubation, 20 µl of MTT (5 mg/ml)
solution was added to each well and incubation was carried out for
4 h at 37°C. The dimethyl sulfoxide (DMSO) was used to dissolve the
formazan product for 10 min at room temperature, and then the
absorbance at 490 nm was measured using a microplate reader. Each
condition was determined in quintuplicates, and all experiments
were repeated at least 3 times.
Migration and invasion assays
According to the manufacturer's instructions, the
migration and invasive abilities of the CRC cells were measured in
a 24-well Transwell cell culture chamber (Corning Costar, Inc.,
Corning, NY, USA) in vitro. Complete medium was added to the
lower chamber, and serum-free medium containing 200 µl cells
(2.5×105/100 µl) was added to the upper chamber. After
24 h of incubation, the cells with migratory ability migrated
through the chamber. The medium in the upper chamber was removed.
The cells in the lower chambers were fixed with 4% paraformaldehyde
and stained with 0.1% crystal violet solution. The number of cells
was counted in five areas of constant size per well under the
microscope using a 20X objective. Ten microliters 1:3 diluted
Matrigel (BD Biosciences, San Diego, CA, USA) was added into the
upper chamber for the invasion assays. All experiments were
achieved in triplicate.
Western blot analysis
The western blot analysis was carried out as
previously described (14). Cells
were lysed in a lysis buffer, and the concentrations of total
proteins were detected using a BCA Protein Assay kit (Thermo Fisher
Scientific, Inc.). Heat-denatured proteins (30 µg) were separated
and transferred onto polyvinylidene fluoride (PVDF) membranes
(Millipore, Billerica, MA, USA). The transferred membranes were
incubated with primary antibodies with a proper dilution at 4°C
overnight. On the following day, the PVDF membranes with proteins
were incubated with the secondary antibody. The signal was detected
using the enhanced chemiluminescence (ECL) substrate kit and
detection system (both from Amersham Biosciences, Piscataway, New
Jersey, USA). The primary antibodies mouse anti-E-cadherin
(1:5,000) and mouse anti-vimentin (1:4,000) were purchased from BD
Biosciences. The primary antibody rabbit anti-p53 monoclonal
antibody (1:1,000) was purchased from Abcam (Cambridge, UK). The
rabbit anti-GAPDH monoclonal antibody (1:5,000; Cell Signaling
Technology, Beverly, MA, USA) was used as an internal control.
Dual-luciferase reporter assay
The database TargetScan (http://www.targetscan.org) was used to predict
potential targets for miR-300. DNA fragments of the TP53 3'UTR
containing the putative miR-300 binding site (or mutated miR-300
binding site) were amplified by PCR from HEK293T cell genomic DNA
and inserted into the PmeI/SpeI sites of the firefly
luciferase coding region of the pMIR-report vector (Invitrogen Life
Technologies). The plasmids were named wild-type
(pMIR-report-p53-wt) and mutated (pMIR-report-p53-mut) sequences,
respectively. Mutation from AGCCAG to CACUAU was introduced in the
potential miR-300 binding sites. HEK293T cells (3×105)
were seeded in a 24-well plate and cotransfected with
pMIR-report-p53 3'UTR-wt or pMIR-report-p53 3'UTR-mut and miR-300
expression plasmids (a Renilla plasmid as internal
reference) using Lipofectamine 3000 for 48 h. The luciferase
activities were assessed using the Dual-Luciferase Reporter assay
kit (Promega Corp., Madison, WI, USA) according to the
manufacturer's instructions. Each transfection was repeated twice
in triplicate.
Statistical analysis
SPSS 15.0 software was used to analyze the data. The
differences between individual groups were analyzed by t-tests. The
Pearson's correlation algorithm was used to analyze the correlation
coefficient and the significance between the expression of miR-300
and p53 mRNA. All data are shown as the means ± SD. P<0.05 was
considered to indicate a statistically significant result.
Results
miR-300 is significantly increased in
CRC tissues compared with that in adjacent colorectal tissues
The expression level of miR-300 was detected in 93
pairs of CRC tissues and corresponding adjacent colorectal mucosal
tissues by qRT-PCR. Our results demonstrated that the expression of
miR-300 in CRC tissues was significantly higher than that observed
in the corresponding adjacent colorectal mucosal tissues
(P<0.001; Fig. 1A). Our results
also showed that miR-300 was significantly correlated with
lymphatic metastasis and TNM stage (P=0.013) by clinicopathological
analysis, but it was not significantly correlated with age, gender,
invasion, and distal metastasis (P>0.05; Table I). Furthermore, in our study, the
area under the ROC curve of miR-300 was 0.597, P=0.023 (Fig. 1B).
