Introduction
Superoxide anion (O2•−), hydroxyl radical
(•OH) and hydrogen peroxide (H2O2) are
unstable and highly reactive oxygen species (ROS). Although ROS are
conventionally harmful or detrimental to cells, they specifically
regulate a variety of cellular procedures such as cell
proliferation, differentiation and apoptosis (1,2). ROS
are persistently produced during the respiratory chain reaction
during oxidative phosphorylation in the form of O2•−
and/or are intentionally generated by specific oxidase enzymes
(3). O2•− is converted
into H2O2 by superoxide dismutase (4). H2O2 is further
metabolized into O2 and H2O by catalase or
glutathione (GSH) peroxidase (5).
Compared to other types of ROS, H2O2 is
nonradical and soluble in both lipid and aqueous surroundings. It
freely diffuses all the way through the cell membrane to reach
remote cells and interacts with ferrous iron (Fenton reaction),
resulting in the generation of the tremendously violent and
short-lived •OH. Since the production of different forms of ROS at
numerous levels can be either useful or harmful to cells and
tissues, a redox state is steadfastly controlled to avoid cell and
tissue damage. Enhanced oxidative stress as a consequence of either
overproduction of ROS and/or downregulation of antioxidants leads
to apoptotic cell death via injury to cellular DNA, proteins and
lipids and is associated with a variety of pathological conditions
such as inflammation and immune responses (6,7).
Mitogen-activated protein kinases (MAPKs) are
evolutionarily conserved signaling proteins that mediate responses
to assorted stimuli. Extracellular signal regulated kinases
(ERK1/2), the c-Jun N-terminal kinase/stress-activated protein
kinases (JNK/SAPK) and the p38 kinases are the three main MAPK
groups in mammalians/eukaryotes (8). Each MAPK pathway has comparatively
unrelated upstream activators and specific substrates (9). The trigger for multiple MAPK pathways
includes major constituents of signaling pathways in cell growth,
cell death and differentiation (10). MAPKs can discriminate the cellular
redox status and are common targets for ROS themselves. For
instance, JNK and p38 are normally activated by a soft oxidative
stress and their activation induces apoptosis in cells (11,12).
Furthermore, ROS can stimulate the ERK pathway by specific
phosphorylation of the ERK enzyme (13). As a general rule, the activation of
ERK is involved in cell survival rather than cell death (14). In addition, the activity of MAPKs is
persistent by means of the subordinate activity of MAPK
phosphatases, which are directly controlled by
H2O2 (15).
Lung cancer is a major cause of cancer-related
mortality in developed countries. The carcinogenesis of lung cancer
is closely related to tissue inflammation mediated by ROS. During
inflammation, the concentration of H2O2 in
tissues is anticipated to reach approximately millimolar levels
(16,17). Moderate H2O2
levels may adjust important cellular events, such as cell growth
and differentiation, by altering signaling cascades and gene
expression and its upper levels can elicit cell death via apoptosis
or necrosis. The lung is particularly prone to an assortment of
blood- and air-borne damage, which may consequently induce lung
fibrosis and cancer (18).
Exogenous H2O2 is frequently used to provoke
oxidative stress in cells and tissues. Because different ROS levels
and cellular roles of MAPKs modulated by ROS may have opposing
properties, even within the same cell type, the relationship
between ROS and MAPK signaling with respect to cell growth and
death needs to be further clarified. Using specific MAPK inhibitors
[JNK inhibitor (SP600125), MEK inhibitor (PD98059) and p38
inhibitor (SB203580)], the present study sought to elucidate the
roles of different MAPKs in H2O2-treated
Calu-6 and A549 lung cancer cells in relation to cell growth and
death as well as investigate changes in ROS and GSH levels.
Materials and methods
Cell culture
The human lung cancer cell lines, Calu-6 and A549,
were obtained from the Korean Cell Line Bank (Seoul, Korea) and
cultured in RPMI-1640 medium (GE Healthcare Life Sciences, Logan,
UT, USA) with 10% fetal bovine serum (FBS; Sigma-Aldrich; Merck
KGaAA, Darmstadt, Germany) and 1% penicillin-streptomycin (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). The cells were
routinely cultivated in 100-mm plastic tissue culture dishes (Nalge
Nunc International, Penfield, NY, USA) in a humidified incubator
containing 5% CO2, at 37°C and harvested in a solution
of trypsin-EDTA (Gibco; Thermo Fisher Scientific, Inc.) while in a
logarithmic phase of growth.
Reagents
H2O2 was purchased from
Sigma-Aldrich; Merck KGaAA. The MEK inhibitor (PD98059) as well as
the JNK (SP600125) and p38 (SB203580) inhibitors were obtained from
Calbiochem (San Diego, CA, USA). All reagents were dissolved in
dimethyl sulfoxide (DMSO; Sigma-Aldrich; Merck KGaAA) at 10 mM. The
cells were pretreated with each MAPK inhibitor for 30 min prior to
treatment with H2O2. Based on previous
experiments (19–21), 10 µM of each MAPK inhibitor was
applied as an optimal dose in all experiments.
Cell growth assay
The effect of the drugs on lung cancer cell growth
was determined by evaluating
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma-Aldrich; Merck KGaAA) dye absorbance as previously described
(19–21). The cells were exposed to 75 or 100
µM H2O2 with or without 10 µM MEK, JNK or p38
inhibitor for 24 h.
Sub-G1 cell analysis
Sub-G1 cells were detected using propidium iodide
dye (PI; Sigma-Aldrich; Merck KGaA) as previously described
(20,21). The cells were exposed to 75 or 100
µM H2O2 in the presence or absence of 10 µM
MEK, JNK or p38 inhibitor for 24 h. The cell DNA content was
assessed by a FACStar flow cytometer (BD Biosciences, Franklin
Lakes, NJ, USA) and analyzed using Lysis II and Cellfit software
version 2.0 (BD Biosciences).
Annexin V staining for cell death
detection
Apoptotic cell death was evaluated by measuring cell
stained with Annexin V-fluorescein isothiocyanate (FITC; Molecular
Probes; Thermo Fisher Scientific, Inc.) as previously described
(19–21). The cells were exposed to 75 or 100
µM H2O2 with or without 10 µM MEK, JNK, or
p38 inhibitor for 24 h. Annexin V staining was analyzed with a
FACStar flow cytometer (BD Biosciences).
Measurement of mitochondrial membrane
potential (MMP; ΔΨm)
MMP was assessed using Rhodamine 123
mitochondrial-specific fluorescent dye (Sigma-Aldrich; Merck KGaAA)
as previously described (19–21).
The cells were exposed to 75 or 100 µM H2O2
in the presence or absence of 10 µM MEK, JNK, or p38 inhibitor for
24 h. Rhodamine 123 staining intensity was assessed by a FACStar
flow cytometer (BD Biosciences). An absence of Rhodamine 123 from
the cells indicated a loss of MMP in lung cancer cells. Similarly
to previous experiments (19–21),
the MMP levels in the cells excluding MMP-loss cells were expressed
as the mean fluorescence intensity (MFI), which was estimated by
CellQuest Pro software (version 5.1; BD Biosciences).
Measurement of intracellular ROS
levels
The intracellular ROS levels were evaluated by a
fluorescent probe dye, 2′,7′-dichlorodihydrofluorescein diacetate
(H2DCFDA; Molecular Probes; Thermo Fisher Scientific,
Inc.) at 1 or 24 h as previously described (19–21).
Dihydroethidium (DHE; Molecular Probes; Thermo Fisher Scientific,
Inc.) is a fluorogenic probe specific to O2•− in ROS. In
brief, the cells were treated with 75 or 100 µM
H2O2 in the presence or absence of 10 µM MEK,
JNK or p38 inhibitor in the presence of 20 µM H2DCFDA or
DHE. The levels of DCF (ROS) and DHE (O2•−) fluorescence
were assessed using a FACStar flow cytometer (BD Biosciences) at 1
h and expressed as MFI, as calculated by CellQuest Pro software
(version 5.1; BD Biosciences). Additionally, the cells were
incubated with 75 or 100 µM H2O2 in the
presence or absence of each MAPK inhibitor for 24 h. The cells were
incubated with 20 µM H2DCFDA or DHE at 37°C for 30 min.