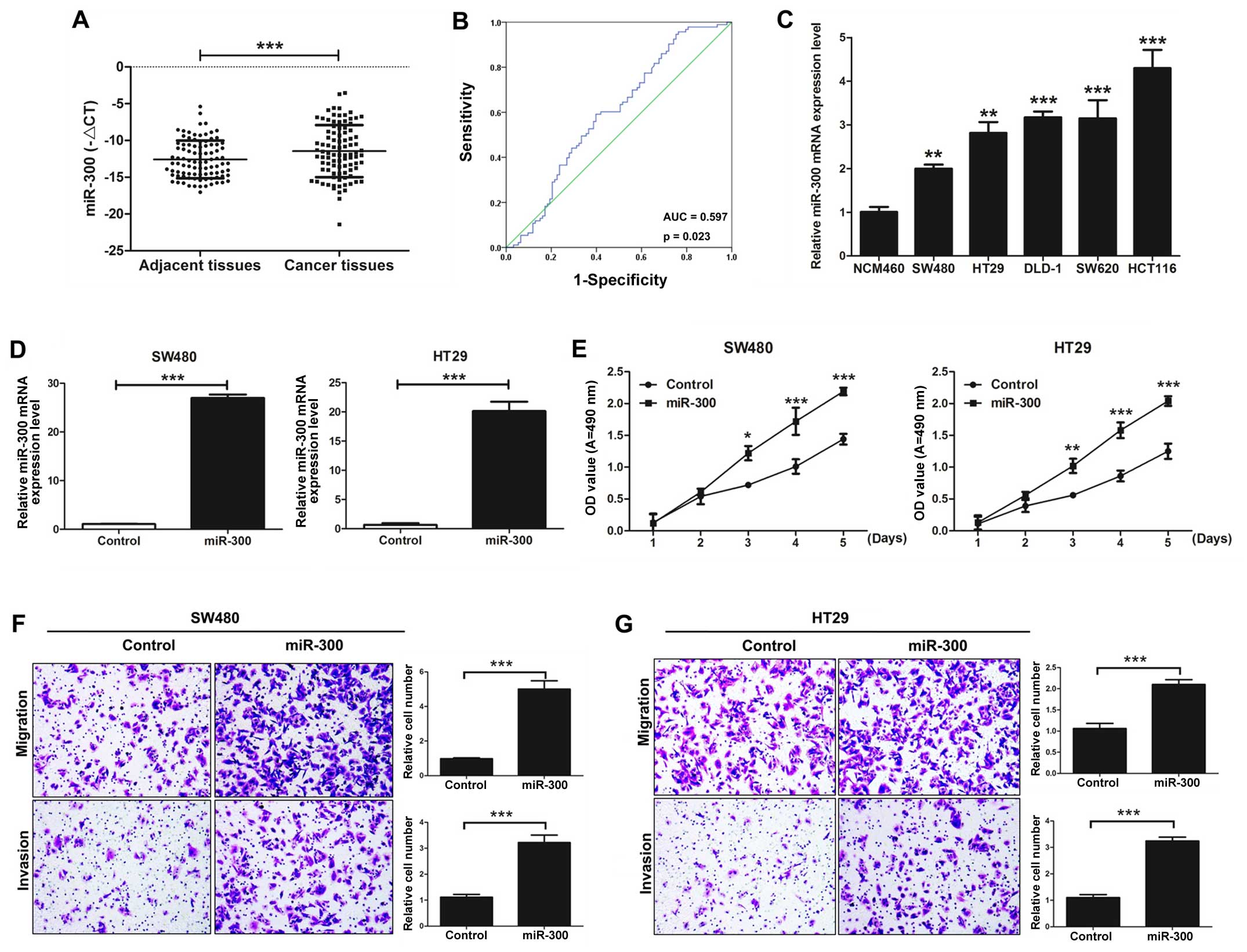 | Figure 1.miR-300 is significantly increases in
colorectal cancer (CRC) tissues and promotes CRC cell
proliferation, migration, and invasion in vitro. (A)
Quantitative RT-PCR (qRT-PCR) was used to detected the the mRNA
expression level of miR-300 in CRC tissues and adjacent colorectal
mucosal tissues. (B) Receiver operating characteristic (ROC) curve
indicates the prediction of prognosis for performance of miR-300.
(C) The mRNA expression level of miR-300 was detected by qRT-PCR in
human-derived colonic epithelial NCM460 cells and CRC cell lines
(SW480, HT29, DLD-1, SW620, and HCT116). (D) The expression of
miR-300 was detected after SW480 and HT29 cells were transfected
with miR-300 or control. (E) The MTT assay was used to determine
the ability of proliferation in the SW480 and HT29 cells
transfected with miR-300 or control. (F) Transwell assays of SW480
cells transfected with miR-300 or negative control were performed.
Magnification, ×200; scale bar, 10 µm. (G) Transwell assays of HT29
cells transfected with miR-300 or negative control were performed.
Magnification, ×200; scale bar, 10 µm. All data are expressed as
mean ± SD of three independent experiments (*P<0.05,
**P<0.01, ***P<0.001). |
 | Table I.Correlation analysis between the
expression level of miR-300 and clinicopathological characteristics
of the colorectal cancer patients. |
Table I.
Correlation analysis between the
expression level of miR-300 and clinicopathological characteristics
of the colorectal cancer patients.
| Characteristics | No. of patients n
(%) | miR-300 (mean ±
SD) | P-value |
|---|
| Total no. of
patients | 93 (100.0) |
|
|
| Age (years) |
|
|
|
|
>60 | 51 (54.8) | 10.11±1.88 | 0.267 |
| ≤60 | 42 (45.2) | 9.64±1.62 |
|
| Gender |
|
|
|
| Male | 46 (49.5) | 9.96±1.97 | 0.750 |
|
Female | 47 (50.5) | 9.82±1.38 |
|
| Invasion |
|
|
|
|
T0-T2 | 41 (44.1) | 9.68±1.70 | 0.345 |
|
T3-T4 | 52 (55.9) | 10.09±1.84 |
|
| Lymphatic
metastasis |
|
|
|
| N0 | 62 (66.7) | 10.84±2.60 |
0.013a |
|
N1-N3 | 31 (33.3) | 9.13±1.06 |
|
| Distal
metastasis |
|
|
|
| M0 | 71 (76.3) | 9.87±1.80 | 0.472 |
| M1 | 22 (23.7) | 10.47±1.38 |
|
| TNM stage |
|
|
|
| 0, I
and II | 62 (66.7) | 10.84±2.60 |
0.013a |
| III and
IV | 31 (33.3) | 9.13±1.06 |
|
miR-300 promotes CRC cell
proliferation, migration, and invasion in vitro
The effects of miR-300 on the capacity of CRC cell
growth, migration, and invasion were investigated. The mRNA
expression level of miR-300 was detected in colonic epithelial
NCM460 cells and CRC cell lines (SW480, HT29, DLD-1, SW620, and
HCT116) by qRT-PCR. The expression level of miR-300 in the CRC cell
lines was significantly higher than that noted in the NCM460 cells
(Fig. 1C). SW480 and HT29 cell
lines were transfected with miR-300 or control vectors.
Overexpression of miR-300 observably increased the expression of
miR-300 (Fig. 1D). The cell
proliferation capacity of the SW480 and HT29 cells transfected with
miR-300 was significantly upregulated compared with the control
group as detected by MTT assay (Fig.
1E). The cell migration and invasion capacities of the SW480
cells transfected with miR-300 were also significantly upregulated
as assessed by Transwell assays in vitro (Fig. 1F). Similar results were found in the
HT29 cells (Fig. 1G). Taken
together, we indicated that miR-300 promotes CRC cell
proliferation, migration and invasion in vitro.
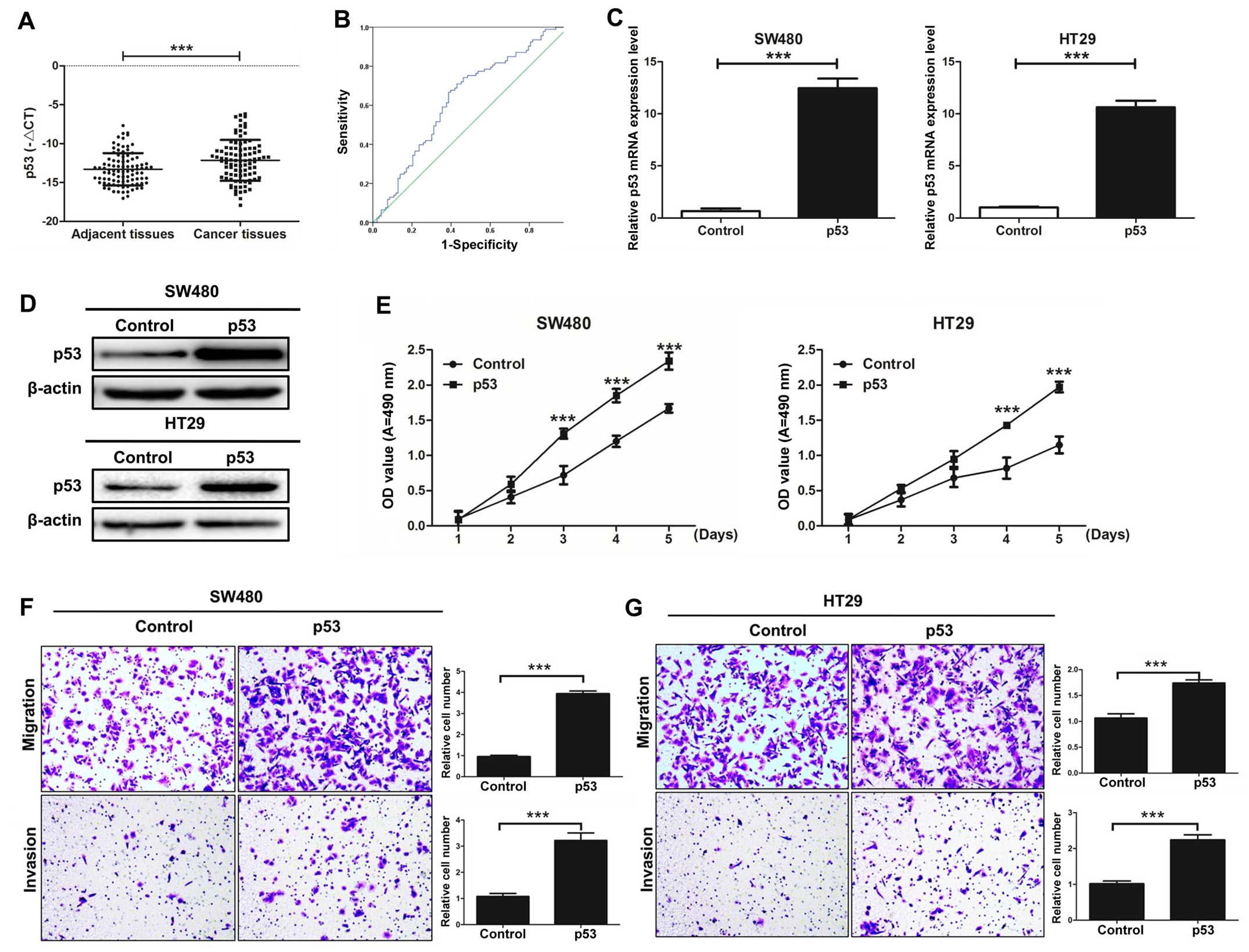
p53 is significantly increased in CRC
tissues compared with that in adjacent colorectal mucosal
tissues