H2DCFDA and DHE fluorescence was analyzed using a
FACStar flow cytometer (BD Biosciences).
Detection of intracellular GSH
The GSH levels were evaluated by means of a
5-chloromethylfluorescein diacetate dye (CMFDA; Molecular Probes;
Thermo Fisher Scientific, Inc.) at 1 or 24 h, as previously
described (19,21). In brief, the cells were treated with
75 or 100 µM H2O2 with or without 10 µM MEK,
JNK, or p38 inhibitor in the presence of 5 µM CMFDA. The CMF
fluorescence was calculated using a FACStar flow cytometer (BD
Biosciences) at 1 h. The CMF (GSH) levels were expressed as MFI,
which was assessed by CellQuest Pro software (version 5.1; BD
Biosciences). In addition, the cells were incubated with 75 or 100
µM H2O2 in the presence or absence of each
MAPK inhibitor for 24 h. The CMF fluorescence intensity was
measured by the FACStar flow cytometer (BD Biosciences). Negative
CMF staining (GSH-depleted) cells were expressed as the percentage
of cells. Similar to previous experiments (19,21),
the CMF levels in the cells excluding GSH-depleted cells were
expressed as MFI.
Statistical analysis
The results represent the mean of at least two
independent experiments (mean ± SD). The Student's t-test or
one-way ANOVA with post hoc analysis using Tukey's multiple
comparison tests was used for parametric data. The results were
considered statistically significant at P<0.05.
Results
MAPK inhibitors affect cell growth and
death in H2O2-treated lung cancer cells
The cell growth and death effects of MAPK inhibitors
(MEK, JNK and p38 inhibitors) were examined in
H2O2-treated lung cancer cells. The same
inhibitors were used to block MAPK signaling pathways in previous
studies (19–21). After exposure to
H2O2 for 24 h, the half maximal inhibitory
concentration (IC50) in Calu-6 and A549 cells was ~50 and 100 µM
based on MTT assay, respectively. In addition,
H2O2 dose-dependently increased the number of
Annexin V-FITC-positive Calu-6 and A549 cells (data not shown).
Doses of 75 or 100 µM H2O2 were chosen to
distinguish the differences in cell growth inhibition and death in
each cell line in the presence or absence of each MAPK inhibitor.
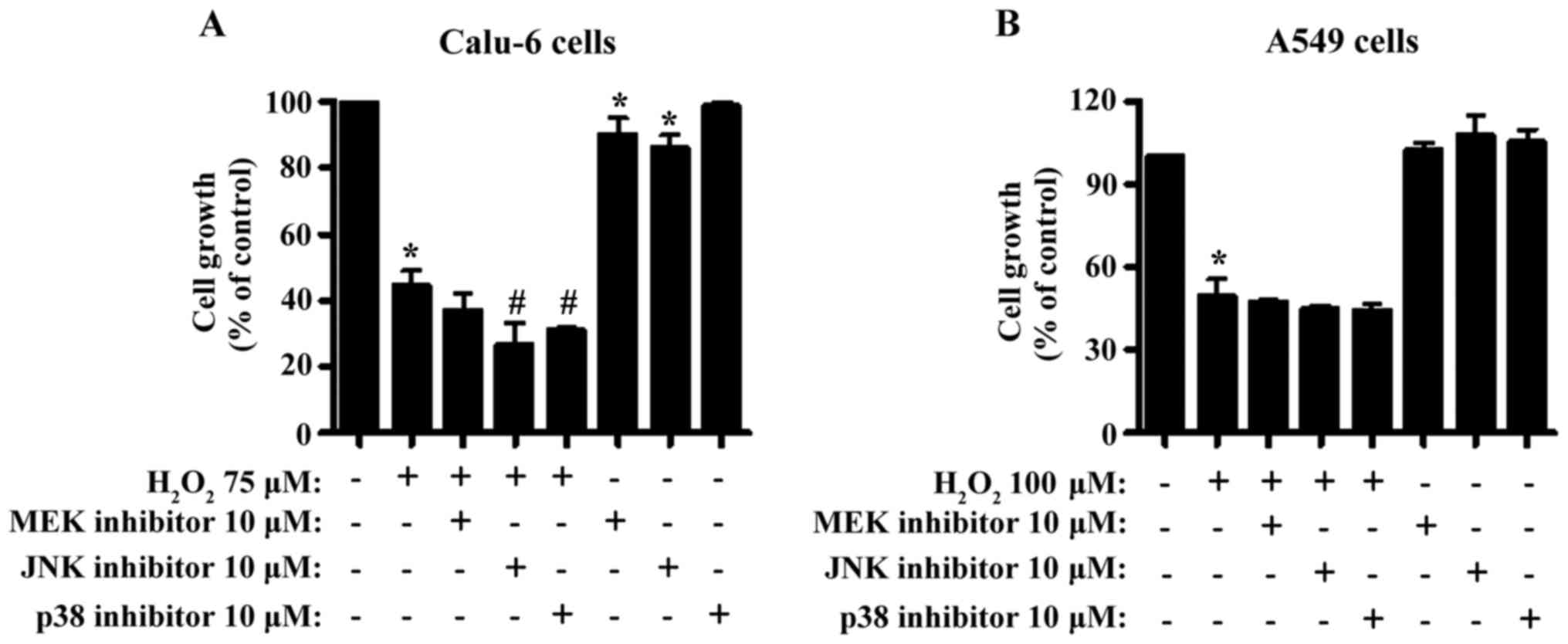
Treatment with 75 µM H2O2 caused ~60% growth
inhibition in Calu-6 cells within 24 h (Fig. 1A). All MAPK inhibitors enhanced
growth inhibition and JNK and p38 inhibitors exhibited significant
effects (Fig. 1A). Both MEK and JNK
inhibitors significantly diminished the growth of Calu-6 control
cells (Fig. 1A). Treatment with 75
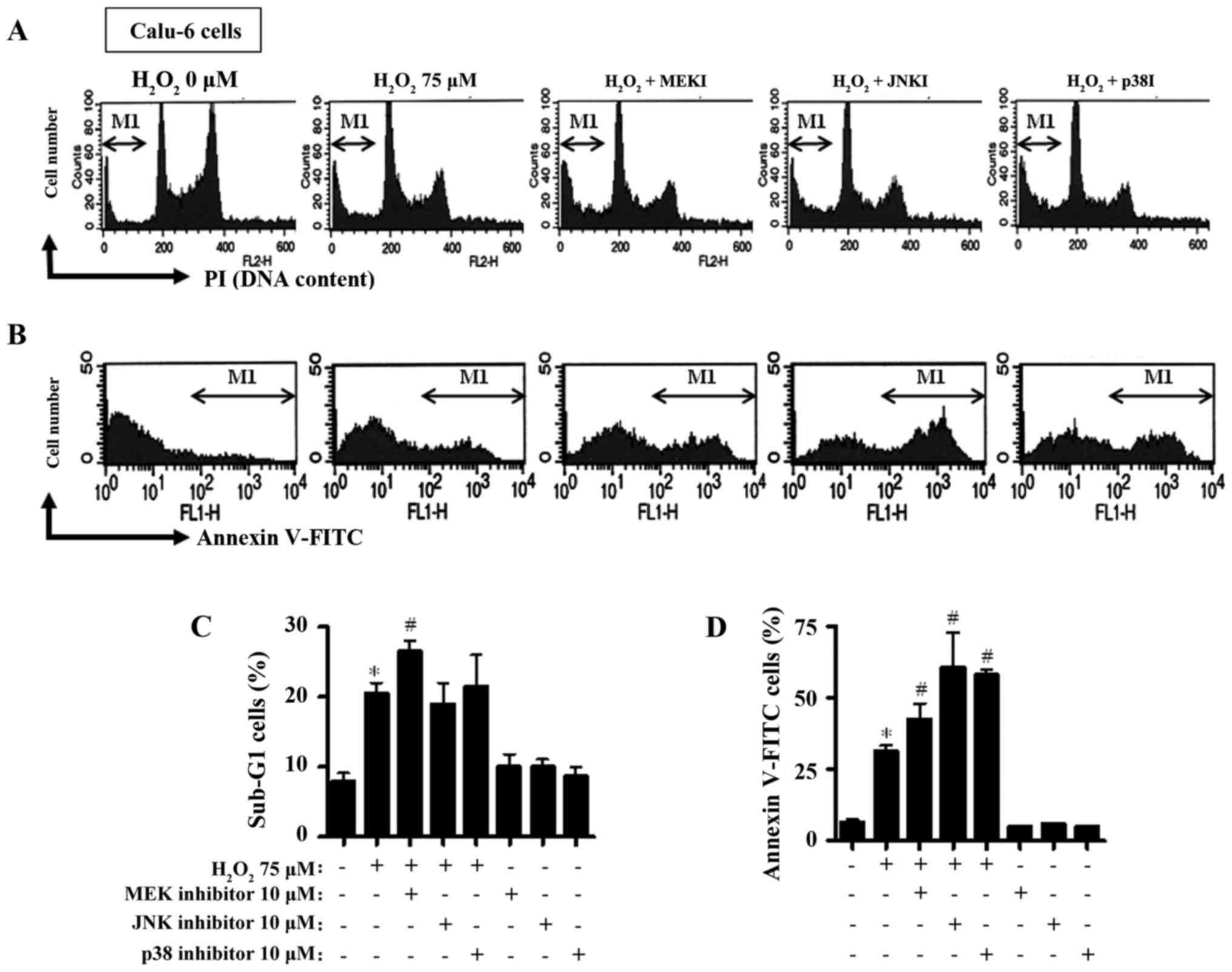
µM H2O2 increased the percentage of the
sub-G1 cells to ~20% (Fig. 2A and
C). The MEK inhibitor exhibited a robust trend towards an
increased number of sub-G1 cells in the
H2O2-treated Calu-6 cells (Fig. 2A and C). Neither the JNK nor the p38
inhibitor significantly altered the numbers of the sub-G1 cells in
these cells (Fig. 2A and C). In
addition, the H2O2 treatment increased the
percentage of Annexin V-FITC-positive stained Calu-6 cells
(Fig. 2B and D). All the inhibitors
increased the percentage of Annexin V-FITC-positive
H2O2-treated Calu-6 cells (Fig. 2B and D).