The expression level of p53 was detected in 93 pairs
of CRC tissues and corresponding adjacent colorectal mucosal
tissues by qRT-PCR. Our results demonstrated that the expression of
p53 in CRC tissues was significantly higher than that observed in
the corresponding adjacent colorectal mucosal tissues (P<0.001;
Fig. 2A). Our results also showed
that p53 was significantly correlated with lymphatic metastasis and
TNM stage (P=0.019) by clinicopathological analysis, but it was not
significantly correlated with age, gender, invasion, and distal
metastasis (P>0.05; Table II).
Furthermore, in our study, the area under the ROC curve of miR-300
was 0.635, P=0.002 (Fig. 2B).
| Table II.Correlation analysis between the
expression level of p53 and clinicopathological characteristics of
the colorectal cancer patients. |
Table II.
Correlation analysis between the
expression level of p53 and clinicopathological characteristics of
the colorectal cancer patients.
|
Characteristics | No. of patients n
(%) | p53 (mean ±
SD) | P-value |
|---|
| Total no. of
patients | 93 (100.0) |
|
|
| Age (years) |
|
|
|
|
>60 | 56 (60.2) | 12.97±2.62 | 0.441 |
|
≤60 | 37 (39.8) | 13.55±2.04 |
|
| Gender |
|
|
|
|
Male | 49 (52.7) | 13.34±2.34 | 0.098 |
|
Female | 44 (47.3) | 12.67±2.77 |
|
| Invasion |
|
|
|
|
T0-T2 | 42 (45.2) | 13.53±2.41 | 0.573 |
|
T3-T4 | 51 (54.8) | 12.79±2.54 |
|
| Lymphatic
metastasis |
|
|
|
| N0 | 52 (55.9) | 13.43±2.40 |
0.0190a |
|
N1-N2 | 41 (44.1) | 12.69±2.58 |
|
| Distal
metastasis |
|
|
|
| M0 | 76 (81.7) | 13.18±2.52 | 0.078 |
| M1 | 17 (18.3) | 12.21±2.00 |
| TNM stage |
|
|
|
| 0, I
and II | 52 (55.9) | 13.43±2.40 |
0.0190a |
| III and
IV | 41 (44.1) | 12.69±2.58 |
|
p53 promotes CRC cell proliferation,
migration, and invasion in vitro
The effects of p53 on the capacity of CRC cell
growth, migration, and invasion were investigated. SW480 and HT29
cell lines were transfected with p53 or control vectors.
Overexpression of p53 observably increased the mRNA and protein
expression levels of p53 (Fig. 2C and
D). The cell proliferation capacity of SW480 and HT29 cells
transfected with p53 was significantly increased compared with the
control group as detected by MTT assay (Fig. 2E). The cell migration and invasion
capacities of SW480 and HT29 cells transfected with p53 were also
significantly enhanced as detected by Transwell assays in
vitro (Fig. 2F and G). Taken
together, we demonstrated that p53 promotes CRC cell proliferation,
migration and invasion in vitro.
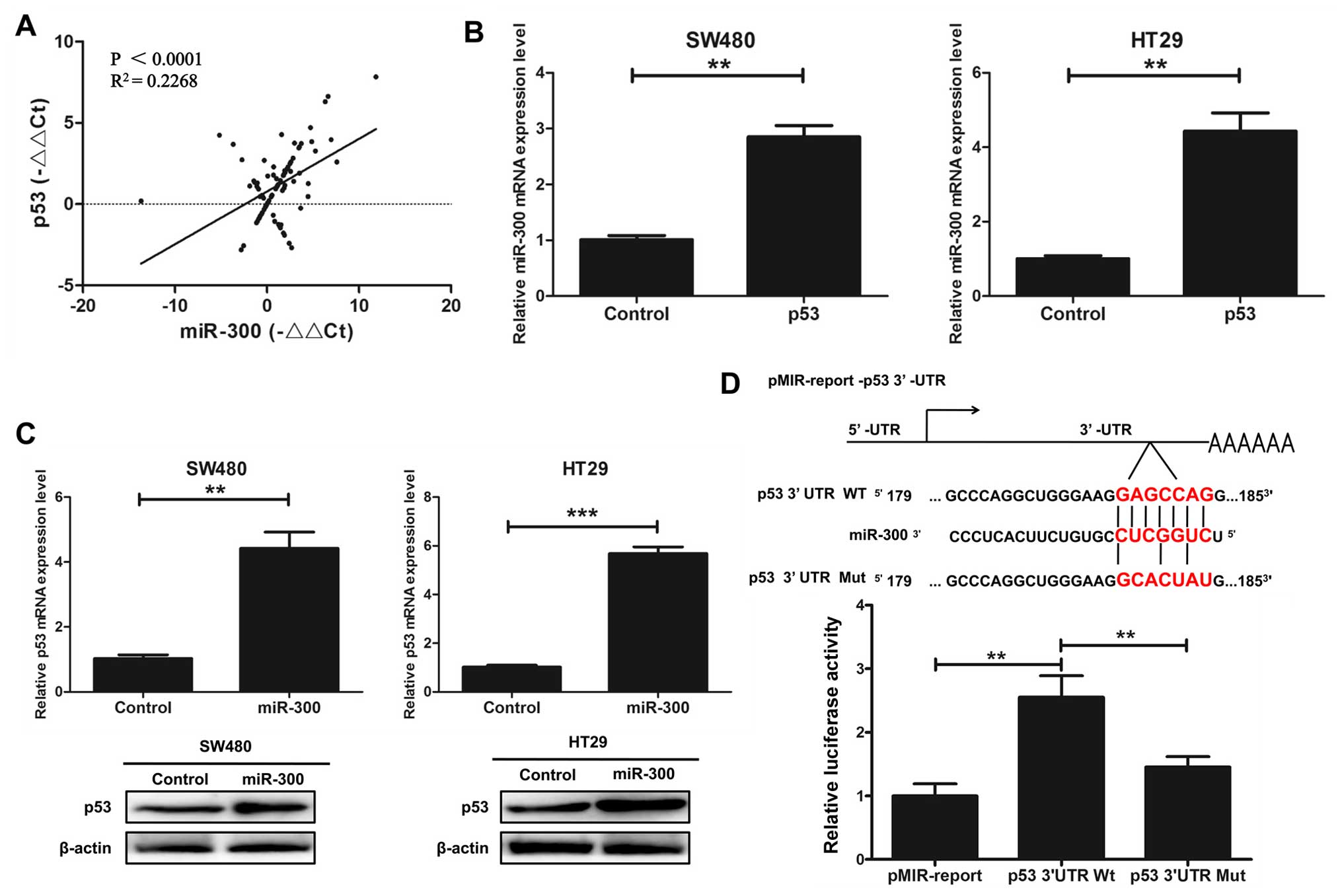
miR-300 regulates p53 gene
transcription in human CRC cell lines
We investigated whether miR-300 physically interacts
with p53. The correlation between miR-300 and p53 gene expression
in CRC tissues was assayed by qRT-PCR. The results indicated that
there was a positive correlation between miR-300 and p53 gene
expression in the CRC tissues (P<0.0001, R2=0.2268;
Fig. 3A). We indicated that
overexpression of p53 increased the mRNA expression level of
miR-300 by qRT-PCR assay in the SW480 and HT29 cells transfected
with p53 or control (Fig. 3B).
qRT-PCR and western blot analysis were used to detect the relative
expression level of p53 in the SW480 and HT29 cells transfected
with miR-300 or the control. The results indicated that
overexpression of miR-300 increased the mRNA and protein expression
of p53 (Fig. 3C). Next, we
hypothesized that miR-300 may directly regulate the transcriptional
level of p53. We cloned the promoter region of wild-type p53 and
mutant p53 into the pMIR-report. The results showed that miR-300
increased the promoter activity of p53 in HEK293T cells by
luciferase reporter gene assays (Fig.