Using another lung cancer cell line, the A549 cells,
a 100 µM H2O2-treatment induced growth
inhibition to ~50% within 24 h (Fig.
1B). None of the MAPK inhibitors significantly changed the
growth of the H2O2-treated and untreated
control cells (Fig. 1B). Treatment
with 100 µM H2O2 alone increased the
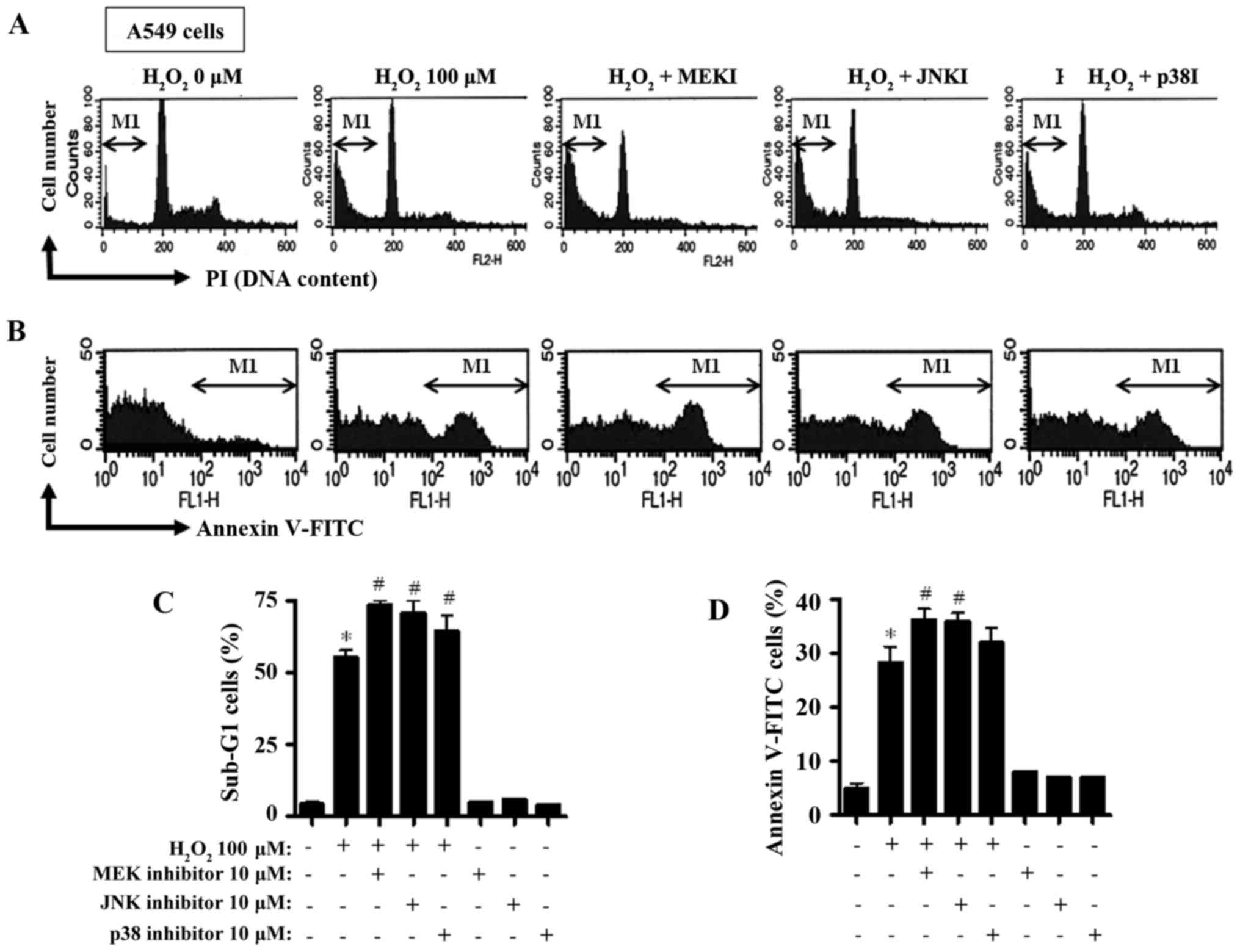
percentage of the sub-G1 cells to ~50% compared to the
H2O2-untreated control cells (Fig. 3A and C). All inhibitors further
augmented the percentage of the sub-G1 cells in the
H2O2-treated cells (Fig. 3A and C), with the MEK inhibitor
being the most potent. Furthermore, the H2O2
treatment increased the percentage of Annexin V-FITC-positive
stained A549 cells (Fig. 3B and D).
All inhibitors augmented the percentage of Annexin V-FITC-positive
H2O2-treated cells (Fig. 3B and D), and both MEK and JNK
inhibitors exhibited significant enhancement (Fig. 3B and D).