3D). Taken together, we indicated that miR-300 regulates p53
gene transcription in human CRC cell lines.
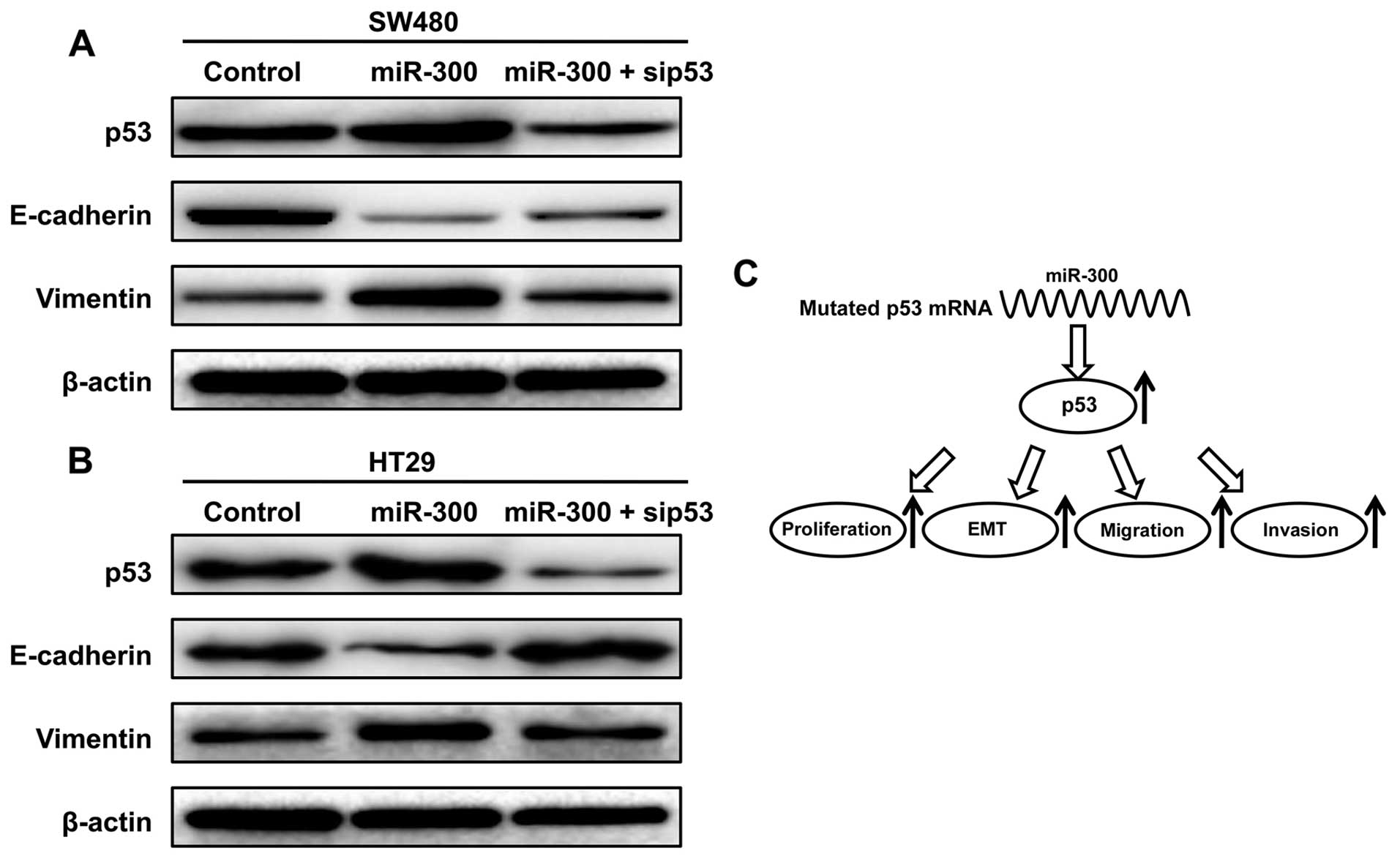
Expression levels of p53 influences
CRC cell EMT
We further investigated whether miR-300 and p53
influence CRC cell EMT. Western blot analysis showed that the level
of E-cadherin was decreased and the levels of the p53 and vimentin
were increased after overexpression of miR-300. The level of
E-cadherin was increased and the levels of p53 and vimentin were
decreased after silencing of p53 by siRNAs compared with the SW480
cells transfected with miR-300 separately (Fig. 4A). Similar results were found in the
HT29 cells (Fig. 4B).