MAPK inhibitors influence MMP in
H2O2-treated lung cancer cells
Cell death is closely correlated with the collapse
of MMP (22). Thus, MMP in
H2O2-treated Calu-6 and A549 cells was
determined in the presence or absence of each MAPK inhibitor using
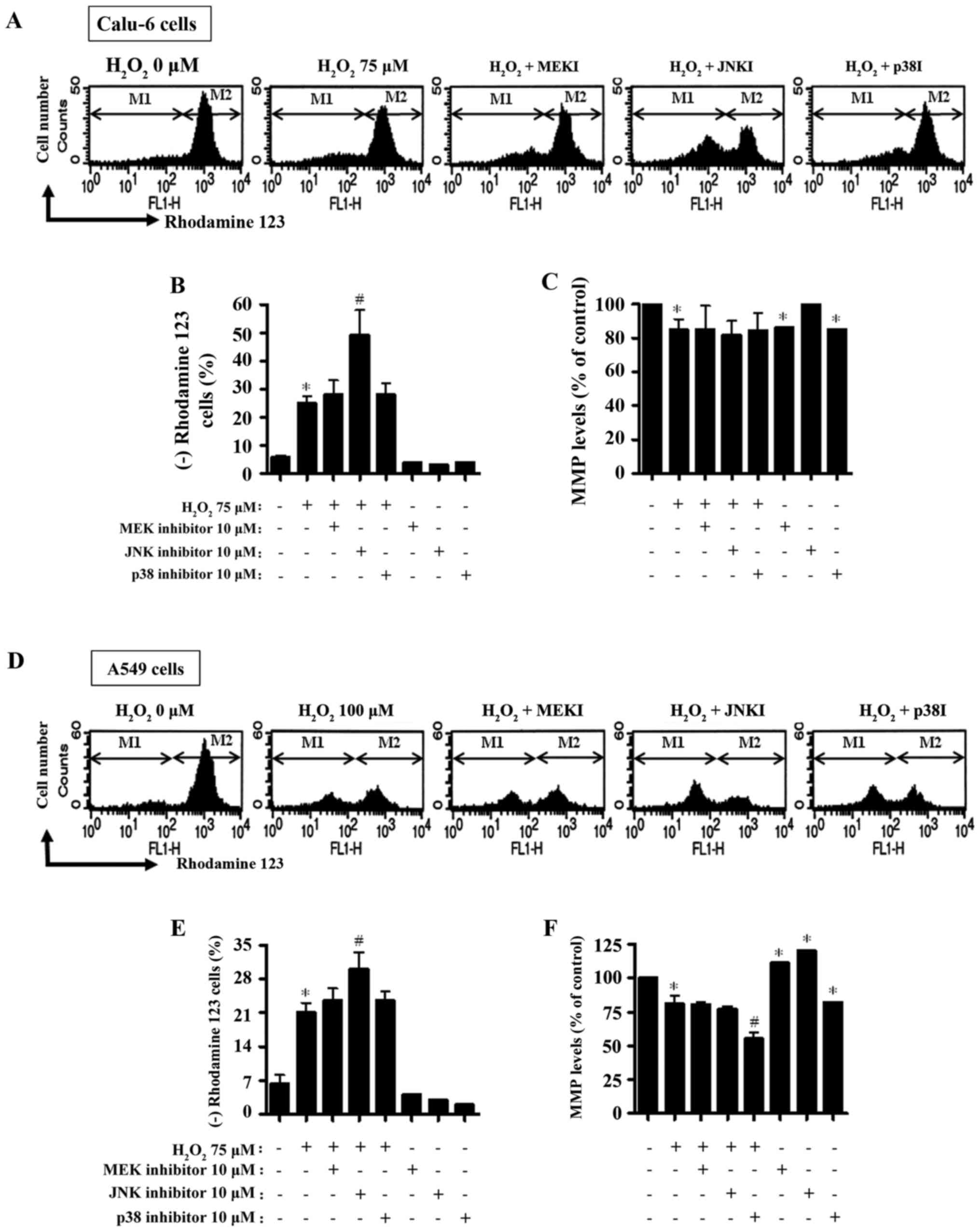
Rhodamine 123 dye at 24 h. As expected, the loss of MMP was
significantly detected in H2O2-treated Calu-6
cells (Fig. 4A and B). The MEK and
p38 inhibitors slightly increased the loss of MMP in
H2O2-treated Calu-6 cells, whereas the JNK
inhibitor significantly boosted the loss of MMP (Fig. 4A and B). With regard to the MMP
levels in lung cancer cells not including negative Rhodamine 123
staining cells, H2O2 decreased the MMP level
in Calu-6 cells (Fig. 4A and C).
None of the MAPK inhibitors influenced the MMP levels in
H2O2-treated Calu-6 cells (Fig. 4A and C). The MEK and p38 inhibitors
significantly reduced the MMP levels in Calu-6 control cells
(Fig. 4A and C). Regarding the A549
cells, significant loss of MMP was observed in
H2O2-treated cells (Fig. 4D and E). All inhibitors augmented
the loss of MMP in H2O2-treated A549 cells
and the JNK inhibitor had a significant effect (Fig. 4D and F). While the MEK and JNK
inhibitors did not alter the MMP levels in
H2O2-treated A549 cells, the p38 inhibitor
significantly enhanced the decrease in MMP levels in these cells
(Fig. 4D and F). MEK and JNK
inhibitors both significantly increased the MMP levels in
H2O2-untreated A549 control cells, but the
p38 inhibitor significantly decreased the MMP level in A549 control
cells (Fig. 4D and F).
MAPK inhibitors alter ROS levels
including O2•− in H2O2-treated
lung cancer cells
Changes in ROS levels were assessed in Calu-6 and
A549 cells treated with H2O2 with or without
each MAPK inhibitor. To determine whether the intracellular ROS
levels in H2O2-treated lung cancer cells were
altered by treatment with each MAPK inhibitor, ROS levels were
assessed at the early time-point of 1 h (Fig. 5) and at the later time-point of 24 h
(Fig. 6).
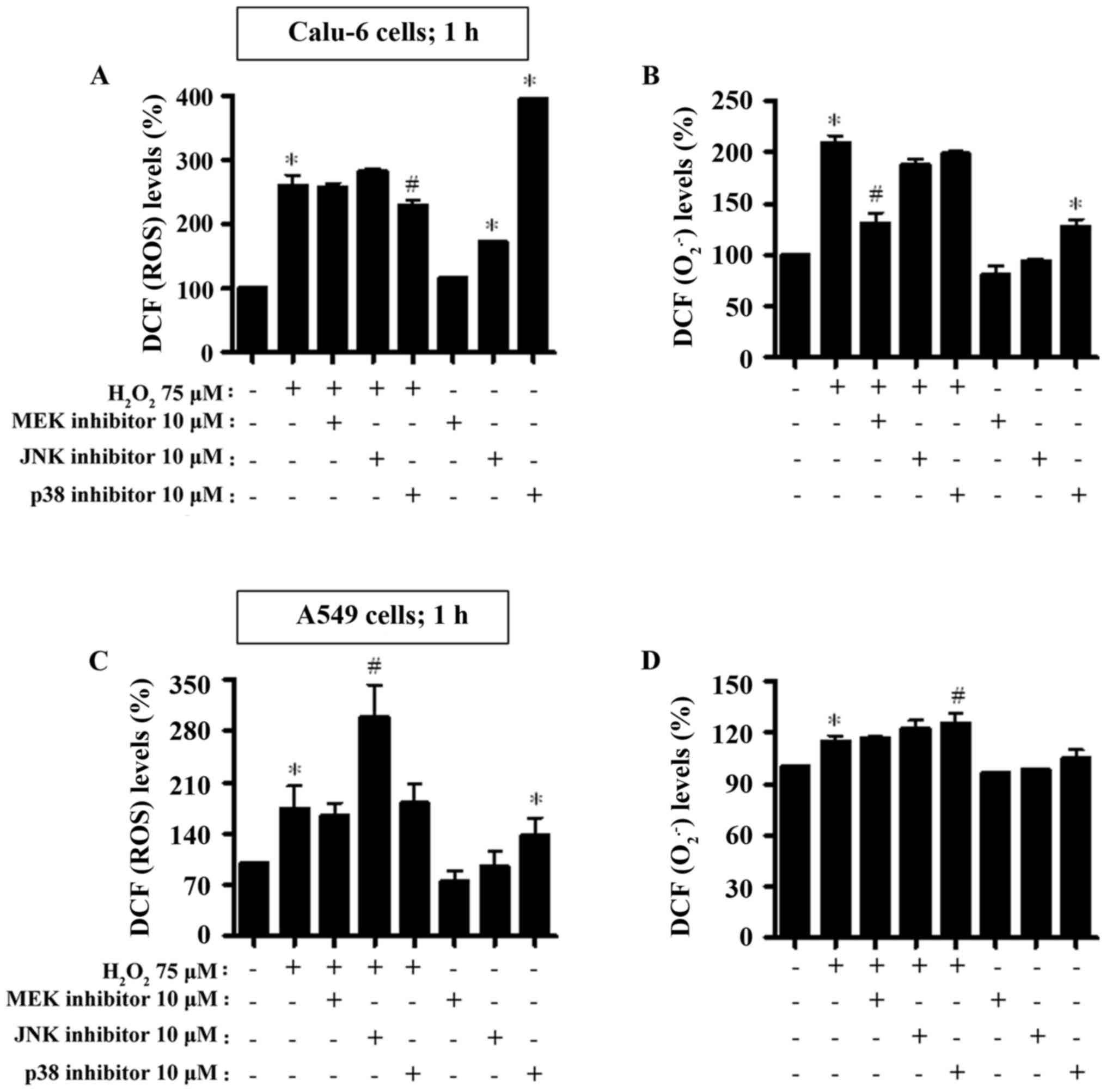
As depicted in Fig. 5A
and C, intracellular ROS (DCF) levels were significantly
increased in Calu-6 and A549 cells treated with
H2O2 at 1 h. The JNK inhibitor increased the
ROS (DCF) level in H2O2-treated Calu-6 cells,
whereas the p38 inhibitor decreased the level in these cells
(Fig. 5A). Both the JNK and p38
inhibitors significantly increased basal ROS (DCF) levels in Calu-6
control cells (Fig. 5A). In
H2O2-treated A549 cells, the JNK inhibitor
exhibited a strong significant increase in ROS (DCF) level
(Fig. 5C). The MEK inhibitor
decreased the basal ROS (DCF) level in A549 control cells, but the
p38 inhibitor significantly increased the basal level (Fig. 5C). When the O2•− levels
in H2O2-treated Calu-6 cells were assessed,
the level of red fluorescence derived from DHE reflecting
intracellular O2•− was significantly increased (Fig. 5B). All MAPK inhibitors, especially
the MEK inhibitor, exhibited a strong reduction of DHE
(O2•−) levels in H2O2-treated
Calu-6 cells (Fig. 5B). In
addition, the MEK inhibitor decreased the basal level of DHE
(O2•−) in Calu-6 control cells and the p38 inhibitor
increased the basal level (Fig.
5B). Treatment with 100 µM H2O2 slightly
increased DHE (O2•−) levels in the A549 cells at 1 h
(Fig. 5D). JNK and p38 inhibitors
appeared to augment the DHE (O2•−) levels in
H2O2-treated A549 cells (Fig. 5D). The MEK and JNK inhibitors
reduced basal levels in H2O2-untreated A549
control cells (Fig. 5D).
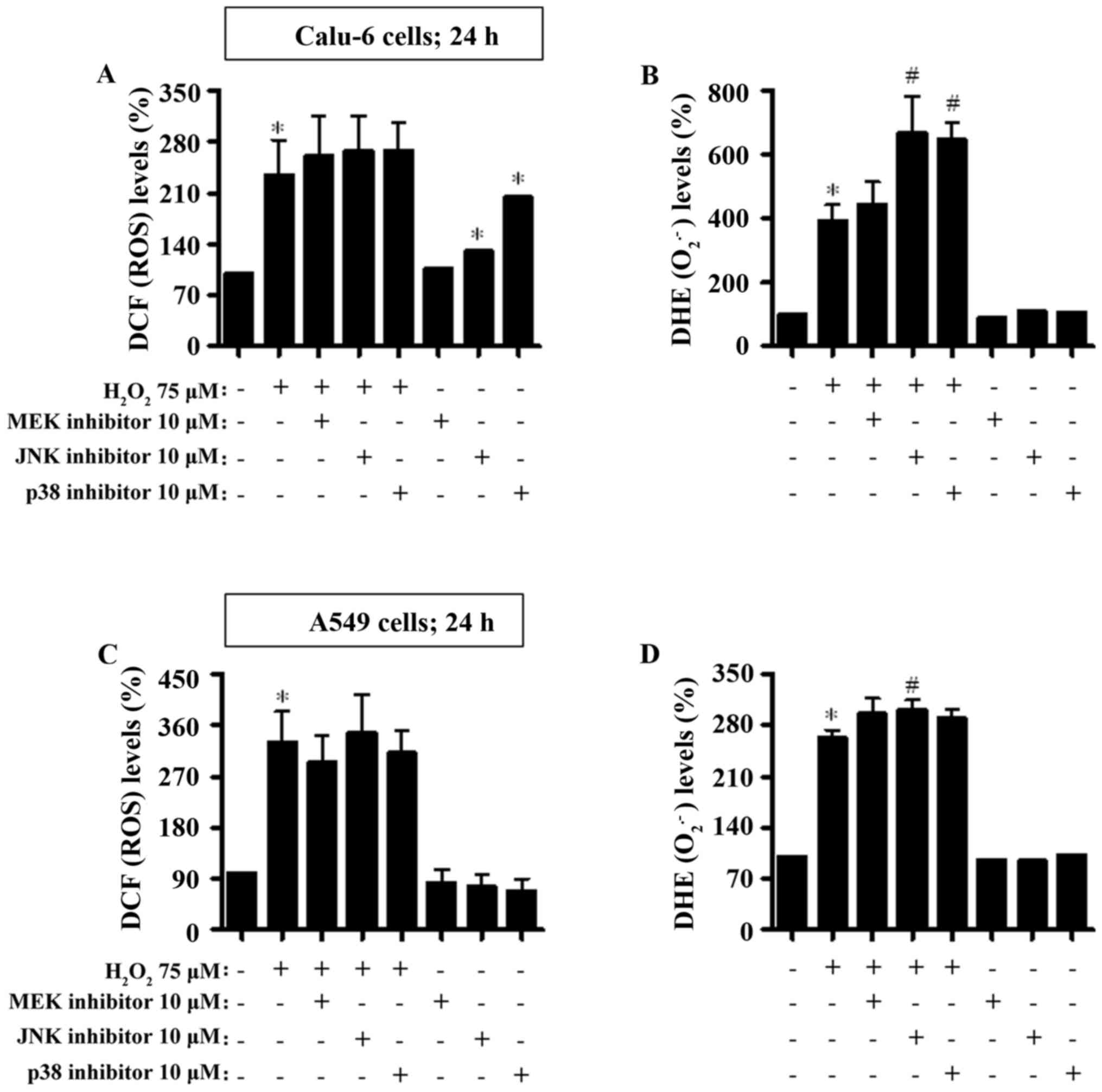
Treatment with 75 and 100 µM
H2O2 increased the intracellular ROS (DCF and
DHE) levels in Calu-6 and A549 cells at 24 h (Fig. 6). None of the MAPK inhibitors
significantly affected ROS (DCF) levels in
H2O2-treated Calu-6 and A549 cells (Fig. 6A and C). The JNK and p38 inhibitors
increased ROS (DCF) levels in H2O2-untreated
Calu-6 control cells (Fig. 6A), but
both inhibitors decreased the levels in
H2O2-untreated A549 control cells (Fig. 6C). Furthermore, the JNK and p38
inhibitors significantly augmented DHE (O2•−) levels in
H2O2-treated Calu-6 cells (Fig. 6B). All MAPK inhibitors increased the
DHE (O2•−) levels in H2O2-treated
A549 cells (Fig. 6D).