A schematic model of miR-300 functions
in mutant p53-expressing human CRC
As shown in Fig. 4C,
miR-300 increased the expression level of p53 by targeting the p53
promoter and promoted EMT, migration and invasion of CRC.
Specifically, miR-300 was found to be a direct positive regulator
of p53 through binding to the binding site in 3'UTR of the p53 gene
in human CRC cells. miR-300 promoted CRC cell proliferation, EMT,
migration and invasion by targeting mutational p53 in
vitro.
Discussion
miRNAs are small non-coding RNAs that silence mRNAs.
In recent years, numerous studies have shown the effects of miRNAs
on cancer-related proliferation, apoptosis, inflammation,
migration, invasion and angiogenesis through regulation of the
expression of target mRNAs (15–19).
More than 60% of human protein-coding genes include conserved
miRNA-binding sites and most protein-coding genes are regulated by
miRNAs (20). Therefore, we
hypothesized that there may be specific miRNAs which may directly
regulate p53 expression. In the present study, we found that both
miR-300 and p53 were significantly increased in CRC tissues
relative to levels noted in the adjacent colorectal mucosal
tissues. Both miR-300 and p53 were significantly correlated with
lymphatic metastasis and TNM stage by clinicopathological analysis.
Furthermore, we identified miR-300 as a direct positive regulator
of p53 by binding to the 3'UTR of the human p53 gene.
It has been reported that the steps of metastasis
involve the EMT process (21,22).
EMT is a process in which cells are transformed from an epithelial
non-motile morphology to displaying mesenchymal characteristics,
such a fibroblast shape and the enhanced ability of cell migration
and invasion (21,23,24).
E-cadherin is related to tumor invasiveness, cancer metastasis, and
poor prognosis (25,26). The impact of E-cadherin on
metastasis has been studied in various models (27–29).
The interaction of E-cadherin molecules generates the core of the
epithelial adherens junction (30).
E-cadherin has been considered an epithelial marker. Vimentin
regulates cell migration. Vimentin results in migration through
recycling of endocytosed cell adhesion receptors (31). Vimentin regulates EMT induction and
promotes migration in breast cancer (32). Vimentin has been considered a
mesenchymal marker. In the present study, we found that the level
of E-cadherin was decreased and the levels of the p53 and vimentin
were increased after overexpression of miR-300. The level of
E-cadherin was increased and the levels of the p53 and vimentin
were decreased after silencing of p53 by siRNAs. Therefore, miR-300
and p53 induced CRC cell EMT. Furthermore, we aslso indicated that
both miR-300 and p53 promoted CRC cell proliferation, migration and
invasion in vitro.
In conclusion, we demonstrated that miR-300 is a
direct positive regulator of p53 through binding to the binding
site in the 3'UTR of the p53 gene in human CRC cells. Both miR-300
and p53 were significantly increased in CRC tissues. miR-300 was
significantly correlated with lymphatic metastasis and TNM stage.
Both miR-300 and p53 promoted CRC cell proliferation, migration and
invasion in vitro. Taken together, we propose that miR-300
promotes proliferation and EMT-mediated CRC migration and invasion
by targeting p53. These findings lay the foundation for exploring
the mechanism of CRC metastasis and seeking effective therapeutic
targets.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Boyle P and Langman JS: ABC of colorectal
cancer: Epidemiology. BMJ. 321:805–808. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Glade MJ: Food, nutrition, and the
prevention of cancer: a global perspective. American Institute for
Cancer Research/World Cancer Research Fund, American Institute for
Cancer Research, 1997. Nutrition. 15:523–526. 1999.PubMed/NCBI
|
|
4
|
Siegel R, Desantis C and Jemal A:
Colorectal cancer statistics, 2014. CA Cancer J Clin. 64:104–117.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Muller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Y, Suh Y-A, Fuller MY, Jackson JG,
Xiong S, Terzian T, Quintás-Cardama A, Bankson JA, El-Naggar AK and
Lozano G: Restoring expression of wild-type p53 suppresses tumor
growth but does not cause tumor regression in mice with a p53
missense mutation. J Clin Invest. 121:893–904. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hollstein M, Sidransky D, Vogelstein B and
Harris CC: p53 mutations in human cancers. Science. 253:49–53.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Creighton CJ, Gibbons DL and Kurie JM: The
role of epithelial-mesenchymal transition programming in invasion
and metastasis: A clinical perspective. Cancer Manag Res.
5:187–195. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sheppard D: Epithelial-mesenchymal
interactions in fibrosis and repair. Transforming growth factor-β
activation by epithelial cells and fibroblasts. Ann Am Thorac Soc.