MAPK inhibitors change GSH levels in
H2O2-treated lung cancer cells
Changes in GSH levels were assessed in Calu-6 and
A549 cells treated with H2O2 with or without
each MAPK inhibitor at 1 h (Fig. 7)
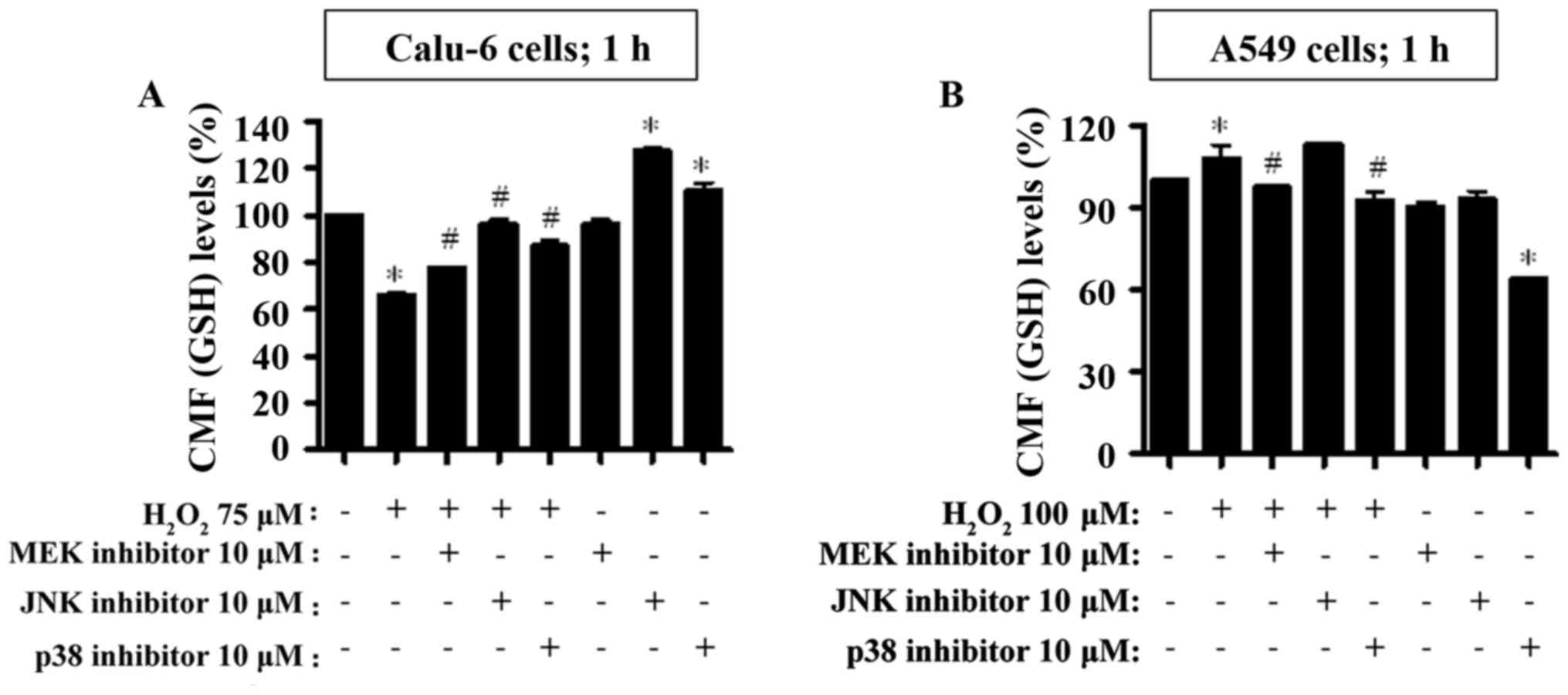
and at 24 h (Fig. 8). Treatment
with 75 µM H2O2 strongly decreased the GSH
levels in Calu-6 cells at 1 h (Fig.
7A). All MAKP inhibitors significantly increased the GSH levels
in the H2O2-treated Calu-6 cells (Fig. 7A). In addition, the JNK and p38
inhibitors augmented the basal levels of GSH in Calu-6 control
cells (Fig. 7A). However, 100 µM
H2O2 increased the GSH levels in A549 cells
at 1 h (Fig. 7B). The MEK and p38
inhibitors attenuated the GSH levels in
H2O2-treated A549 cells (Fig. 7B). All MAPK inhibitors decreased the
GSH levels in H2O2-untreated A549 control
cells and the decrease was more significant upon treatment with the
p38 inhibitor (Fig. 7B).
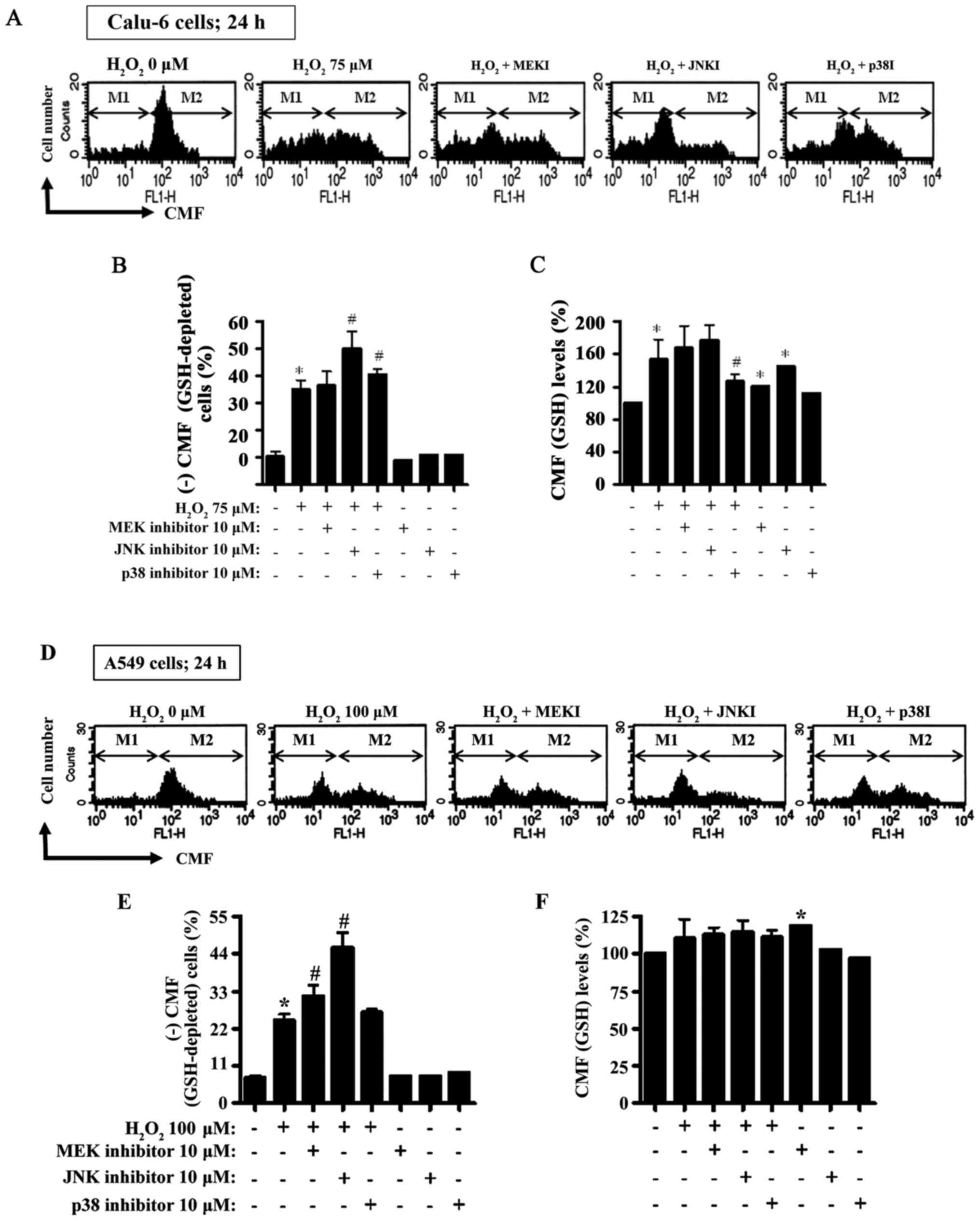
Treatment with 75 µM H2O2 for
24 h resulted in an increase to ~25% in the GSH-depleted Calu-6
cells compared to the H2O2-untreated control
cells (Fig. 8A and B). Unlike the
MEK inhibitor, both the JNK and p38 inhibitors significantly
increased GSH-depleted cell numbers in
H2O2-treated Calu-6 cells (Fig. 8A and B). Furthermore, when the CMF
(GSH) levels in Calu-6 cells (not including negative CMF-staining
cells) were assessed at 24 h, the GSH levels exhibited an increase
in H2O2-treated Calu-6 cells (Fig. 8A and C). Both the MEK and JNK
inhibitors did not significantly alter GSH levels in
H2O2-treated Calu-6 cells, but the p38
inhibitor significantly attenuated the GSH level in these cells
(Fig. 8A and C). All inhibitors
increased the GSH levels in H2O2-untreated
Calu-6 control cells and especially the JNK inhibitor expressed a
strong increase (Fig. 8A and C). In
relation to A549 cells, 100 µM H2O2 resulted
in an increase in the number of GSH-depleted A549 cells to ~16% at
24 h compared to the H2O2-untreated control
cells (Fig. 8D and E). The MEK and
JNK inhibitors significantly enhanced the GSH deletion in
H2O2-treated A549 cells (Fig. 8D and E). Treatment with 100 µM
H2O2 did not significantly change the level
of GSH at 24 h (Fig. 8D and F).
None of the MAPK inhibitors altered the level of GSH in
H2O2-treated A549 cells (Fig. 8D and F). The MEK inhibitor
significantly increased the basal level of GSH in A549 control
cells, whereas the p38 inhibitor reduced the level of GSH (Fig. 8D and F).
Discussion
Treatment with 75 and 100 µM
H2O2 increased the number of sub-G1 and
Annexin V-FITC-positive cells in Calu-6 and A549 lung cancer cells,
accompanied by the downregulation of Bcl-2 and procaspase-3
proteins and activation of caspase-3 and −8 (data not shown). These
results indicated that H2O2 induced cell
death in these lung cancer lines via caspase-dependent apoptosis.
The present study focused on evaluating the effects of MAPK
inhibitors on cell growth and cell death as well as intracellular
ROS and GSH levels in H2O2-treated lung
cancer cells.
Usually, the ERK signaling pathway is pro-survival
rather than pro-apoptotic (14).
The MEK inhibitor, which presumably inactivates ERK, appeared to
enhance growth inhibition in H2O2-treated
Calu-6 cells. The MEK inhibitor significantly increased the number
of sub-G1 and Annexin V-FITC-positive
H2O2-treated Calu-6 and A549 cells. Thus,
H2O2 seemed to inactivate ERK in lung cancer
cells, resulting in growth inhibition and apoptosis. In most cases,
the JNK and p38 activities are stimulated by ROS or a mild
oxidative change in the intracellular thiol/disulfide redox
condition, which is positively involved in the induction of
apoptosis (11,12). However, both JNK and p38 inhibitors
augmented the growth inhibition in
H2O2-treated Calu-6 cells. In addition, both
inhibitors significantly increased cell death in
H2O2-treated Calu-6 and A549 cells.
Therefore, in H2O2-treated lung cancer cells,
the JNK and p38 signaling pathways are probably associated with
cell growth or survival rather than cell death. Similar to these
results, the JNK and p38 inhibitors enhanced the death of HeLa
cervical cancer cells treated with 100 µM
H2O2 (23).
Interestingly, JNK and p38 inhibitors exhibited a significant
increase in Annexin V-FITC-positive cells but did not increase the
number of sub-G1 cells in H2O2-treated Calu-6
cells. Thus, the suppression of the JNK and p38 signaling pathways
by each inhibitor induced necrotic cell death in
H2O2-treated Calu-6 cells. In addition, none
of the MAPK inhibitors significantly affected cell growth
inhibition in H2O2-treated A549 cells,
indicating that the MAPK signaling pathways are more associated
with cell survival rather than cell growth. Furthermore, the MEK
and JNK inhibitors reduced the growth of the Calu-6 control but not
of the A549 control cells. These inhibitors differentially
influenced the growth of Calu-6 and A549 lung cancer cells.
ROS can augment the disturbance of redox status in
cells by triggering a breakdown in MMP (24). Correspondingly,
H2O2 induced the loss of MMP in lung cancer
cells. Similar to the percentage of Annexin V-FITC-positive cells,
the JNK inhibitor strongly increased the loss of MMP in
H2O2-treated lung cancer cells. However, the
MEK and p38 inhibitors slightly enhanced the loss in these cells.
These results indicated that JNK signaling is tightly involved in
the maintenance of an intact MMP in lung cancer cells. The MMP
levels in Rhodamine 123-positive cells revealed that
H2O2 reduced the MMP levels in lung cancer
cells. Only the p38 inhibitor enhanced a decrease in MMP levels in
H2O2-treated A549 cells. In addition, the MEK
inhibitor decreased the basal MMP level in
H2O2-untreated Calu-6 cells but increased the
basal level in A549 control cells. The JNK inhibitor increased the
basal MMP level in H2O2-untreated A549 cells.
The p38 inhibitor reduced the basal MMP levels in both Calu-6 and
A549 control cells. These results indicated that each MAPK
signaling pathway had different and specific effects on MMP in
Calu-6 and A549 lung cancer cells.
The main ROS related to cell signaling pathways are
O2•− and H2O2. Intracellular ROS
levels, including O2•−,were significantly increased in
both lung cancer cells treated with H2O2 at 1
and 24 h. Treatment with 75 and 100 µM H2O2
directly produced O2•− by impairing the mitochondrial
membrane function and both H2O2 and
O2•− can be efficiently converted into toxic •OH via the
Fenton reaction to destroy these cancer cells. However,
H2O2 slightly increased O2•− (DHE)
levels in A549 cells compared to Calu-6 cells at 1 h, indicating
that it does not have a strong effect on both mitochondrial
respiratory transport chain and the activity of various oxidases to
generate O2•− in A549 cells within this early
time-point. Only the JNK inhibitor significantly enhanced the
increased ROS (DCF) levels in H2O2-treated
Calu-6 and A549 cells at 1 h. Both JNK and p38 inhibitors increased
O2•− (DHE) levels in H2O2-treated
A549 cells at 1 h. In contrast, the MEK inhibitor decreased
O2•− (DHE) levels in H2O2-treated
Calu-6 cells at 1 h and the p38 inhibitor decreased ROS (DCF)
levels in these cells at 1 h. In addition, none of the MAPK
inhibitors significantly altered ROS (DCF) levels in
H2O2-treated lung cancer cells at 24 h.
However, all MAPK inhibitors, especially the JNK and p38 inhibitors
augmented DHE (O2•−) levels in
H2O2-treated Calu-6 and A549 cells at 24 h.
Thus, it is plausible that the enhancement of
H2O2-induced lung cancer cell death by MAPK
inhibitors, especially the JNK inhibitor, is more related to the
levels of O2•− (DHE) rather than ROS (DCF). Furthermore,
although the JNK and p38 inhibitors significantly increased ROS
(DCF) levels in Calu-6 control cells at 1 and 24 h, these
inhibitors did not significantly provoke cell death and MMP loss.
Additionally, each MAPK inhibitor had different effects on basal
ROS (DCF) and O2•− (DHE) levels in
H2O2-untreated Calu-6 and A549 control cells
regardless of cell death. Since changes in O2•− and
H2O2 levels by these MAPK inhibitors and the
outcomes of their corresponding signaling pathways influenced by
these types of ROS are complex in cells and different in each cell
type, the detailed molecular mechanisms underlying the effects of
MAPK inhibitors and their relationship between ROS and cell death
require further study.
GSH is an important tripeptide (non-protein)
antioxidant in cells. GSH content has a vital effect on cell death
(25–27). Similarly, H2O2
increased the percentage of GSH-depleted cells in both Calu-6 and
A549 lung cancer cells at 24 h. In addition, the JNK inhibitor
effects of enhancing cell death and MMP loss in
H2O2-treated lung cancer cells significantly
increased the number of GSH-depleted cells in these cells.
Additionally, the p38 inhibitor augmented GSH depletion in
H2O2-treated Calu-6 cells and the MEK
inhibitor enhanced the depletion in
H2O2-treated A549 cells. These results
supported the hypothesis that cell death effects are inversely
proportional to GSH content (26,28,29).
However, the MEK inhibitor did not increase GSH depletion in
H2O2-treated Calu-6 cells and the p38
inhibitor did not enhance GSH depletion in
H2O2-treated A549 cells. Therefore, the loss
of GSH content is necessary, but not sufficient for the accurate
prediction of cell death in lung cancer cells. JNK signaling, among
other MAPK signaling pathways, appeared to be more significant for
the relationship between cell death and GSH depletion.