12:(Suppl 1). S21–S23. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kovacic JC, Mercader N, Torres M, Boehm M
and Fuster V: Epithelial-to-mesenchymal and
endothelial-to-mesenchymal transition: From cardiovascular
development to disease. Circulation. 125:1795–1808. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fraga CH, True LD and Kirk D: Enhanced
expression of the mesenchymal marker, vimentin, in hyperplastic
versus normal human prostatic epithelium. J Urol. 159:270–274.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang L, Lai YK, Zhang J, Wang H, Lin MC,
He ML and Kung HF: Targeting S100P inhibits colon cancer growth and
metastasis by lentivirus-mediated RNA interference and proteomic
analysis. Mol Med. 17:709–716. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang L, Chen Y, Chan CY, Wang X, Lin L,
He ML, Lin MC, Yew DT, Sung JJ, Li JC, et al: Down-regulation of
stathmin is required for TGF-beta inducible early gene 1 induced
growth inhibition of pancreatic cancer cells. Cancer Lett.
274:101–108. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Peacock O, Lee AC, Cameron F, Tarbox R,
Vafadar-Isfahani N, Tufarelli C and Lund JN: Inflammation and
MiR-21 pathways functionally interact to downregulate PDCD4 in
colorectal cancer. PLoS One. 9:e1102672014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang J, Fei B, Wang Q, Song M, Yin Y,
Zhang B, Ni S, Guo W, Bian Z, Quan C, et al: MicroRNA-638 inhibits
cell proliferation, invasion and regulates cell cycle by targeting
tetraspanin 1 in human colorectal carcinoma. Oncotarget.
5:12083–12096. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang J, Paris PL, Chen J, Ngo V, Yao H,
Frazier ML, Killary AM, Liu CG, Liang H, Mathy C, et al: Next
generation sequencing of pancreatic cyst fluid microRNAs from low
grade-benign and high grade-invasive lesions. Cancer Lett.
356:404–409. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kasinski AL and Slack FJ: Epigenetics and
genetics. MicroRNAs en route to the clinic: Progress in validating
and targeting microRNAs for cancer therapy. Nat Rev Cancer.
11:849–864. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stahlhut C and Slack FJ: MicroRNAs and the
cancer phenotype: Profiling, signatures and clinical implications.
Genome Med. 5:1112013. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Friedman RC, Farh KK-H, Burge CB and
Bartel DP: Most mammalian mRNAs are conserved targets of microRNAs.
Genome Res. 19:92–105. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Christofori G: New signals from the
invasive front. Nature. 441:444–450. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Berx G, Raspé E, Christofori G, Thiery JP
and Sleeman JP: Pre-EMTing metastasis? Recapitulation of
morphogenetic processes in cancer. Clin Exp Metastasis. 24:587–597.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelial-mesenchymal transition: New insights in
signaling, development, and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schipper JH, Frixen UH, Behrens J, Unger
A, Jahnke K and Birchmeier W: E-cadherin expression in squamous
cell carcinomas of head and neck: Inverse correlation with tumor
dedifferentiation and lymph node metastasis. Cancer Res.
51:6328–6337. 1991.PubMed/NCBI
|
|
26
|
Oka H, Shiozaki H, Kobayashi K, Inoue M,
Tahara H, Kobayashi T, Takatsuka Y, Matsuyoshi N, Hirano S,
Takeichi M, et al: Expression of E-cadherin cell adhesion molecules
in human breast cancer tissues and its relationship to metastasis.
Cancer Res. 53:1696–1701. 1993.PubMed/NCBI
|
|
27
|
Perl A-K, Wilgenbus P, Dahl U, Semb H and
Christofori G: A causal role for E-cadherin in the transition from
adenoma to carcinoma. Nature. 392:190–193. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vleminckx K, Vakaet L Jr, Mareel M, Fiers
W and van Roy F: Genetic manipulation of E-cadherin expression by
epithelial tumor cells reveals an invasion suppressor role. Cell.
66:107–119. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Frixen UH, Behrens J, Sachs M, Eberle G,
Voss B, Warda A, Löchner D and Birchmeier W: E-cadherin-mediated
cell-cell adhesion prevents invasiveness of human carcinoma cells.
J Cell Biol. 113:173–185. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gumbiner BM: Regulation of
cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol.
6:622–634. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ivaska J, Vuoriluoto K, Huovinen T, Izawa
I, Inagaki M and Parker PJ: PKCepsilon-mediated phosphorylation of
vimentin controls integrin recycling and motility. EMBO J.
24:3834–3845. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vuoriluoto K, Haugen H, Kiviluoto S,
Mpindi JP, Nevo J, Gjerdrum C, Tiron C, Lorens JB and Ivaska J:
Vimentin regulates EMT induction by Slug and oncogenic H-Ras and
migration by governing Axl expression in breast cancer. Oncogene.
30:1436–1448. 2011. View Article : Google Scholar : PubMed/NCBI
|