H2O2 decreased CMF (GSH) levels in Calu-6
cells at 1 h, probably as a result of its use to scavenge
intracellular ROS. All MAPK inhibitors, especially the JNK and p38
inhibitors, increased GSH levels in
H2O2-treated and untreated Calu-6 cells at 1
h, indicating that these inhibitors positively preserved GSH
content in Calu-6 cells. However, 75 µM H2O2
increased CMF (GSH) levels in Calu-6 cells excluding GSH-depleted
cells at 24 h. It is likely that this increase was required to
reduce the intracellular ROS levels increased by
H2O2 to protect cells from immediate cell
death. The p38 inhibitor significantly diminished GSH levels in
H2O2-treated Calu-6 cells at 24 h. Both the
MEK and JNK inhibitors raised the basal levels of GSH in
H2O2-treated Calu-6 cells at 24 h. In
addition, 100 µM H2O2 did not significantly
alter GSH levels in A549 cells at 1 and 24 h. MEK and p38
inhibitors reduced GSH in H2O2-treated A549
cells at 1 h. All MAPK inhibitors, especially the p38 inhibitor,
reduced the basal levels of GSH in A549 control cells at 1 h. None
of the MAPK inhibitors influenced GSH levels in
H2O2-treated A549 cells at 24 h. However, the
MEK inhibitor increased basal GSH levels in A549 control cells at
24 h. Consequently, these results revealed that each MAPK signaling
pathway differently influences intracellular GSH levels
irrespective of GSH-depleted cells dependent on cell types,
incubation times and the presence or absence of
H2O2. The exact functions of each MAPK
inhibitor in H2O2-induced lung cell death
still need to be further defined with respect to changes in ROS and
GSH.
In conclusion, exogenous H2O2
induced growth inhibition and cell death in Calu-6 and A549 lung
cancer cells by increasing intracellular ROS and depleting GSH. All
MAPK inhibitors generally amplified
H2O2-induced lung cancer cell death.
Especially, the enhanced cell death and MMP loss effects of the JNK
inhibitor in H2O2-treated lung cancer cells
were related to increased O2•− (DHE) levels and GSH
depletion.
Acknowledgements
This study was supported by a grant from the
National Research Foundation of Korea (NRF) funded by the Korean
government (MSIP; 2016R1A2B4007773) and supported by the ‘Research
Base Construction Fund Support Program’ funded by Chonbuk National
University in 2017.
Glossary
Abbreviations
Abbreviations:
|
H2O2
|
hydrogen peroxide
|
|
ROS
|
reactive oxygen species
|
|
GSH
|
glutathione
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MEK
|
MAP kinase or ERK kinase
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
JNK
|
c-Jun N-terminal kinase
|
|
MMP (ΔΨm)
|
mitochondrial membrane potential
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-
diphenyltetrazolium bromide
|
|
FITC
|
fluorescein isothiocyanate
|
|
H2DCFDA
|
2′,7′-dichlorodihydrofluorescein
diacetate
|
|
DHE
|
dihydroethidium
|
|
CMFDA
|
5-chloromethylfluorescein
diacetate
|
|
PI
|
propidium iodide
|
References
|
1
|
Gonzalez C, Sanz-Alfayate G, Agapito MT,
Gomez-Niño A, Rocher A and Obeso A: Significance of ROS in oxygen
sensing in cell systems with sensitivity to physiological hypoxia.
Respir Physiol Neurobiol. 132:17–41. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baran CP, Zeigler MM, Tridandapani S and
Marsh CB: The role of ROS and RNS in regulating life and death of
blood monocytes. Curr Pharm Des. 10:855–866. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial ROS-induced ROS release: An update and review.
Biochim Biophys Acta. 1757:509–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zelko IN, Mariani TJ and Folz RJ:
Superoxide dismutase multigene family: A comparison of the CuZn-SOD
(SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures,
evolution, and expression. Free Radic Biol Med. 33:337–349. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wilcox CS: Reactive oxygen species: Roles
in blood pressure and kidney function. Curr Hypertens Rep.
4:160–166. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Blaser H, Dostert C, Mak TW and Brenner D:
TNF and ROS crosstalk in inflammation. Trends Cell Biol.
26:249–261. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Reuter S, Gupta SC, Chaturvedi MM and
Aggarwal BB: Oxidative stress, inflammation, and cancer: How are
they linked? Free Radic Biol Med. 49:1603–1616. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Genestra M: Oxyl radicals, redox-sensitive
signalling cascades and antioxidants. Cell Signal. 19:1807–1819.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kusuhara M, Takahashi E, Peterson TE, Abe
J, Ishida M, Han J, Ulevitch R and Berk BC: p38 Kinase is a
negative regulator of angiotensin II signal transduction in
vascular smooth muscle cells: Effects on Na+/H+ exchange and
ERK1/2. Circ Res. 83:824–831. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Blenis J: Signal transduction via the MAP
kinases: Proceed at your own RSK. Proc Natl Acad Sci USA. 90:pp.
5889–5892. 1993; View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hsin YH, Chen CF, Huang S, Shih TS, Lai PS
and Chueh PJ: The apoptotic effect of nanosilver is mediated by a
ROS- and JNK-dependent mechanism involving the mitochondrial
pathway in NIH3T3 cells. Toxicol Lett. 179:130–139. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mao X, Yu CR, Li WH and Li WX: Induction
of apoptosis by shikonin through a ROS/JNK-mediated process in
Bcr/Abl-positive chronic myelogenous leukemia (CML) cells. Cell
Res. 18:879–888. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guyton KZ, Liu Y, Gorospe M, Xu Q and
Holbrook NJ: Activation of mitogen-activated protein kinase by
H2O2. Role in cell survival following oxidant injury. J Biol Chem.
271:4138–4142. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Henson ES and Gibson SB: Surviving cell
death through epidermal growth factor (EGF) signal transduction
pathways: Implications for cancer therapy. Cell Signal.
18:2089–2097. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Latimer HR and Veal EA: Peroxiredoxins in
regulation of MAPK signalling pathways; Sensors and barriers to
signal transduction. Mol Cells. 39:40–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rhee SG, Kang SW, Jeong W, Chang TS, Yang
KS and Woo HA: Intracellular messenger function of hydrogen
peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol.
17:183–189. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vilhardt F and van Deurs B: The phagocyte
NADPH oxidase depends on cholesterol-enriched membrane microdomains
for assembly. EMBO J. 23:739–748. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hinz B, Phan SH, Thannickal VJ, Prunotto
M, Desmoulière A, Varga J, De Wever O, Mareel M and Gabbiani G:
Recent developments in myofibroblast biology: Paradigms for
connective tissue remodeling. Am J Pathol. 180:1340–1355. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Han YH and Park WH: The effects of MAPK
inhibitors on a proteasome inhibitor, MG132-induced HeLa cell death
in relation to reactive oxygen species and glutathione. Toxicol
Lett. 192:134–140. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Han YH and Park WH: Pyrogallol-induced
As4.1 juxtaglomerular cell death is attenuated by MAPK inhibitors
via preventing GSH depletion. Arch Toxicol. 84:631–640. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Han YH, Moon HJ, You BR and Park WH: The
effects of MAPK inhibitors on pyrogallol-treated Calu-6 lung cancer
cells in relation to cell growth, reactive oxygen species and
glutathione. Food Chem Toxicol. 48:271–276. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: Release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Park WH: The effect of MAPK inhibitors and
ROS modulators on cell growth and death of H2O2-treated HeLa cells.
Mol Med Rep. 8:557–564. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Campo ML, Kinnally KW and Tedeschi H: The
effect of antimycin A on mouse liver inner mitochondrial membrane
channel activity. J Biol Chem. 267:8123–8127. 1992.PubMed/NCBI
|
|
25
|
Han YH, Kim SH, Kim SZ and Park WH:
Carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) as an
O2(*-) generator induces apoptosis via the depletion of
intracellular GSH contents in Calu-6 cells. Lung Cancer.
63:201–209. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Estrela JM, Ortega A and Obrador E:
Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci.
43:143–181. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Han YH, Kim SZ, Kim SH and Park WH:
Intracellular GSH level is a factor in As4.1 juxtaglomerular cell
death by arsenic trioxide. J Cell Biochem. 104:995–1009. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Han YH, Kim SZ, Kim SH and Park WH:
Enhancement of arsenic trioxide-induced apoptosis in HeLa cells by
diethyldithiocarbamate or buthionine sulfoximine. Int J Oncol.
33:205–213. 2008.PubMed/NCBI
|
|
29
|
Han YH, Kim SZ, Kim SH and Park WH:
Suppression of arsenic trioxide-induced apoptosis in HeLa cells by
N-acetylcysteine. Mol Cells. 26:18–25. 2008.PubMed/NCBI
|